
Abstract
There exists a complex, dynamic interaction between mechanical ventilation and the splanchnic vasculature that contributes to a myriad of gastrointestinal tract complications that arise during critical illness. Positive pressure-induced splanchnic hypoperfusion appears to play a pivotal role in the pathogenesis of these complications, the most prevalent of which are stress-related mucosal damage, gastrointestinal hypomotility and diarrhea. Furthermore, characteristics of the splanchnic vasculature make the gastrointestinal tract vulnerable to adverse effects related to positive pressure ventilation. While most of these complications seen in mechanically ventilated patients are reflections of altered gastrointestinal physiology, some may be attributed to medical interventions instituted to treat critical illness.
Since maintenance of normal hemodynamics cannot always be achieved, pharmacologic prophylactic therapy has become a mainstay in the prevention of gastrointestinal complications in the intensive care unit. Improved understanding of the systemic effects of mechanical ventilation and greater application of lung-protective ventilatory strategies may potentially minimize positive pressure-induced reductions in splanchnic perfusion, systemic cytokine release and, consequently, reduce the incidence of gastrointestinal complications associated with mechanical ventilation. Herein, we discuss the pathophysiology of gastrointestinal complications associated with mechanical ventilation, summarize the most prevalent complications and focus on preventive strategies and available treatment options for these complications.
The most common causes of gastrointestinal hemorrhage in mechanically ventilated patients are bleeding from stress-related mucosal damage and erosive esophagitis. In general, histamine H2 receptor antagonists and proton pump inhibitors prevent stress-related mucosal disease by raising the gastric fluid pH. Proton pump inhibitors tend to provide more consistent pH control than histamine H2 receptor antagonists. There is no consensus on the drug of choice for stress ulcer prophylaxis with several meta-analyses providing conflicting results on the superiority of any medication. Prevention of erosive esophagitis include careful use of nasogastric tubes and institution of strategies that improve gastric emptying. Many mechanically ventilated patients have gastrointestinal hypomotility and diarrhea. Treatment options for gastrointestinal motility are limited, thus, preventive measures such as correction of electrolyte abnormalities and avoidance of medications that impair gastrointestinal motility are crucial. Treatment of diarrhea depends on the underlying cause. When associated with Clostridium difficile infection antibacterial therapy should be discontinued, if possible, and treatment with oral metronidazole should be initiated.
More studies are warranted to better understand the systemic effects of mechanical ventilation on the gastrointestinal tract and to investigate the impact of lung protective ventilatory strategies on gastrointestinal complications.
Similar content being viewed by others


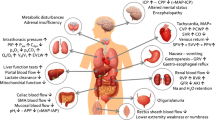
The outcome of critically ill patients depends not only on the treatment of underlying disease, but also on prevention and treatment of complications that occur in the intensive care unit (ICU). The characteristics of the splanchnic vasculature place the gastrointestinal (GI) tract at particularly high risk for complications in critically ill patients.[1] Figure 1 summarizes the possible GI complications that may be encountered in critically ill patients. While most of these complications are reflections of altered GI physiology, some may be attributed to medical interventions instituted to treat critical illness.
Mechanical ventilation, although life saving, is associated with a number of complications that may hinder recovery from critical illness.[1] The complexity of interactions between critical illness and mechanical ventilation, and their effects on the GI tract necessitate better understanding of the pathogenesis of GI complications. It is conceivable that critical illness ‘primes’ the GI tract thereby creating an environment conducive to the development of mechanical ventilation-induced complications. In this article, we discuss the pathophysiology of GI complications associated with mechanical ventilation and focus on preventive strategies and available treatment options for these complications.
1. Pathophysiology
Current knowledge about the influence of mechanical ventilation on the GI tract is limited. While some GI complications may be directly attributable to mechanical ventilation, most occur as a consequence of the underlying pathophysiologic changes associated with critical illness.[2] The complexity and heterogeneity of critical illness makes it impossible to specifically implicate mechanical ventilation as being the immediate cause of GI complications. Nevertheless, as mechanical ventilation can contribute to the pathogenesis of GI complications in much the same way as critical illness, it is reasonable to conclude that mechanical ventilation may potentiate the adverse effects of an underlying critical illness and worsen GI physiology.
1.1 Splanchnic Hypoperfusion
Impaired splanchnic perfusion plays a pivotal role in the pathogenesis of GI complications associated with mechanical ventilation (figure 2). The splanchnic vasculature lacks vasomotor autoregulation, which leads to persistent vasoconstriction following resolution of hemodynamic instability. The vascular architecture of the gut mucosa is similar to the renal medulla in permitting oxygen shunting which, under normal conditions, can cause hypoxia at the tips of villi.[3,4] Additionally, gut mucosal oxygen content is low due to a hematocrit of approximately 10% that results from dilutional effects of absorbed fluid and nutrients.[5] All of these factors place the GI tract at increased risk of ischemic events.

Organ-specific gastrointestinal (GI) complications in critically ill patients on mechanical ventilation. Both hollow and solid organs in the GI system can be affected by mechanical ventilation. CIRC=critical illness-related colonic hypomotility

Proposed mechanisms for the development of gastrointestinal (GI) complications associated with mechanical ventilation. Similar to underlying critical illness, mechanical ventilation can contribute to the pathogenesis of GI complications by altering splanchnic blood flow and leading to increased release of proinflammatory mediators. MODS = multiple organ dysfunction syndrome; PEEP=positive end-expiratory pressure; SIRS=systemic inflammatory response syndrome; SNS=sympathetic nervous system; Vt=tidal volume; ↑=increase
Mechanical ventilation may lead to splanchnic hypoperfusion by decreasing mean arterial pressure. Mechanical ventilation, especially with high levels of positive end-expiratory pressure (PEEP), decreases venous return by reducing the pressure gradient between systemic veins and the right atrium. Reduced preload results in decreased cardiac output and hypotension, particularly in patients with hypovolemia and impaired venoconstriction. Mesenteric blood flow parallels this reduction in cardiac output. PEEP-induced reductions in mesenteric blood flow can be corrected by restoration of cardiac output following fluid resuscitation.[6]
Another mechanism by which mechanical ventilation may affect splanchnic perfusion is a PEEP-induced increase in intra-abdominal pressure as a result of diaphragmatic descent. Intra-abdominal pressure is normally subatmospheric to 0mm Hg. In animals, gradual increases in intra-abdominal pressure up to 40mm Hg decrease blood flow in all organs in the GI tract.[7] In another animal study, both mesenteric and mucosal blood flow and intramucosal pH (pHi) diminished once intra-abdominal pressure reached 20mm Hg.[8] These reductions in intestinal mucosal blood flow parallel the rise in intra-abdominal pressure. Conceivably, a PEEP-induced increase in intra-abdominal pressure may lead to similar reductions of splanchnic perfusion. It is noteworthy that PEEP-related rises in intra-abdominal pressure may offset the negative effect of PEEP on venous return and cardiac output.[9]
In addition to reduced mean arterial pressure and PEEP-induced increase in intra-abdominal pressure, mechanical ventilation may adversely affect GI perfusion by reflex vasoconstriction of GI vasculature. Positive pressure ventilation is associated with elevated plasma renin-angiotensin-aldosterone activity and increased serum catecholamine levels, as a result of sympathetic activation.[10,11] These neurohormonal alterations contribute to splanchnic hypoperfusion by causing vasoconstriction and redistribution of blood away from the splanchnic vascular bed.[12,13] Whether due to decreased cardiac output and/or increased vascular resistance, splanchnic hypoperfusion produces an imbalance between oxygen supply and demand that may contribute to mucosal damage (e.g. stress ulcer) and/or hypomotility (e.g. ileus).[12,13] Perhaps more concerning than splanchnic hypoperfusion is reperfusion injury to GI epithelial cells that may occur after restoration of blood flow.[14] Repetitive episodes of hypoperfusion followed by reperfusion may contribute to acute nonocclusive mesenteric ischemia in the critical care setting.[15]
1.2 Increased Cytokine Release and Bacterial Translocation
The influence of positive pressure ventilation on the GI tract is not limited to reduced splanchnic blood flow due to impairment of cardiac output. Mechanical ventilation with ’injurious’ ventilatory strategies (i.e. high end-inspiratory lung volumes) may also play an important role in the pathogenesis of GI complications. High tidal volume ventilation leads to an increased release of cytokines, which may amplify inflammatory responses and contribute to the development of multiple organ dysfunction syndrome (MODS).[16] Cytokines may indirectly contribute to splanchnic hypoperfusion and directly impair intestinal smooth muscle function.[12,17,18]
Both high-peak airway pressures and lack of PEEP have been shown to cause translocation of bacteria from the alveolar space into the bloodstream in animals, providing another mechanism by which mechanical ventilation can produce systemic manifestations.[19] Growing evidence regarding increased cytokine release during ‘injurious’ ventilatory strategies suggests a potentially critical role for mechanical ventilation in the initiation and propagation of a systemic inflammatory response syndrome (SIRS) that may include dysfunction and damage to the GI tract as a part of MODS.[20]
1.3 Impaired Gut Barrier Function
Gut barrier function can be affected by reduced splanchnic blood flow and increased proinflammatory mediators, independent of mechanical ventilation.[5] Gut barrier function is dependent on the function of mucosal cells and intracellular junctions, mucus production, gut-associated lymphoid tissue and secretory immunoglobulin (Ig)A production, all of which may be impaired during critical illness.[21] Other consequences of critical illness, such as malnutrition and altered intestinal microflora, may also threaten GI epithelial cells. Moreover, impaired barrier function may allow the passage of proinflammatory mediators (e.g. endotoxin), and possibly micro-organisms, from the intestinal lumen to the bloodstream.[14] This process can become self-sustaining if the underlying disease that initiates the cascade is not controlled.
1.4 Medications
Medications used to facilitate mechanical ventilation, such as opioids and benzodiazepines, may decrease GI motility and impair venous return via venodilation and/or depressed responsiveness to catecholamines. Opioids slow gastric emptying and cause intestinal hypomotility in critically ill patients.[22,23] Hypotension associated with propofol infusions is dose related and occurs more frequently after bolus dosing. Propofol may lead to hypertriglyceridemia, particularly at high doses or with long-term use,[24,25] and may result in the elevation of pancreatic enzymes.[26,27] Other frequently used medications that may adversely affect the GI tract include vasopressors (particularly dopamine), antibacterials, and additives in oral medications (e.g. sorbitol)
2. Gastrointestinal (GI) Complications in Mechanically Ventilated Patients
2.1 GI Hemorrhage
Acute respiratory failure requiring mechanical ventilation for longer than 48 hours is one of the two strongest independent risk factors for clinically significant GI bleeding in the ICU.[28,29] The most common causes of GI hemorrhage in mechanically ventilated patients are bleeding from stress-related mucosal damage (SRMD) and erosive esophagitis.
Mechanically ventilated patients almost invariably develop SRMD and subepithelial hemorrhage within 24 hours of admission to ICU.[30–32] SRMD occurs within a few hours of critical illness; SRMD can present as lesions ranging from subepithelial petechiae to superficial erosions and can progress into true ulcers. These lesions are usually multiple and occur predominantly in the fundus of stomach, typically sparing the antrum.[32] The mucosa distal to the fundus (antrum and duodenum) can also be affected, although these lesions typically appear later, tend to be deeper, and may be associated with a higher incidence of bleeding.[31,33]
The majority of lesions associated with SRMD are asymptomatic and clinically insignificant (figure 3). Patients with clinically evident bleeding present with frank hemorrhages in the form of hematemesis, melena, coffee-ground material or hematochezia. Clinically significant or life-threatening bleeding is defined as bleeding that causes hemodynamic changes or necessitates transfusion.[34] Mechanically ventilated patients who develop clinically significant bleeding generally do so within the first 2 weeks of their ICU stay.[35]

Features of stress ulcer bleeding in mechanically ventilated patients.The majority of critically ill patients with stress ulcer are asymptomatic. Clinically evident bleeding is found in up to 25% of patients who do not receive prophylactic therapy. Clinically significant bleeding occurs in fewer than 5% of patients. IV=intravenous; PPI=proton pump inhibitor
Clinically evident bleeding due to SRMD occurs in up to 25% of critically ill patients who do not receive prophylactic therapy.[29,32,36] Of those, approximately 20% (corresponding to ∼5% of all patients) are clinically significant in that they require intervention and/or transfusion.[29,37–39] Not surprisingly, clinically significant stress ulcer bleeding is associated with an increased length of ICU stay (up to 11 more days), morbidity and mortality.[28,29,35]
Erosive esophagitis occurs in almost 50% of mechanically ventilated patients and accounts for 25% of all upper GI bleeding in ICU patients.[40] The etiopathogenesis of esophageal injury in critically ill patients is multifactorial and includes the use of nasogastric tubes, gastroesophageal reflux and duodenogastroesophageal (bile) reflux.[41,42] Nasogastric tubes cause mechanical irritation and interfere with normal esophageal motility and sphincter function leading to a higher incidence of gastroesophageal reflux.[41,42] It is noteworthy that gastroesophageal reflux occurs independent of body position and is not influenced by the size of the nasogastric tubes.[43] The standard use of stress ulcer prophylaxis makes acid-induced mucosal damage a less likely explanation,[41,44] suggesting that erosive esophagitis probably results from bile-induced damage from duodenogastroesophageal reflux, combined with direct trauma caused by nasogastric tubes.[40] The severity of esophagitis correlates with the volume of residual gastric contents. Gastric colonization with bacteria, which alters bile composition and increases the percentage of injurious unconjugated bile, may contribute to esophageal damage.
2.2 Non-Hemorrhagic Complications
2.2.1 GI Hypomotility
Many mechanically ventilated patients have abnormal upper GI motility[45] that affects the stomach more than the duodenum. Although dysfunction of antral Cajal cells, which regulate gastric contractility, has been proposed as the mechanism responsible for gastric dysmotility,[22,23] it is more likely due to a complex interaction between local enteric afferent neurotransmitter imbalance, autonomous (especially vagal) dysfunction, and abnormal central processing.
In general, patients with hypomotility present with decreased bowel sounds or abdominal distention[46] (table I). In one study, many ICU patients experienced some degree of intolerance to enteral feeding, manifested as high gastric residuals (39%) and constipation (16%).[47] Furthermore, abnormal peristalsis favors the development of duodenogastric reflux and subsequent colonization of the stomach by Enterobacteriacea species.

Motility-related gastrointestinal complications in mechanically ventilated patients
Other less common, but more challenging, clinical problems include critical illness-related colonic hypomotility (CIRC) and Ogilvie’s syndrome.[50] Both are characterized by the absence of defecation in combination with colonic distension in Ogilvie’s syndrome or no physical or radiographic abnormalities in CIRC. Patients with CIRC lack colonic prokinetic movements, whereas the upper GI tract functions properly. It has been suggested that Ogilvie’s syndrome is preceded by CIRC, or may be a variant of CIRC.
2.2.2 Diarrhea
Diarrhea affects up to half of critically ill patients, and those with acute respiratory failure appear to be particularly at risk.[46,51] The etiology of diarrhea in the ICU is multifactorial and, as such, is probably a reflection of the severity of the underlying illness and gut dysmotility (table II).

Common causes of diarrhea in mechanically ventilated patients
Diarrhea associated with enteral feeding affects up to 25% of ICU patients.[52,53] The incidence has been shown to be higher in patients who receive infusion rates >50 mL/hour.[52] Contrary to the common perception, the impact of formula osmolality on the incidence of diarrhea remains uncertain. Liberal use of antibacterials predisposes ICU patients to diarrhea, which accounts for 20–50% of all cases of nosocomial diarrhea.[54] Broad-spectrum antibacterials alter colonic bacteria, thereby altering the fermentation of enterally administered carbohydrates to nonabsorbable metabolites, which cause a malabsorption syndrome and osmotic diarrhea. Almost 40% of patients receiving antibacterials develop antibiotic-associated diarrhea,[55] 15–25% of which is attributable to Clostridium difficile toxin. Unlike other forms of diarrhea, C. difficile is associated with significant morbidity and even mortality if toxic megacolon develops.[56] The number and duration of use of antibacterials are determinants for diarrhea associated with C. difficile infection. Diagnosis of C. difficile diarrhea requires a high index of suspicion and is typically made by detection of cytotoxins in the stool, although tissue culture toxin assays remain the gold standard. Newer rapid enzyme immunoassays can detect C. difficile with fair sensitivity (69–87%) and good specificity (99–100%).[57] While there are no guidelines as to how many assays are needed, the stool should be checked for toxin at least twice before C. difficile can be excluded. Importantly, subsequent episodes of diarrhea must be evaluated for C. difficile-associated diarrhea in the same manner and with the same vigilance as initial episodes.
Relative luminal excess of bile acids[58] as a result of decreased absorption related to atrophy of the terminal ileum following starvation (as early as 4 days)[59] and hypoalbuminemia also contribute to diarrhea in the ICU.[60,61] Although the precise role of hypoalbuminemia as a risk factor has been challenged,[58] it can lead to gut edema and impaired nutrient absorption. Albumin levels less than 2.6 g/dL have been associated with an increased risk of diarrhea[60] although chronicity, rather than severity, of hypoalbuminemia is a more important contributor to diarrhea.
2.2.3 Other Complications
Mechanical ventilation not only affects hollow organs of the GI tract, but can also impact on solid organs, such as the liver, pancreas and gallbladder. It is noteworthy that most available evidence regarding the effects of positive pressure ventilation on solid organs comes from animal studies and has been extrapolated to humans.
Pancreas
In animals, PEEP may decrease blood flow to the pancreas and stomach to a greater extent than to the intestines. Furthermore, hemodynamic consequences of PEEP in the pancreas can persist despite the maintenance of mean arterial pressure.[62] In animals, high levels of PEEP (>15cm H2O) cause pancreatitis, particularly when the pancreas is stimulated.[63] Whether mechanical ventilation alone may cause elevation in pancreatic enzymes and pancreatitis in humans is unknown. However, these concerns, while theoretic, are worthy of consideration in critically ill patients with otherwise unexplained pancreatitis.
Liver
Positive pressure ventilation with PEEP may reduce both portal venous and hepatic arterial blood flow along with hepatic venous oxygen saturation.[64–67] These effects occur in parallel with changes in cardiac output. Improvement of cardiac output improves hepatic blood flow[65,66] similar to that seen with enteral feeding.[68] PEEP also elevates both portal and hepatic venous pressures and causes hepatic congestion in animals.[66,69] This has been speculated to be due to increased portal transmural pressure as a result of a rise in hepatic venous pressure. Positive pressure ventilation also mediates its adverse effects on portal blood flow by raising intrathoracic venous pressure,[64] increasing hepatic sinusoidal resistance via mechanical compression of the liver by the descending diaphragm[65,67] and diminishing mesenteric blood flow.[64]
Collectively, a mismatch between hepatic metabolic demand and PEEP-induced impairment in blood and oxygen supply can result in abnormal liver function. In patients with septic shock, PEEP impairs gluconeogenesis (a marker for hepatic metabolic performance) in parallel to reductions in cardiac output and hepatic venous oxygen saturation.[70] Conceivably, high PEEP may impair the clearance of drugs that are hepatically metabolized. In view of current evidence, it is reasonable to hypothesize that PEEP contributes to liver dysfunction, especially in the presence of hypoxemia, hypotension or any other condition that compromises hepatic oxygen supply.
Gallbladder
Acute acalculous cholecystitis is an insidious complication that affects ∼1% (0.2–3%) of ICU patients.[71,72] The pathogenesis is most likely multifactorial, involving both ischemic and chemical (biliary) injuries to the gallbladder epithelium. Mechanical ventilation as well as shock, sepsis, multiple transfusions, dehydration, prolonged enteral fasting, total parenteral nutrition and medications (e.g. sedatives and opiates) have been implicated in the pathogenesis of acute acalculous cholecystitis. Prolonged fasting and resultant biliary sludge are known risk factors for acalculous cholecystitis,[73] and are frequently encountered in critically ill patients. Biliary sludge occurs as a result of decreased cholecystokinin (CCK)-mediated gallbladder emptying (which normally occurs several times a day) and stagnation of concentrated bile.[74] Mechanical ventilation may affect the gallbladder epithelium both directly, by causing hypoperfusion and ischemia of the wall, and indirectly, by leading to poor contractility with consequent biliary stasis and sludge formation similar to prolonged fasting.[75] Motility changes can be detected as early as 24 hours after admission to the ICU. For the same duration of starvation following major abdominal surgery, patients requiring mechanical ventilation had a higher degree of gallbladder atony compared with those who were spontaneously breathing. Early diagnosis of acalculous cholecystitis is crucial because of the high morbidity and mortality (up to 50%) associated with this condition.[76]
3. Prevention and Treatment
Splanchnic hypoperfusion appears to play a key role in the pathogenesis of many GI complications associated with mechanical ventilation. Thus, maintenance of normal hemodynamics, and splanchnic blood flow and oxygen delivery are crucial in the prevention of these complications. In fact, early aggressive hemodynamic support decreases the incidence of bleeding from stress-related mucosal damage to negligible levels.[39] In addition to early hemodynamic stabilization, advances in ICU care and use of prophylaxis for SRMD have resulted in a decline in the incidence of SRMD-associated bleeding in the last decade.[77,78]
The impact on the GI tract of protective ventilatory strategies with low tidal volume ventilation has yet to be reported. However, in light of current knowledge regarding the probable association between ‘injuriousrs ventilatory strategies (causing volutrauma and biotrauma) and SIRS/MODS, the avoidance of high tidal volumes and the use of optimal PEEP levels to minimize repetitive opening and collapse of alveoli may prove beneficial and should be part of a strategy for the prevention of GI complications.
3.1 GI Hemorrhage
3.1.1 Stress ulcer prophylaxis
SRMD occurs when the injurious effects of gastric acid overwhelm the protective and reparative mucosal defense mechanisms that are impaired by the ischemia that frequently occurs during critical illness and mechanical ventilation[37,79,80] (figure 4). Mucosal ischemia decreases the capacity to neutralize hydrogen ions and contributes to intramural acidosis, cell death and ulceration.[81,82] Ischemia also compromises gastric energy utilization and impairs protective processes (e.g. mucus production), especially in the fundus, where most SRMD develops.[37,82] Although luminal hyperacidity is not required, gastric acid is an essential factor in the pathogenesis of SRMD.[30,83] Therefore, it is not surprising that therapies targeting gastric acid or improving mucosal defense have become the mainstay of prevention (table III).

Proposed mechanisms for the development of stress-related mucosal damage (SRMD) in mechanically ventilated patients. SRMD occurs when the harmful effects of gastric acid overwhelm the protective and reparative mechanisms of the mucosal defense mechanisms which are impaired due to local mucosal ischemia that frequently occurs during critical illness and mechanical ventilation. GI=gastrointestinal; PG=prostaglandin; ↓=decrease; ↑= increase

Medications used for stress ulcer prophylaxis in mechanically ventilated patients (reproduced from Mutlu et al.,[2 with permission)
Pharmacologic Approaches
Medications that target gastric acid secretion, such as histamine H2-receptor antagonists and proton pump inhibitors, prevent SRMD by raising the gastric fluid pH (ideally above 4.0) and reducing the diffusion of hydrogen ions back into the mucosa. Histamine H2 receptor antagonists provide prophylaxis against SRMD by inhibiting gastric acid secretion. The major concern with the use of histamine H2 receptor antagonists is tolerance or tachyphylaxis. Continuous administration of these agents may provide more effective acid inhibition compared with intermittent dosing, but the relevance of this practice remains unclear.[84] Although routine measurements of gastric pH (especially within the first 24 hours) have been recommended when histamine H2 receptor antagonists are used, until now no studies have proved the superiority of pH-adjusted dosing over standard regimens.
Proton pump inhibitors provide more consistent pH control than histamine H2 receptor antagonists. Compared with oral proton pump inhibitors, intravenous (IV) forms have the advantages of ease of administration, increased drug delivery to parietal cells and more rapid onset of action. Newer proton pump inhibitors are also devoid of the unwanted adverse effects on cytochrome P450, making them attractive alternatives for stress ulcer prophylaxis. In critically ill patients, IV administration of proton pump inhibitors, achieves and maintains gastric pH ≥4 within hours of initiation of therapy, with a progressive increase of pH within the first 48 hours.[85,86] On the other hand, continuous IV administration of histamine H2-receptor antagonists is unable to maintain pH control by day two, despite achieving a gastric pH ≥4 initially. Until now, no large-scale study has specifically investigated the role of IV proton pump inhibitors in SRMD prophylaxis or whether more consistent increases in pH translate into better outcomes. Thus, the growing use of proton pump inhibitors in the ICU is without strong clinical data to support this practice in every critically ill patient.
As in the case with histamine H2 receptor antagonists, antacids neutralize gastric acid in a dose-dependent fashion. Other beneficial effects include bile acid binding and increased mucosal prostaglandin production (particularly with antacids containing aluminum hydroxide). Frequent administration and pH monitoring render the use of antacids cumbersome and, consequently, antacids have become historical footnotes in stress ulcer prophylaxis in most ICUs.
Pirenzepine is another pH-altering drug that acts via activation of muscarinic (M1) receptors and has been successfully used for stress ulcer prophylaxis in Europe. Other preventive strategies (e.g. sucralfate, misoprostol) provide cytoprotection via augmentation of mucosal defensive mechanisms and normalization of gastric mucosal microcirculation.[87]
Concerns About Prophylactic Therapy
Short-term use of either pH-altering or cytoprotective medications is relatively safe (incidence of adverse effects <1%).[88] The major concern with the use of pH-altering medications is their association with gastric colonization with Enterobacteriacea (due to increased gastric pH) and subsequent retrograde gastro-oropharyngeal contamination leading to an increased risk of ventilator-associated pneumonia (VAP).[89–91] When all studies that evaluated sequential colonization from stomach to trachea were considered, gastric colonization preceded tracheal colonization in 4–24% and VAP in 0–15% of patients.[92] Acidification of enteral feeding reduces the incidence of gastric colonization significantly, but not VAP.[93] Earlier reports[94–96] showing a lower incidence of VAP with sucralfate have been challenged by studies that reported only a trend[34,38] or no difference.[97–100] This lack of consensus can be attributed to the differences in study design, gastric pH measurement methods, medication dosage, timing of VAP (early vs late), gastric volume, simultaneous administration of enteral feeding and, more importantly, body position (supine vs semirecumbent), which may predispose an individual to gastro-oropharyngeal colonization. Based on available data, the risk of VAP attributable to pH-altering drugs can be minimized if clinicians carry out preventive measures, including keeping the patient in a semi-recumbent position, avoiding high gastric residuals and administering the enteral feeds into the small bowel whenever possible.
Supine positioning is an independent predictor of VAP, thus proper positioning may be more important than gastric pH.[101,102] Semi-recumbent positioning has been strongly recommended for the prevention of nosocomial pneumonia.[103] Thus, until the contribution of positional effects on reflux is determined, no firm conclusions can be made regarding whether gastric colonization leads to VAP.
Choice of Medication
There is no consensus on the drug of choice for stress ulcer prophylaxis. Several meta-analyses provide conflicting results on the superiority of any medication.[34,104–108] Discrepancies result from methodological problems in trials, inclusion of non-randomized studies, and differences in evaluated endpoints (asymptomatic versus clinically evident versus important bleeding). Nevertheless, both pH-altering agents (histamine H2 receptor antagonists, proton pump inhibitors [enteral form] and antacids) and sucralfate decrease the incidence of clinically significant bleeding by approximately 50% (from ∼4% to 1.7–2%) and are effective in the prevention of clinically evident as well as significant stress ulcer bleeding.[34,107] Limited studies on combination therapy suggest better pH control, but no additional benefit in clinical outcomes compared with single agent therapy.[109,110]
Most deaths in patients with stress ulcer bleeding do not result from GI hemorrhage. Thus, the contribution of stress ulcer bleeding to overall ICU mortality does not appear to be significant in unselected ICU populations and routine prophylaxis in all patients is not warranted. Identification of patients at risk of stress ulcer bleeding appears to be more important than the particular medication used and can reduce unnecessary medication use and cost. Respiratory failure requiring mechanical ventilation for more than 48 hours and coagulopathy (defined as a platelet count of <50000mm3, an international normalized ratio of >1.5, or a partial thromboplastin time of more than twice the control value) are the two most important risk factors for stress ulcer bleeding. On the contrary, when neither of these risk factors are present, the incidence of clinically significant stress ulcer bleeding is negligible (0.1%).[28] Among mechanically ventilated patients, those who develop organ dysfunction (particularly renal failure) at any time during the ICU stay appear to be especially at high risk of stress ulcer bleeding.[35] Additional risk factors for which stress ulcer prophylaxis should be considered include sepsis, hypotension, hepatic failure, renal failure, major trauma, extensive burns and intracranial hypertension.
A functional GI tract is important in choosing regimens for ulcer prophylaxis (figure 5). When the enteral route is available, histamine H2 receptor antagonists or sucralfate can be administered for the prevention of SRMD. Oral proton pump inhibitors should be considered as a second-line agent because of their lack of superiority over histamine H2 receptor antagonists and difficulties encountered with their administration (i.e. clogging of enteral tubes). When the GI tract is not an option, intravenous histamine H2 receptor antagonists should be used as a first-line agent. Routine use of proton pump inhibitors administered IV is not justified because there is a lack of evidence showing their superiority over histamine H2 receptor antagonists in stress ulcer prophylaxis, and also because of their higher costs and the low incidence of clinically significant bleeding with aggressive hemodynamic support. Therefore, their use should be reserved for patients who have active GI bleeding prior to the development of critical illness. Proton pump inhibitors may also be used in patients in whom histamine H2 receptor antagonists are contraindicated (e.g. severe thrombocytopenia, undesired drug interactions) or are ineffective (e.g. history of GI bleeding with histamine H2-receptor antagonists or sucralfate). We believe that proton pump inhibitors administered IV should also be considered for patients with multiple risk factors for stress ulcer bleeding, such as coagulopathy, renal failure, or prolonged shock in addition to respiratory failure requiring mechanical ventilation. Patients with high gastric acid production (i.e. head trauma patients) may also potentially benefit from IV proton pump inhibitors. Among patients with visible vessels that are nonbleeding or adherent clots, who do not undergo endoscopic therapy, acid inhibition with proton pump inhibitors significantly reduces the rebleeding rate and the need for surgery.[111] Similarly, even after endoscopic therapy, proton pump inhibitors provide a beneficial effect on hemostasis, which is a pH-dependent process.[112] Platelet aggregation and plasma coagulation are nearly abolished below a pH of 5.4 and fibrin clots may dissolve when the pH falls below 4.0. Therefore, in clinically evident or significant stress ulcer bleeding, proton pump inhibitors administered IV are preferable to histamine H2-receptor antagonists as they maintain a more reliable pH control, which is essential for local hemostatic mechanisms.

Stress ulcer prophylaxis in mechanically ventilated patients. Choice of medication depends on the availability of the enteral route. When IV medications are considered, PPIs should be reserved for patients with active gastrointestinal bleeding and for those with the highest risk for hemorrhage. H2RA=histamine H2 receptor antagonist; IV=intravenous; PO=oral; PPI=proton pump inhibitor
Enteral Feeding
Enteral feeding may also decrease the risk of clinically evident GI bleeding.[35,48,49] The beneficial effects of enteral feeding are probably multifactorial, including dilutional alkalinization of gastric fluid and mucosal cytoprotection (by restoration of gastric epithelial energy stores).[35,113] The latter is a more likely, but unproven, explanation since the alkalizing effects of enteral feeding are variable and parenteral nutrition is also associated with reduced stress ulcer bleeding.[113] In a multicenter trial, histamine H2 receptor antagonists provided stress ulcer prophylaxis regardless of whether or not patients were receiving enteral nutrition.[35] Furthermore, in another study, patients who were receiving only enteral nutrition for stress ulcer prophylaxis had a higher incidence of stress ulcer bleeding compared with those who were receiving prophylaxis with histamine H2 receptor antagonists and/or antacids.[36] All studies that investigated the impact of enteral nutrition on stress ulcer prophylaxis were limited in several aspects, including differences in study designs, patient populations, type and amount of enteral formula administered, and lack of definition of stress ulcer bleeding. Therefore, in view of the limited data, the use of enteral nutrition as the only therapy for stress ulcer prophylaxis should be discouraged until more definitive studies comparing enteral nutrition with pharmacologic prevention are available. Initiation and discontinuation of pharmacologic prophylaxis should be independent of enteral nutrition.
3.1.2 Erosive Esophagitis
Prevention of erosive esophagitis requires maintenance of semi-recumbent positioning, judicious use of nasogastric tubes and the institution of strategies that improve gastric emptying and prevent gastroesophageal and duodenogastric reflux (e.g. metoclo-pramide).
3.2 Non-Hemorrhagic Complications
3.2.1 GI Hypomotility
Treatment options for GI hypomotility are limited; thus, preventive measures such as correction of electrolyte abnormalities (e.g. hypokalemia) and avoidance of medications that impair GI motility (i.e. opiates) are crucial.[22] β-Adrenoceptor stimulation with catecholamines delays orocecal and duodenocecal transit time.[114] A β-adrenoceptor agonist, isoproterenol (isoprenaline), inhibits contractility in the antrum and small intestine.[115] On the contrary, β-adrenoceptor blockade with atenonol or propranolol hastens both transit times.[114] In addition to β-adrenergic agonists and dopamine that may cause GI hypomotility at doses as low as 5 μg/kg/min, phenothiazines, diltiazem, verapamil, and drugs with anticholinergic adverse effects should also be avoided if possible.[116] If necessary, nasogastric suction and/or rectal tubes and, in intractable cases, colonoscopy, can be used to decompress the GI tract if significant dilation develops. Rectal tubes have been associated with complications, including discomfort, local ulceration, infection and perforation of the rectum, and should not be used unless necessary.
Prokinetic agents can be considered once mechanical obstruction is excluded (table IV). Withdrawal of cisapride from the US market two years ago has limited the treatment options to metoclopramide and erythromycin. The precise mechanism of action of metoclopramide is unclear, but it improves antroduodenal coordination and reverses the inhibitory effects of dopamine on GI motility. Similar improvement is achieved with erythromycin 200mg once daily via its action on motilin receptors.[117,118] It is noteworthy that an intact vagal pathway is necessary for erythromycin’s beneficial effects on the GI tract.[119] Although not proven, the use of erythromycin could conceivably contribute to overgrowth of resistant bacteria (due to subinhibitory concentrations of antibacterials that may promote bacterial resistance). Prucalopride is a novel enterokinetic that enhances gastric, small bowel and colonic motility via its excitatory effect on serotonin 5-HT4 receptors. It may prove useful in improving GI hypomotility in critically ill patients.[120]

Treatment options for gastrointestinal hypomotility in mechanically ventilated patients
Domperidone, a peripheral type 2 dopamine receptor antagonist, regulates the motility of gastric and small intestinal smooth muscle with some effects on the motor function of the esophagus.[121] It has been shown to increase the duration of antral and duodenal contractions and increase gastric emptying. It also has antiemetic activity as a result of blockade of dopamine D2 receptors in the chemoreceptor trigger zone of the brainstem. Because only a small amount of domperidone crosses the blood-brain barrier, adverse effects in the CNS (i.e. extrapyramidal adverse effects) are rare. Domperidone is not available in the US, but can be obtained in Canada or Mexico.
Bethanechol is a synthetic parasympathomimetic that acts directly on muscarinic acetylcholinergic receptors. Stimulation of muscarinic receptors restores GI peristalsis, increases motility, and increases the resting lower esophageal sphincter pressure. However, its use is significantly limited by cholinergic adverse effects.[1]
Neostigmine has been successfully used as a therapeutic tool for CIRC and Ogilvie’s syndrome.[50,122] Major concerns regarding the use of neostigmine include bradycardia, increased airway secretions and bronchial reactivity. Concomitant treatment with neostigmine and the anticholinergic agent glycopyrrolate has been reported to diminish the central cholinergic effects of neostigmine without diminishing its effect on colonic motility.[123]
3.2.2 Diarrhea
Treatment of diarrhea depends on the underlying cause. The inability to identify the exact cause often complicates the picture and limits care. Clostridium difficile should always be considered in the differential diagnosis. When diarrhea is present, antibacterial therapy should be discontinued, if possible, otherwise oral metronidazole should be initiated. Oral vancomycin is effective, but should be reserved for patients who cannot tolerate or do not respond to metronidazole or for those who are in the first 20 weeks of pregnancy. Diarrhea associated with enteral feeding is generally self-limited and subsides with a reduction in feeding rate. If possible, hyperosmolar formulas should be replaced with isotonic tube feedings. Neither routine use of peptide-based enteral formulas nor the addition of fiber offer any benefit over standard formulas (with whole protein) in terms of reducing the incidence of diarrhea.[124–128] Nonetheless, peptide-based enteral formulas may be considered in patients with severe hypoalbuminemia (albumin <2.6 g/dL) and diarrhea.[124–126]
3.2.3 Acalculous Cholecystitis
The only strategy that has been proposed in prevention of acalculous cholecystitis is daily stimulation of gallbladder contraction with IV CCK, which has been shown to be effective in patients receiving total parenteral nutrition.[129] Because prolonged fasting and resultant biliary sludge are known risk factors for acalculous cholecystitis,[73] we believe that early enteral nutrition and maintenance of normal hemodynamics are important in the prevention of acalculous cholecystitis. Although CCK may potentially be beneficial in critically ill patients, particularly those who cannot be fed enterally, its efficacy and cost effectiveness have not been well established.
Early diagnosis is critical in preventing the high morbidity and mortality (up to 50%) associated with acalculous cholecystitis.[76] The diagnosis may often be unrecognized due to the complexity of underlying medical and surgical problems and the lack of reproducible signs and biochemical parameters. Aspiration of the gallbladder has a limited role in the diagnosis of acalculous cholecystitis because of its low sensitivity.[130] Diagnosis relies on imaging studies, particularly ultrasonography, which has become the modality of choice. While lacking specificity, major ultrasonographic criteria include biliary sludge, gallbladder distention (hydrops) and gallbladder wall thickening in the absence of ascites and hypoalbuminemia. Because of high false-positive rates in critically ill patients, who frequently are fasting and have viscous bile, hepatobiliary scintigraphy is better at excluding than confirming the diagnosis of acalculous cholecystitis.[131]
Although cholecystectomy has been the traditional approach, it is not always feasible because of the severity of underlying disease in ICU patients. In high-risk patients, percutaneous cholecystostomy is an acceptable option with a low procedure-related risk and a success rate >60%.[76,132] Transpapillary endoscopic cholecystostomy is another treatment modality for those who are poor candidates for the percutaneous approach.
4. Conclusions
Mechanically ventilated patients frequently develop GI complications, some of which can be seen in up to 50% of patients. While it remains unclear to what extent these complications influence mortality, they are undoubtedly associated with significant morbidity, rendering the care of critically ill patients more difficult and increasing the length of ICU stay and costs. The properties of splanchnic vasculature put mechanically ventilated patients at risk of a variety of GI complications that can impact on the outcome of critically ill patients. Maintenance of splanchnic perfusion appears to be important in the prevention of these complications. Since normal hemodynamics cannot always be achieved, pharmacologic prophylaxis remains the mainstay of preventive strategies.
Improved understanding of the systemic effects of mechanical ventilation and greater application of lung-protective ventilatory strategies may potentially minimize positive pressure-induced reductions in splanchnic perfusion, and systemic cytokine release, and consequently reduce the incidence of GI complications associated with mechanical ventilation. Unquestionably, more studies are warranted to better understand the systemic effects of mechanical ventilation on the GI tract and to investigate the impact of lung-protective ventilatory strategies on GI complications. In the meantime, preventive, evidence-based strategies remain important in reducing the impact of these complications and improving outcomes in critical illness.
References
Mutlu GM, Factor P. Complications of mechanical ventilation. Respir Care Clin N Am 2000; 6: 213–52
Mutlu GM, Mutlu EA, Factor P. GI complications in patients receiving mechanical ventilation. Chest 2001; 119: 1222–41
Haglund U. The gastrointestinal and hepatic systems: normal physiology: the gastrointestinal system. In: Webb AR, Shapiro MJ, Singer M, Suter PM, editors. Oxford textbook of critical care. New York: Oxford University Press, 1999: 297–300
Bohlen GH. Tissue oxygenation and splanchnic blood flow. In: Shepherd AP, Granger DN, editors. Physiology of intestinal circulation. New York: Raven Press, 1984: 143–51
Bion JF. Multiple organ failure. In: Webb AR, Shapiro MJ, Singer M, Suter PM, editors. Oxford textbook of critical care. New York: Oxford University Press, 1999: 923–6
Love R, Choe E, Lippton H, et al. Positive end-expiratory pressure decreases mesenteric blood flow despite normalization of cardiac output. J Trauma 1995; 39: 195–9
Caldwell CB, Ricotta JJ. Changes in visceral blood flow with elevated intraabdominal pressure. J Surg 1987; 43: 14–20
Diebel LN, Dulchavsky SA, Wilson RF. Effect of increased intraabdominal pressure on mesenteric and intestinal mucosal blood flow. J Trauma 1992; 33: 45–9
van den Berg PC, Jansen JR, Pinsky MR. Effect of positive pressure on venous return in volume-loaded cardiac surgical patients. J Appl Physiol 2002; 92: 1223–31
Tanaka S, Sagawa S, Miki K, et al. Changes in muscle sympathetic nerve activity and renal function during positive-pressure breathing in humans. Am J Physiol 1994; 266: R1220
Chernow B, Soldano S, Cook D, et al. Positive end-expiratory pressure increases plasma catecholamine levels in non-volume loaded dogs. Anaesth Intensive Care 1986; 14: 421–5
Cullen JJ, Ephgrave KS, Caropreso DK. Gastrointestinal myoelectric activity during endotoxemia. Am J Surg 1996; 171: 596–9
Aneman A, Ponten J, Fandriks L, et al. Hemodynamic, sympathetic and angiotensin II responses to PEEP ventilation before and during administration of isoflurane. Acta Anaesthesiol Scand 1997; 41: 41–8
Spain DA, Kawabe T, Keelan PC, et al. Decreased alpha-adrenergic response in the intestinal microcirculation after “two-hit”; hemorrhage/resuscitation and bacteremia. J Surg Res 1999; 84: 180–5
Bassiouny HS. Nonocclusive mesenteric ischemia. Surg Clin North Am 1997; 77: 319–26
Schwartz MD, Moore EE, Moore FA, et al. Nuclear factor-kappa B is activated in alveolar macrophages from patients with acute respiratory distress syndrome. Crit Care Med 1996; 24: 1285–92
Lodato RF, Khan AR, Zembowicz MJ, et al. Roles of IL-1 and TNF in the decreased ileal muscle contractility induced by lipopolysaccharide. Am J Physiol 1999; 276: G1356
Cullen JJ, Caropreso DK, Ephgrave KS, et al. The effect of endotoxin on canine jejunal motility and transit. J Surg Res 1997; 67: 54–7
Nahum A, Hoyt J, Schmitz L, et al. Effect of mechanical ventilation strategy on dissemination of intratracheally instilled Escherichia coli in dogs. Crit Care Med 1997; 25: 1733–43
Ranieri VM, Suter PM, Tortorella C, et al. Effect of mechanical ventilation on inflammatory mediators in patients with acute respiratory distress syndrome: a randomized controlled trial. JAMA 1999; 282: 54–61
Sedman PC, Macfie J, Sagar P, et al. The prevalence of gut translocation in humans. Gastroenterology 1994; 107: 643–9
Heyland DK, Tougas G, King D, et al. Impaired gastric emptying in mechanically ventilated, critically ill patients. Intensive Care Med 1996; 22: 1339–44
Bosscha K, Nieuwenhuijs VB, Vos A, et al. Gastrointestinal motility and gastric tube feeding in mechanically ventilated patients. Crit Care Med 1998; 26: 1510–7
Barrientos-Vega R, Mar Sanchez-Soria M, Morales-Garcia C, et al. Prolonged sedation of critically ill patients with midazolam or propofol: impact on weaning and costs. Crit Care Med 1997; 25: 33–40
Carrasco G, Molina R, Costa J, et al. Propofol vs midazolam in short-, medium-,and long-term sedation of critically ill patients: a cost-benefit analysis. Chest 1993; 103: 557–64
Kumar AN, Schwartz DE, Lim KG. Propofol-induced pancreatitis: recurrence of pancreatitis after rechallenge. Chest 1999; 115: 1198–9
Possidente CJ, Rogers FB, Osler TM, et al. Elevated pancreatic enzymes after extended propofol therapy. Pharmacotherapy 1998; 18: 653–5
Cook DJ, Fuller HD, Guyatt GH, et al. Risk factors for gastrointestinal bleeding in critically ill patients: Canadian Critical Care Trials Group. N Engl J Med 1994; 330: 377–81
Schuster DP, Rowley H, Feinstein S, et al. Prospective evaluation of the risk of upper gastrointestinal bleeding after admission to a medical intensive care unit. Am J Med 1984; 76: 623–30
Lucas CE, Sugawa C, Riddle J, et al. Natural history and surgical dilemma of “stress” gastric bleeding. Arch Surg 1971; 102: 266–73
Czaja AJ, McAlhany JC, Pruitt Jr BA. Acute gastroduodenal disease after thermal injury: an endoscopic evaluation of incidence and natural history. N Engl J Med 1974; 291: 925–9
Peura DA, Johnson LF. Cimetidine for prevention and treatment of gastroduodenal mucosal lesions in patients in an intensive care unit. Ann Intern Med 1985; 103: 173–7
Terdiman JP, Ostroff JW. Gastrointestinal bleeding in the hospitalized patient: a case-control study to assess risk factors, causes, and outcome. Am J Med 1998; 104: 349–54
Cook DJ, Reeve BK, Guyatt GH, et al. Stress ulcer prophylaxis in critically ill patients: resolving discordant meta-analyses. JAMA 1996; 275: 308–14
Cook D, Heyland D, Griffith L, et al. Risk factors for clinically important upper gastrointestinal bleeding in patients requiring mechanical ventilation: Canadian Critical Care Trials Group. Crit Care Med 1999; 27: 2812–7
Gurman G, Samri M, Sarov B, et al. The rate of gastrointestinal bleeding in a general ICU population: a retrospective study. Intensive Care Med 1990; 16: 44–9
Silen W. Pathogenetic factors in erosive gastritis. Am J Med 1985; 79: 45–8
Cook D, Guyatt G, Marshall J, et al. A comparison of sucralfate and ranitidine for the prevention of upper gastrointestinal bleeding in patients requiring mechanical ventilation: Canadian Critical Care Trials Group. N Engl J Med 1998; 338: 791–7
Zandstra DF, Stoutenbeek CP. The virtual absence of stress-ulceration related bleeding in ICU patients receiving prolonged mechanical ventilation without any prophylaxis: a prospective cohort study. Intensive Care Med 1994; 20: 335–40
Wilmer A, Tack J, Frans E, et al. Duodenogastroesophageal reflux and esophageal mucosal injury in mechanically ventilated patients. Gastroenterology 1999; 116: 1293–9
Ibanez J, Penafiel A, Raurich JM, et al. Gastroesophageal reflux in intubated patients receiving enteral nutrition: effect of supine and semirecumbent positions. J Parenter Enteral Nutr 1992; 16: 419–22
Orozco-Levi M, Torres A, Ferrer M, et al. Semirecumbent position protects from pulmonary aspiration but not completely from gastroesophageal reflux in mechanically ventilated patients. Am J Respir Crit Care Med 1995; 152: 1387–90
Ferrer M, Bauer TT, Torres A, et al. Effect of nasogastric tube size on gastroesophageal reflux and microaspiration in intubated patients. Ann Intern Med 1999; 130: 991–4
Torres A, Serra-Batlles J, Ros E, et al. Pulmonary aspiration of gastric contents in patients receiving mechanical ventilation: the effect of body position. Ann Intern Med 1992; 116: 540–3
Dive A, Moulart M, Jonard P, et al. Gastroduodenal motility in mechanically ventilated critically ill patients: a manometric study. Crit Care Med 1994; 22: 441–7
Dark DS, Pingleton SK. Nonhemorrhagic gastrointestinal complications in acute respiratory failure. Crit Care Med 1989; 17: 755–8
Montejo JC. Enteral nutrition-related gastrointestinal complications in critically ill patients: a multicenter study: The Nutritional and Metabolic Working Group of the Spanish Society of Intensive Care Medicine and Coronary Units. Crit Care Med 1999; 27: 1447–53
Pingleton SK, Hadzima SK. Enteral alimentation and gastrointestinal bleeding in mechanically ventilated patients. Crit Care Med 1983; 11: 13–6
Raff T, Germann G, Hartmann B. The value of early enteral nutrition in the prophylaxis of stress ulceration in the severely burned patient. Burns 1997; 23: 313–8
Ponec RJ, Saunders MD, Kimmey MB. Neostigmine for the treatment of acute colonic pseudo-obstruction. N Engl J Med 1999; 341: 137–41
Kelly TW, Patrick MR, Hillman KM. Study of diarrhea in critically ill patients. Crit Care Med 1983; 11:7–9
Smith CE, Marien L, Brogdon C, et al. Diarrhea associated with tube feeding in mechanically ventilated critically ill patients. Nurs Res 1990; 39: 148–52
Jones BJ, Lees R, Andrews J, et al. Comparison of an elemental and polymeric enteral diet in patients with normal gastrointestinal function. Gut 1983; 24: 78–84
Liolios A, Oropello JM, Benjamin E. Gastrointestinal complications in the intensive care unit. Clin Chest Med 1999; 20: 329–45
Cleary RK. Clostridium difficile-associated diarrhea and colitis: clinical manifestations, diagnosis, and treatment. Dis Colon Rectum 1998; 41: 1435–49
Jobe BA, Grasley A, Deveney KE, et al. Clostridium difficile colitis: an increasing hospital-acquired illness. Am J Surg 1995; 169: 480–3
Kelly CP, Pothoulakis C, LaMont JT. Clostridium difficile colitis. N Engl J Med 1994; 330: 257–62
DeMeo M, Kolli S, Keshavarzian A, et al. Beneficial effect of a bile acid resin binder on enteral feeding induced diarrhea. Am J Gastroenterol 1998; 93: 967–71
Hernandez G, Velasco N, Wainstein C, et al. Gut mucosal atrophy after a short enteral fasting period in critically ill patients. J Crit Care 1999; 14: 73–7
Brinson RR, Kolts BE. Hypoalbuminemia as an indicator of diarrheal incidence in critically ill patients. Crit Care Med 1987; 15: 506–9
Hwang TL, Lue MC, Nee YJ, et al. The incidence of diarrhea in patients with hypoalbuminemia due to acute or chronic malnutrition during enteral feeding. Am J Gastroenterol 1994; 89: 376–8
Halden E, Jakobson S, Janeras L, et al. Effects of positive end-expiratory pressure on cardiac output distribution in the pig. Acta Anaesthesiol Scand 1982; 26: 403–8
Kahle M, Lippert J, Willemer S, et al. Effects of positive end-expiratory pressure (PEEP) ventilation on the exocrine pancreas in minipigs. Res Exp Med (Berl) 1991; 191: 309–25
Bredenberg CE, Paskanik AM. Relation of portal hemodynamics to cardiac output during mechanical ventilation with PEEP. Ann Surg 1983; 198: 218–22
Matuschak GM, Pinsky MR, Rogers RM. Effects of positive end-expiratory pressure on hepatic blood flow and performance. J Appl Physiol 1987; 62: 1377–83
Fujita Y. Effects of PEEP on splanchnic hemodynamics and blood volume. Acta Anaesthesiol Scand 1993; 37: 427–31
Brienza N, Revelly JP, Ayuse T, et al. Effects of PEEP on liver arterial and venous blood flows. Am J Respir Crit Care Med 1995; 152: 504–10
Purcell PN, Branson RD, Hurst JM, et al. Gut feeding and hepatic hemodynamics during PEEP ventilation for acute lung injury. J Surg Res 1992; 53: 335–41
Risoe C, Hall C, Smiseth OA. Splanchnic vascular capacitance and positive end-expiratory pressure in dogs. J Appl Physiol 1991; 70: 818–24
Trager K, Radermacher P, Georgieff M. PEEP and hepatic metabolic performance in septic shock. Intensive Care Med 1996; 22: 1274–5
Rady MY, Kodavatiganti R, Ryan T. Perioperative predictors of acute cholecystitis after cardiovascular surgery. Chest 1998; 114: 76–84
Savino JA, Scalea TM, Del Guercio LR. Factors encouraging laparotomy in acalculous cholecystitis. Crit Care Med 1985; 13: 377–80
Roslyn JJ, Pitt HA, Mann LL, et al. Gallbladder disease in patients on long-term parenteral nutrition. Gastroenterology 1983; 84: 148–54
Lee SP. Pathogenesis of biliary sludge. Hepatology 1990; 12: 200S–3S
Savoca PE, Longo WE, Pasternak B, et al. Does visceral ischemia play a role in the pathogenesis of acute acalculous cholecystitis? J Clin Gastroenterol 1990; 12: 33–6
Melin MM, Sarr MG, Bender CE, et al. Percutaneous cholecystostomy: a valuable technique in high-risk patients with presumed acute cholecystitis. Br J Surg 1995; 82: 1274–7
Peura DA, Koretz RL. Controversies, dilemmas, and dialogues: prophylactic therapy of stress-related mucosal damage: why, which, who, and so what? Am J Gastroenterol 1990; 85: 935–7
Laine L. Acute and chronic gastrointestinal bleeding. In: Feldman M, Scharschmidt BF, Sleisenger MH, editors. Sleisenger & Fordtran’s gastrointestinal and liver disease. Vol. 1. Philadelphia: WB Saunders, 1998: 198–219
Bresalier RS. The clinical significance and pathophysiology of stress-related gastric mucosal hemorrhage. J Clin Gastroenterol 1991; 13: S35
Peura DA. Stress-related mucosal damage: an overview. Am J Med 1987; 83: 3–7
Cheung LY. Gastric mucosal blood flow: its measurement and importance in mucosal defense mechanisms. J Surg Res 1984; 36: 282–8
Menguy R, Desbaillets L, Masters YF. Mechanism of stress ulcer: influence of hypovolemic shock on energy metabolism in the gastric mucosa. Gastroenterology 1974; 66: 46–55
Skillman JJ, Gould SA, Chung RS, et al. The gastric mucosal barrier: clinical and experimental studies in critically ill and normal man, and in the rabbit. Ann Surg 1970; 172: 564–84
Baghaie AA, Mojtahedzadeh M, Levine RL, et al. Comparison of the effect of intermittent administration and continuous infusion of famotidine on gastric pH in critically ill patients: results of a prospective, randomized, crossover study. Crit Care Med 1995; 23: 687–91
Aris R, Karlstadt R, Paoletti V, et al. Intermittent intravenous pantoprazole achieves a similar onset time to pH = 4 in ICU patients as continuous infusion H2-receptor antagonist, without tolerance. Am J Gastroenterol 2001; 96: S148 (A147)
Morris J, Karlstadt R, Blatcher D, et al. Intermittent intravenous pantoprazole rapidly achieves and maintains gastric pH =4 compared with continuous infusion H2-receptor antagonist in intensive care unit [presentation]. 31st Annual Congress of the Society of Critical Care Medicine (SSCM); 2002 Jan 26–30; San Diego (CA)
McCarthy DM. Sucralfate. N Engl J Med 1991; 325: 1017–25
ASHP Therapeutic guidelines on stress ulcer prophylaxis. Am J Health Syst Pharm 1999; 56: 347–79
du Moulin GC, Paterson DG, Hedley-Whyte J, et al. Aspiration of gastric bacteria in antacid-treated patients: a frequent cause of postoperative colonisation of the airway. Lancet 1982; I: 242–5
Craven DE, Steger KA, Barat LM, et al. Nosocomial pneumonia: epidemiology and infection control. Intensive Care Med 1992; 18: S3
Heyland D, Mandell LA. Gastric colonization by Gram-negative bacilli and nosocomial pneumonia in the intensive care unit patient: evidence for causation. Chest 1992; 101: 187–93
Bonten MJ, Gaillard CA, de Leeuw PW, et al. Role of colonization of the upper intestinal tract in the pathogenesis of ventilator-associated pneumonia. Clin Infect Dis 1997; 24: 309–19
Heyland DK, Cook DJ, Schoenfeld PS, et al. The effect of acidified enteral feeds on gastric colonization in critically ill patients: results of a multicenter randomized trial: Canadian Critical Care Trials Group. Crit Care Med 1999; 27: 2399–406
Driks MR, Craven DE, Celli BR, et al. Nosocomial pneumonia in intubated patients given sucralfate as compared with antacids or histamine type 2 blockers: the role of gastric colonization. N Engl J Med 1987; 317: 1376–82
Prod’hom G, Leuenberger P, Koerfer J, et al. Nosocomial pneumonia in mechanically ventilated patients receiving antacid, ranitidine, or sucralfate as prophylaxis for stress ulcer: a randomized controlled trial. Ann Intern Med 1994; 120: 653–62
Tryba M. Sucralfate versus antacids or H2-antagonists for stress ulcer prophylaxis: a meta-analysis on efficacy and pneumonia rate. Crit Care Med 1991; 19: 942–9
Bonten MJ, Gaillard CA, van Tiel FH, et al. The stomach is not a source for colonization of the upper respiratory tract and pneumonia in ICU patients. Chest 1994; 105: 878–84
Bonten MJ, Gaillard CA, van der Geest S, et al. The role of intragastric acidity and stress ulcer prophylaxis on colonization and infection in mechanically ventilated ICU patients: a stratified, randomized, double-blind study of sucralfate versus antacids. Am J Respir Crit Care Med 1995; 152: 1825–34
Reusser P, Zimmerli W, Scheidegger D, et al. Role of gastric colonization in nosocomial infections and endotoxemia: a prospective study in neurosurgical patients on mechanical ventilation. J Infect Dis 1989; 160: 414–21
de Latorre FJ, Pont T, Ferrer A, et al. Pattern of tracheal colonization during mechanical ventilation. Am J Respir Crit Care Med 1995; 152: 1028–33
Kollef MH. Ventilator-associated pneumonia: a multivariate analysis. JAMA 1993; 270: 1965–70
Drakulovic MB, Torres A, Bauer TT, et al. Supine body position as a risk factor for nosocomial pneumonia in mechanically ventilated patients: a randomised trial. Lancet 1999; 354: 1851–8
Centers for Disease Control and Prevention. Guidelines for prevention of nosocomial pneumonia. Morb Mortal Wkly Rep 1997; 46: 1–79
Lacroix J, Infante-Rivard C, Jenicek M, et al. Prophylaxis of upper gastrointestinal bleeding in intensive care units: a meta-analysis. Crit Care Med 1989; 17:862–9
Shuman RB, Schuster DP, Zuckerman GR. Prophylactic therapy for stress ulcer bleeding: a reappraisal. Ann Intern Med 1987; 106: 562–7
Tryba M. Prophylaxis of stress ulcer bleeding: a meta-analysis. J Clin Gastroenterol 1991; 13: S44
Cook DJ, Witt LG, Cook RJ, et al. Stress ulcer prophylaxis in the critically ill: a meta-analysis. Am J Med 1991; 91: 519–27
Messori A, Trippoli S, Vaiani M, et al. Bleeding and pneumonia in intensive care patients given ranitidine and sucralfate for prevention of stress ulcer: meta-analysis of randomised controlled trials. BMJ 2000; 321: 1103–6
Koelz HR, Aeberhard P, Hassler H, et al. Prophylactic treatment of acute gastroduodenal stress ulceration: low-dose antacid treatment without and with additional ranitidine. Scand J Gastroenterol 1987; 22: 1147–52
Krakamp B, Rommel T, Edelmann M, et al. Prevention of stress-induced hemorrhage of the upper gastrointestinal tract in neurosurgical intensive care patients: a controlled, randomized double-blind study with ranitidine alone and in combination with pirenzepine. Med Klin 1989; 84: 133–4, 172
Khuroo MS, Yattoo GN, Javid G, et al. A comparison of omeprazole and placebo for bleeding peptic ulcer. N Engl J Med 1997; 336: 1054–8
Lau JY, Sung JJ, Lee KK, et al. Effect of intravenous omeprazole on recurrent bleeding after endoscopic treatment of bleeding peptic ulcers. N Engl J Med 2000; 343: 310–6
Ruiz-Santana S, Ortiz E, Gonzalez B, et al. Stress-induced gastroduodenal lesions and total parenteral nutrition in critically ill patients: frequency, complications, and the value of prophylactic treatment: a prospective, randomized study. Crit Care Med 1991; 19: 887–91
McIntyre AS, Thompson DG, Day S, et al. Modulation of human upper intestinal nutrient transit by a beta adrenoreceptor mediated pathway. Gut 1992; 33: 1062–70
Thollander M, Svensson TH, Hellstrom PM. Stimulation of beta-adrenoceptors with isoprenaline inhibits small intestinal activity fronts and induces a post-prandial-like motility pattern in humans. Gut 1997; 40: 376–80
Levein NG, Thorn SE, Lindberg G, et al. Dopamine reduces gastric tone in a dose-related manner. Acta Anaesthesiol Scand 1999; 43: 722–5
Dive A, Miesse C, Galanti L, et al. Effect of erythromycin on gastric motility in mechanically ventilated critically ill patients: a double-blind, randomized, placebo-controlled study. Crit Care Med 1995; 23: 1356–62
Annese V, Janssens J, Vantrappen G, et al. Erythromycin accelerates gastric emptying by inducing antral contractions and improved gastroduodenal coordination. Gastroenterology 1992; 102: 823–8
Feighner SD, Tan CP, McKee KK, et al. Receptor for motilin identified in the human gastrointestinal system. Science 1999; 284: 2184–8
Poen AC, Felt-Bersma RJ, Van Dongen PA, et al. Effect of prucalopride, a new enterokinetic agent, on gastrointestinal transit and anorectal function in healthy volunteers. Aliment Pharmacol Ther 1999; 13: 1493–7
Barone JA. Domperidone: a peripherally acting dopamine2-receptor antagonist. Ann Pharmacother 1999; 33(4): 429–40
van der Spoel JI, Oudemans-van Straaten HM, Stoutenbeek CP, et al. Neostigmine resolves critical illness-related colonic ileus in intensive care patients with multiple organ failure -a prospective, double-blind, placebo-controlled trial. Intensive Care Med 2001; 27: 822–7
Child CS. Prevention of neostigmine-induced colonic activity: a comparison of atropine and glycopyrronium. Anaesthesia 1984; 39: 1083–5
Brinson RR, Kolts BE. Diarrhea associated with severe hypoalbuminemia: a comparison of a peptide-based chemically defined diet and standard enteral alimentation. Crit Care Med 1988; 16: 130–6
Heimburger DC, Geels WJ, Thiesse KT, et al. Randomized trial of tolerance and efficacy of a small-peptide enteral feeding formula versus a whole-protein formula. Nutrition 1995; 11: 360–4
Mowatt-Larssen CA, Brown RO, Wojtysiak SL, et al. Comparison of tolerance and nutritional outcome between a peptide and a standard enteral formula in critically ill, hypoalbuminemic patients. J Parenter Enteral Nutr 1992; 16: 20–4
Dobb GJ, Towler SC. Diarrhoea during enteral feeding in the critically ill: a comparison of feeds with and without fibre. Intensive Care Med 1990; 16: 252–5
Hart GK, Dobb GJ. Effect of a fecal bulking agent on diarrhea during enteral feeding in the critically ill. J Parenter Enteral Nutr 1988; 12: 465–8
Sitzmann JV, Pitt HA, Steinborn PA, et al. Cholecystokinin prevents parenteral nutrition induced biliary sludge in humans. Surg Gynecol Obstet 1990; 170: 25–31
McGahan JP, Lindfors KK. Acute cholecystitis: diagnostic accuracy of percutaneous aspiration of the gallbladder. Radiology 1988; 167: 669–71
Shuman WP, Rogers JV, Rudd TG, et al. Low sensitivity of sonography and cholescintigraphy in acalculous cholecystitis. AJR Am J Roentgenol 1984; 142: 531–4
Browning PD, McGahan JP, Gerscovich EO. Percutaneous cholecystostomy for suspected acute cholecystitis in the hospitalized patient. J Vasc Interv Radiol 1993; 4: 531–7
Acknowledgments
This work is supported by the RO1 HL-66211, American Heart Association and the Evanston Northwestern Healthcare Research Institute. The authors have provided no information on conflicts of interest directly relevant to the content of this review.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mutlu, G.M., Mutlu, E.A. & Factor, P. Prevention and Treatment of Gastrointestinal Complications in Patients on Mechanical Ventilation. Am J Respir Med 2, 395–411 (2003). https://doi.org/10.1007/BF03256667
Published:
Issue Date:
DOI: https://doi.org/10.1007/BF03256667