
Abstract
There is an urgent need to develop new innovative therapies for the control of cancer. Antigen-specific immunotherapy and the employment of proteasome inhibitors have emerged as two potentially plausible approaches for the control of cancer. In the current study, we explored the combination of the DNA vaccine encoding calreticulin (CRT) linked to human papillomavirus type 16 E7 antigen (CRT/E7) with the proteasome inhibitor, bortezomib, for their ability to generate E7-specific immune responses and antitumor effects in vaccinated mice. We found that the combination of treatment with bortezomib and CRT/E7(detox) DNA generated more potent E7-specific CD8+ T cell immune responses and better therapeutic effects against TC-1 tumors in tumor-bearing mice compared to monotherapy. Furthermore, we found that treatment with bortezomib led to increased apoptosis of TC-1 tumor cells and could render the TC-1 tumor cells more susceptible to lysis by E7-specific CD8+ T cells. Our data have significant implications for future clinical translation.
Similar content being viewed by others


Introduction
Advanced-stage cancers are difficult to control using conventional therapies such as chemotherapy, surgery, and radiation. Therefore, new innovative therapies are urgently required in order to combat the high mortality and morbidity associated with cancers. Antigen-specific immunotherapy has emerged as an attractive approach for the treatment of cancers since it has the ability to specifically eradicate systemic tumors and control metastases without damaging normal cells. DNA vaccination has become a potentially promising approach for antigen-specific immunotherapy due to its safety, stability, and ease of preparation (for review, see [1, 2]). We have previously developed several innovative strategies to enhance DNA vaccine potency by directly targeting the DNA into the dendritic cells (DCs) in vivo via gene gun as well as by modifying the properties of antigen-expressing DCs (for review see [3, 4]).
One of the strategies to enhance DNA vaccine potency uses intracellular targeting strategies to enhance major histocompatibility complex (MHC) class I–II antigen presentation and processing in DCs. Previously, we have studied the linkage of calreticulin (CRT), a Ca2+-binding protein located in the endoplasmic reticulum (for review, see [5]) to several antigens, including human papilloma virus type-16 (HPV-16) E7 [6, 7], E6 [8], and nucleocapsid protein of severe acute respiratory syndrome coronavirus [9]. Intradermal administration of CRT linked to any of these target antigens led to a significant increase in the antigen-specific CD8+ T cell (CTL) immune responses and impressive antitumor effects. Thus, CRT has been shown to be highly potent in enhancing the antigen-specific immune responses and antitumor effects generated by DNA vaccination in several preclinical models.
Another important innovative cancer therapy involves the employment of proteasome inhibitors. Proteasomes play an important role in a number of cellular processes that may contribute to cancer cell growth and survival. This suggests that proteasome inhibitors could provide a new and promising class of anticancer agents (for review, see [10]). The dipeptide boronic acid analog bortezomib, also known as PS341, which inhibits the function of the 26S proteasome, was the first proteasome inhibitor used in a clinical setting [11]. This drug is Food and Drug Administration approved and is currently in clinical trials for the treatment of multiple myeloma [12, 13] as well as in many others hematologic and solid tumors [14] including lymphomas [15], chronic lymphocytic leukemia [16], and cancers of the head and neck [17], prostate [18], and breast [19].
In the current study, we hypothesize that the combination of the DNA vaccine encoding CRT linked to HPV-16 E7 (CRT/E7) with the proteasome inhibitor; bortezomib, will lead to enhanced antitumor effects against E7-expressing tumors in a preclinical model. We observed that the combination of treatment with bortezomib and CRT/E7(detox) DNA generated potent E7-specific CD8+ T cell immune responses and significant therapeutic effects against TC-1 tumors in tumor-bearing mice. Furthermore, treatment with bortezomib led to increased apoptosis of TC-1 tumor cells in tumor-bearing mice. In addition, treatment with bortezomib could render the TC-1 tumor cells more susceptible to lysis by E7-specific CTLs in vitro. The clinical implications of the current study are discussed.
Materials and methods
Mice
Six- to 8-week-old female C57BL/6 mice were purchased from the National Cancer Institute (Frederick, MD, USA) and housed in the oncology animal facility of Johns Hopkins Hospital (Baltimore, MD, USA). All animal procedures were performed according to approved protocols and in accordance with recommendations for the proper use and care of laboratory animals.
Reagents and cell lines
We previously generated an E7-expressing tumorigenic cell line TC-1 [20] and a firefly luciferase-expressing TC-1 cell line (TC-1-luc) [21]. Commercially available bortezomib (PS341; Millennium Pharmaceuticals) was reconstituted according to the manufacturer’s instructions and diluted in 0.9% saline before in vivo administration.
DNA constructs and gene gun vaccination
pNGVL4a-CRT/E7detox (4a-CRT/E7d) was obtained from the National Institutes of Health National Gene Vector Laboratory. A second-generation plasmid from pNGVL-3, pNGVL4a, encodes a kanamycin-resistant gene and a transcription unit consisting of a cytomegalovirus promoter and multiple cloning site, followed by a poly-A tail. It contains two short immunostimulatory DNA sequences (ISS) in the noncoding region of the backbone. The ISS sequences are derived from a bacterial ampicillin-resistant gene and consist of tandem repeats of a CpG dinucleotide in the base context, 5′-AACGTT-3′. It has been demonstrated that an ISS-containing pDNA can elicit the production of interferon (IFN)-γ and interleukin 12 in transfected keratocytes and dermal antigen-presenting cells, which results in a potent T helper cell type 1 (Th1) response. This vector also contains a transcription unit that yields high levels of Ag expression and its DNA adjuvant unit elicits strong immunologic Th1 type responses against the pDNA-encoded protein. E7 oncogenic protein detox form has substituted amino acids at positions 24 (glycine in place of cysteine) and 26 (glycine in place of glutamic acid). These substitutions have been shown to inhibit E7 protein binding to pRb at position 11, abating E7’s ability to transform cells. Thus, it is more adequately used in clinical trials.
For the gene gun-mediated intradermal vaccination, DNA-coated gold particles (1 μg DNA per bullet) were delivered to the shaved abdominal region of C57BL/6 mice using a helium-driven gene gun (BioRad, Hercules, CA, USA) with a discharge pressure of 400 psi, according to manufacturer protocol. C57BL/6 mice were vaccinated via gene gun with two bullets of 4a-CRT/E7d for a total of 2 μg DNA. These mice received two booster vaccinations of the same regimen at 4-day intervals.
In vivo treatment experiments
C57BL/6 mice were inoculated subcutaneously with 7 × 104 per mouse of TC-1 cells on day 0. The tumor-bearing mice were divided to four groups (five per group) based on treatment regimen: control (no treatment), bortezomib, 4a-CRT/E7d, or bortezomib and 4aCRT/E7d. For the administration of bortezomib, 1 mg/kg of bortezomib was injected intraperitoneally on days 2, 5, 8, and 11 after tumor inoculation. For the gene gun administration of 4a-CRT/E7d DNA, 2 μg was injected into the tumor-bearing mice on days 9, 13, and 17 after tumor inoculation.
Intracellular cytokine staining by flow cytometry for immune assays
Immune assays were done in different immune response models. In the nontumor model, the mice were given bortezomib and/or 4a-CRT/E7d DNA vaccine as described in the above in vivo treatments without the tumor inoculation. In the tumor model, the mice were inoculated with 1 × 105 TC-1 tumor cells and treated with the same treatments as described in the above in vivo treatment experiments.
Splenocytes were harvested from different groups of mice 7 days after the last vaccination with 4a-CRT/E7d DNA. Prior to intracellular cytokine staining, 5 × 106 per mouse of pooled splenocytes from each vaccination group were incubated for 16 h with 1 μl/ml of E7 peptide containing an MHC class I (H-2Kb or Db) epitope (aa 50–57) for detecting antigen-specific CD8+ T cell precursors in the presence of GolgiPlug (BD Pharmingen, San Diego, CA, USA). Intracellular IFN-γ staining and flow cytometry analysis were performed as described previously [22]. Analysis was performed on a Becton-Dickinson FACScan with CELLQuest software (Becton-Dickinson Immunocytometry System, Mountain View, CA, USA).
Cell staining with annexin V and 7-AAD for the analysis of apoptosis
In order to induce apoptosis, control TC-1 and bortezomib-treated TC-1 cells used bortezomib (1.5 mg/kg) 300-μl intraperitoneal injection, 2-D interval, started on D7 in tumor model with TC-1 cells 1 × 105 sc challenge on D0. Then mice were sacrificed 24 h after last bortezomib dosage. Tumor cells were prepared by single cell suspension method. Then they were stained with phycoerythrin (PE)-conjugated annexin V and the vital dye 7-AAD as protocol. The cells are then acquired and analyzed on the FACScan cytometry in order to distinguish three different populations. The protocol of staining described as follows. The cells were resuspended in at least 2 × 106 cells per milliliter in 5-ml tubes. Two milliliter of cold phosphate-buffered saline (PBS) was added to tubes and centrifuged for 4.5 min at 1,600 rpm. The pellet was resuspended in 2 ml of cold PBS and centrifuged for 4.5 min at 1,600 rpm. The cells were then resuspended in 1 ml of 1× binding buffer and 100 µl were used in each sample, then 0.5 ml 1× binding buffer were added again for washing. One hundred microliter of incubation solution of PE-conjugated annexin V and 1 and 2.5 µl of 7-AAD were added to each tube and gently vortexed. The tubes were incubated at room temperature for 15 min in the dark; 0.5 ml of 1× binding buffer was added and centrifuged for 4.5 min at 1,600 rpm and the supernatant was removed. Four hundred microliter of 1× binding buffer was added to each sample. The samples were analyzed within 1 h of staining. Flow cytometry analysis was performed on a Becton-Dickinson FACScan with CELLQuest software (Becton-Dickinson Immunocytometry System, Mountain View, CA, USA).
Cytotoxic killing assay
For the in vitro cytotoxic killing assay, 1 × 105 per well of luciferase-expressing TC-1 cells were plated in a 24-well plate. The TC-1 cells were treated with or without 16 nM bortezomib overnight (18 h) at 37°C and were used as target cells; 1 × 106 E7-specific CTLs were incubated with the target cells at a 10:1 (E:T) ratio for 4.5 h. The E7-specific CD8+ T cells have been previously described [23]. The target cells incubated without CTLs served as a negative control. Luciferin (1.3 × 10−3 mg per well) was added to the wells for optical imaging with Xenogen IVIS 200.
Tumor measurement and conditional survival
Tumor size was monitored by measuring the longest dimension (length) and shortest dimension (width) in a 3-day interval using dial calipers. Tumor volume was calculated by the following formula: tumor diameter = 0.5 × (length + width). The mice were killed at any time during the experiment if tumor size was over than 2 cm in the longest dimension.
Statistical analysis
All data are expressed as means ± standard error (SE) and are representative of at least two independent experiments. Comparisons between individual data points were made by Student t test. Kaplan–Meier survival curves for tumor treatment experiments were applied. To determine the significance of differences between curves, p values were calculated using a log-rank test. p of 0.05 was considered significant. Graphs were plotted using SigmaPlot program file (Systat software, Inc.).
Results
Combination of treatment with bortezomib and CRT/E7(detox) DNA generate the best therapeutic effects against TC-1 tumors in tumor-bearing mice
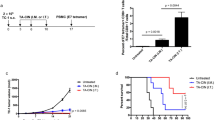
To determine the antitumor effects of treatment with the proteasome inhibitor, bortezomib combined with vaccination with DNA encoding CRT linked to the mutated form of E7 (CRT/E7(detox)), we first challenged groups of C57BL/6 mice (five per group) with TC-1 tumor cells and then treated them with bortezomib and/or the DNA vaccine as illustrated in Fig. 1a. As shown in Fig. 1b, tumor-bearing mice treated with bortezomib combined with CRT/E7(detox) DNA showed significantly lower tumor volumes over time compared to tumor-bearing mice treated with bortezomib alone or the DNA vaccine alone. Furthermore, tumor-bearing mice treated with bortezomib combined with CRT/E7(detox) DNA showed significantly improved survival compared to tumor-bearing mice treated with bortezomib alone or the DNA vaccine alone (Fig. 1c). Thus, our data suggest that the combined treatment using bortezomib and CRT/E7(detox) DNA generates the best therapeutic antitumor effects and long-term survival in TC-1 tumor-bearing mice.

In vivo tumor treatment experiments. Groups of C57BL/6 mice (five per group) were subcutaneously challenged with 7 × 104 per mouse of TC-1 tumor cells on day 0. Tumor-bearing mice were treated with bortezomib at a dose of 1 mg/kg intraperitoneally four times with 3-day intervals starting from day 2. Starting from day 9, mice were vaccinated with DNA encoding 4a-CRT/E7(detox) DNA via gene gun in the amount of 2 μg/mouse three times with 4-day intervals. a Schematic diagram of the treatment regimen of bortezomib and the 4a-CRT/E7(detox) DNA vaccine. b Line graph depicting the tumor volume in TC-1 tumor-bearing mice treated with bortezomib followed by the 4a-CRT/E7(detox) DNA vaccine (mean + SE; p < 0.001). c Kaplan and Meier survival analysis of TC-1 tumor-bearing mice treated with bortezomib followed by the 4a-CRT/E7(detox) DNA vaccine. Data shown are representative of two experiments performed
Combination of treatment with bortezomib and CRT/E7(detox) DNA generates potent E7-specific CD8+ T cell immune responses in tumor-bearing mice
In order to determine the E7-specific CD8+ T cell immune response in TC-1 tumor-bearing mice treated with bortezomib and/or CRT/E7(detox) DNA, we first challenged groups of C57BL/6 mice (five per group) with TC-1 tumor cells and then treated them with DNA vaccine alone, bortezomib alone, or bortezomib combined with DNA vaccination as illustrated in Fig. 1a. Seven days after the last treatment, we harvested splenocytes from vaccinated mice and characterized them for the presence of E7-specific CD8+ T cells using intracellular cytokine staining for IFN-γ followed by flow cytometry analysis. As shown in Fig. 2, tumor-bearing mice that were administered bortezomib combined with CRT/E7(detox) DNA generated significantly higher number of E7-specific CD8+ T cells compared to tumor-bearing mice that were administered CRT/E7(detox) DNA alone. In addition, treatment with bortezomib alone did not significantly increase the number of E7-specific CD8+ T cells in tumor-bearing mice compared to untreated mice (data not shown). Thus, our results suggest that treatment of tumor-bearing mice with bortezomib enhances the E7-specific CD8+ T cell immune responses generated by CRT/E7(detox) DNA vaccination.

Intracellular cytokine staining followed by flow cytometry analysis to determine the number of E7-specific CD8+ T cells in tumor-bearing mice treated with bortezomib and/or CRT/E7(detox) DNA. Groups of C57BL/6 mice (five per group) were challenged with TC-1 tumor cells and treated with bortezomib and/or 4a-CRT/E7(detox) DNA using the regimen as illustrated in Fig. 1a. One week after the last vaccination, splenocytes from tumor-bearing mice were harvested and characterized for E7-specific CD8+ T cells using intracellular IFN-γ staining followed by flow cytometry analysis. a Representative data of intracellular cytokine staining followed by flow cytometry analysis showing the number of E7-specific IFNγ+ CD8+ T cells in the various groups (right upper quadrant). b Bar graph depicting the numbers of E7-specific IFN-γ-secreting CD8+ T cells per 3 × 105 pooled splenocytes (mean + SE). Data shown are representative of two experiments performed
Treatment with bortezomib does not enhance the E7-specific CD8+ T cell immune responses generated by CRT/E7(detox) DNA vaccination in naïve mice
In order to determine whether the E7-specific CD8+ T cell immune responses in naïve mice vaccinated with CRT/E7(detox) DNA can be enhanced by treatment with bortezomib, we treated C57BL/6 mice (five per group) with bortezomib intraperitoneally four times with 3-day intervals. Seven days after the initial administration of bortezomib, mice were vaccinated with CRT/E7(detox) DNA via gene gun three times with 4-day intervals as illustrated in Fig. 3a. One week after the last vaccination, we harvested splenocytes from vaccinated mice and characterized them for the presence of E7-specific CD8+ T cells using intracellular cytokine staining for IFN-γ followed by flow cytometry analysis. As shown in Fig. 3b, naïve mice treated with bortezomib did not enhance the E7-specific CD8+ T cell responses generated by CRT/E7(detox) DNA vaccination. A graphical representation of the number of E7-specific CD8+ T cells is depicted in Fig. 3c. Thus, our results suggest that treatment of naïve mice with bortezomib did not lead to enhanced E7-specific CD8+ T cell immune responses generated by CRT/E7(detox) DNA vaccination. Taken together, these data suggest that treatment with bortezomib only enhances the E7-specific CD8+ T cell responses generated by CRT/E7(detox) DNA vaccination in the presence of tumor.

Intracellular cytokine staining followed by flow cytometry analysis to determine the number of E7-specific CD8+ T cells in mice treated with bortezomib and/or CRT/E7(detox) DNA. a Schematic diagram of the immunization regimen of bortezomib and the 4a-CRT/E7(detox) DNA vaccine. Groups of C57BL/6 mice (five per group) were injected with bortezomib at a dose of 1 mg/kg intraperitoneally four times with 3-day intervals. Starting from day 7, mice were vaccinated with CRT/E7(detox) DNA via gene gun in the amount of 2 μg/mouse three times with 4-day intervals. One week after the last vaccination, splenocytes from mice were harvested and stained for CD8 and intracellular IFN-γ and then characterized for E7-specific CD8+ T cells using intracellular IFN-γ staining followed by flow cytometry analysis. b Representative data of intracellular cytokine staining followed by flow cytometry analysis showing the number of E7-specific IFNγ+ CD8+ T cells in the various groups (right upper quadrant). c Bar graph depicting the numbers of E7-specific IFN-γ-secreting CD8+ T cells per 3 × 105 pooled splenocytes (mean ± SE). Data shown are representative of two experiments performed
Treatment with bortezomib leads to increased apoptosis of TC-1 tumor cells in tumor-bearing mice
Since treatment with bortezomib only enhances the E7-specific CD8+ T cell responses generated by CRT/E7(detox) DNA vaccination in the presence of tumor, we reasoned that bortezomib may lead to increased apoptosis of tumor cells, leading to increased number of E7-specific T cell precursors and enhanced immune response. Thus, in order to determine if treatment with bortezomib leads to increased apoptosis of TC-1 tumor cells in tumor-bearing mice, we first challenged C57BL/6 mice (five per group) with TC-1 tumor cells subcutaneously. One week later, tumor-bearing mice were treated with bortezomib intraperitoneally four times with 2-day intervals. Twenty-four hours later, tumor cells from treated mice were harvested and stained for 7-AAD and annexin V and then characterized for apoptosis using flow cytometry analysis. As shown in Fig. 4a, tumor cells isolated from mice treated with bortezomib generated a significantly higher percentage of apoptotic tumor cells compared to tumor cells isolated from untreated mice. Figure 4b demonstrates a bar graph depicting the percentage of apoptotic tumor cells in tumor cells isolated from mice treated with or without bortezomib. Our results suggest that treatment of tumor-bearing mice with bortezomib leads to significant apoptosis of TC-1 tumor cells.

Flow cytometry analysis to determine the apoptotic cell death in tumors isolated from mice treated with or without bortezomib. Groups of C57BL/6 mice (five per group) were subcutaneously challenged with 1 × 105 per mouse of TC-1 tumor cells on day 0. One week later, tumor-bearing mice were treated with bortezomib 1.5 mg/kg intraperitoneally four times with 3-day intervals. Twenty-four hours later, tumor cells from mice were harvested and stained for 7-AAD and annexin V followed by flow cytometry analysis. a Representative flow cytometry data demonstrating the percentage of apoptotic tumor cells isolated from tumor-bearing mice treated with or without bortezomib (right upper quadrant). b Bar graph depicting the percentage of apoptotic cells per 1 × 104 TC-1 cells (mean ± SE). Data shown are representative of two experiments performed
Treatment with bortezomib renders the TC-1 tumor cells more susceptible to lysis by E7-specific CTLs
In order to determine if treatment of TC-1 tumor cells with bortezomib will render the tumor cell more susceptible to E7-specific T-cell-mediated killing, we performed a cytotoxicity assay using luciferase-expressing TC-1 tumor cells. TC-1 tumor cells were treated with bortezomib with or without E7-specific CTLs. Untreated TC-1 tumor cells and TC-1 cells treated with E7-specific CTLs alone were used as controls. The CTL-mediated killing of the TC-1 tumor cells in each well was monitored using bioluminescent imaging systems. This assay has been used previously by us [24] and others [25] and the degree of CTL-mediated killing of the tumor cells has been shown to correlate with the decrease of luminescence activity. As shown in Fig. 5, the lowest luciferase activity was observed in the wells with TC-1 incubated with bortezomib and E7-specific cytotoxic T cells as compared to the wells incubated with bortezomib alone or E7-specific cytotoxic T cells alone. Thus, our data suggest that the TC-1 tumor cells treated with bortezomib increased the susceptibility of the tumor cells for lysis by the E7-specific cytotoxic T cells. We also observed that treatment with bortezomib alone led to a higher degree of TC-1 tumor cell death compared to CTLs alone using the specified conditions for the in vitro study. However, the observed effect may depend on the concentration of bortezomib and/or the number of CTLs used in the in vitro killing assay. We also performed a CTL killing assay using TC-1 tumors treated with different concentrations of bortezomib. We observed that TC-1 cells treated with increasing doses of bortezomib demonstrated enhanced CTL-mediated killing (Supplementary Fig. 1). Thus, our results indicate that the TC-1 tumor cells treated with increasing doses of bortezomib increased the susceptibility of the tumor cells for lysis by the E7-specific cytotoxic T cells.

In vitro cytotoxicity assay. Luciferase-expressing TC-1 tumor cells were added to 24-well plates at a dose of 1 × 105 per well. Eighteen hours later, TC-1 tumor cells were treated with 16 nM bortezomib overnight followed by incubation with or without 1 × 106 E7-specific cytotoxic T cells (CTL). The E7-specific CD8+ T cells have been previously described [23]. Untreated TC-1 tumor cells and TC-1 cells treated with 1 × 106 E7-specific cytotoxic T cells (CTL) alone were used as controls. The degree of CTL-mediated killing of the tumor cells was indicated by the decrease of luminescence activity using the IVIS luminescence imaging system series 2000. Bioluminescence signals were acquired for 15 s. a Representative luminescence images of 24-well plates showing lysis of the tumor cells. b Bar graph depicting the quantification of luminescence intensity in tumor cells treated with bortezomib and/or E7-specific cytotoxic T cells (mean ± SE). Data shown are representative of two experiments performed
Discussion
In the current study, we observed that the combination of treatment with bortezomib and CRT/E7(detox) DNA generated potent E7-specific CD8+ T cell immune responses, resulting in significant therapeutic effects against TC-1 tumors in tumor-bearing mice. Furthermore, treatment with bortezomib led to increased apoptosis of TC-1 tumor cells and could render the TC-1 tumor cells more susceptible to lysis by E7-specific CTLs. Thus, the combination of treatment with bortezomib and CRT/E7(detox) DNA represents a potentially innovative therapy for the control of E7-expressing tumors.
We selected the HPV-16 E7 antigen for our DNA vaccine development in the current study. It is now clear that persistent infection with HPV is a primary factor in the development of cervical cancer [26]. HPV is one of the most common sexually transmitted diseases in the world and HPV DNA has been detected in 99.7% of cervical cancers. Furthermore, HPV-16 is a “high-risk” type and is associated with more than 50% of all cervical cancers (for review, see [27]). The HPV early viral protein E7 is a potentially ideal target antigen since it is a completely foreign antigen and is expressed early in viral infection. Furthermore, E7 is essential for transformation and is coexpressed in almost all HPV-associated cancer cells but not in normal cells. Thus, DNA vaccines targeting HPV-16 E7 antigen potentially can be used for the control of HPV-associated malignancies.
The timing of administration of bortezomib is potentially important for the enhancement of E7-specific CD8+ T cell immune responses generated by CRT/E7 DNA vaccination. It has been reported that proliferating T cells and not resting T cells are highly sensitive to bortezomib-mediated cytotoxicity [28]. In the current study, we administered bortezomib before the second DNA vaccination. This would increase the susceptibility of the treated TC-1 tumors without compromising the E7-specific CD8+ T cells since the first DNA vaccination in general does not result in significant number of proliferative antigen-specific CD8+ T cells (also see Fig. 3). Thus, for eventual clinical translation, it will be important to determine the timing of administration of bortezomib in order to generate enhanced therapeutic antitumor effects using DNA vaccines.
In our study, we observed that treatment with bortezomib led to an increase in the apoptotic cell death of the TC-1 tumor cells. The apoptotic tumor cells may potentially be taken up by antigen-presenting cells, resulting in the activation of tumor-specific CD8+ T cells (so called cross-priming mechanism). We have previously observed that treatment of the tumor with epigallocatechin-3-gallate (EGCG) led to apoptotic tumor cell death, resulting in higher levels of E7 peptide-loaded dendritic cells in the draining lymph nodes of tumor-bearing mice [29]. This increase in the levels of E7 peptide-loaded dendritic cells resulted in an increased number of E7-specific CD8+ T cell precursors in treated mice. Thus, treatment with EGCG significantly boosted the potency of the CRT/E7 DNA vaccine, by enhancing the E7-specific CD8+ T cell immune responses and antitumor effects. Similarly, treatment with bortezomib also led to apoptotic tumor cell death, thus enhancing CRT/E7 DNA vaccine potency. Therefore, it would be of interest to observe if the degree of apoptotic cell death generated by these reagents correlates with the ability to enhance the DNA vaccine potency.
We observed that treatment with bortezomib rendered the E7-expressing TC-1 tumor cells more susceptible to killing by the E7-specific CD8+ T cells. Several mechanisms may account for the observed phenomenon. For example, bortezomib may lead to upregulation of MHC class I, resulting in increased recognition of antigenic peptide–MHC class I complex by E7-specific CD8+ T cells. However, we have characterized the expression of MHC class I and B-7 on TC-1 tumor cells following treatment with bortezomib. We did not find significant change in the levels of MHC class I and B-7 on TC-1 cells (data not shown). Furthermore, it has been reported that tumor cells treated with bortezomib does not lead to increased expression of MHC class I molecules [30]. Alternatively, treatment with bortezomib may lead to increased expression of E7 protein and/or death receptors (DR). It has been shown that renal cell carcinoma cells treated with bortezomib demonstrated upregulation of DR5 [30]. Furthermore, it has been reported that treatment of tumors with bortezomib can lead to the upregulation of molecules that may sensitize tumors for apoptotic cell death, such as Fas, rendering tumors more susceptible to immune cells expressing death ligands, such as FasL [31]. We have characterized the expression of FasL on the E7-specific CD8+ T cells and we found that our E7-specific CD8+ T cells do express FasL (see Supplementary Fig. 2). In addition, TC-1 cells have been shown to express Fas [32]. Thus, our data indicate that the sensitization of the tumor by bortezomib (presumably by upregulation of Fas on tumor cells) may contribute to the enhanced cytotoxic killing by E7-specific CD8+ T cells. It would be of interest to further characterize and manipulate these molecules in order to illustrate the mechanism for the increased susceptibility of the TC-1 tumors to the killing by E7-specific CD8+ T cells.
In summary, our study demonstrated that the combination of treatment with bortezomib and CRT/E7(detox) DNA generated potent E7-specific CD8+ T cell immune responses, resulting in significant therapeutic effects against TC-1 tumors in tumor-bearing mice. It would be important to further determine the optimal dose and regimen of bortezomib and CRT/E7 DNA vaccine in order to generate the best therapeutic effects in a preclinical model. This information would serve as an important foundation for future clinical translation.
References
Donnelly JJ, Ulmer JB, Liu MA (1997) DNA vaccines. Life Sci 60:163–172
Gurunathan S, Klinman DM, Seder RA (2000) DNA vaccines: immunology, application, and optimization. Annu Rev Immunol 18:927–974
Hung CF, Wu TC (2003) Improving DNA vaccine potency via modification of professional antigen presenting cells. Curr Opin Mol Ther 5:20–24
Tsen SW, Paik AH, Hung CF, Wu TC (2007) Enhancing DNA vaccine potency by modifying the properties of antigen-presenting cells. Expert Rev Vaccines 6:227–239
Gelebart P, Opas M, Michalak M (2005) Calreticulin, a Ca2+-binding chaperone of the endoplasmic reticulum. Int J Biochem Cell Biol 37:260–266
Cheng WF, Hung CF, Chai CY, Hsu KF, He L, Ling M, Wu TC (2001) Tumor-specific immunity and antiangiogenesis generated by a DNA vaccine encoding calreticulin linked to a tumor antigen. J Clin Invest 108:669–678
Kim JW, Hung CF, Juang J, He L, Kim TW, Armstrong DK, Pai SI, Chen PJ, Lin CT, Boyd DA, Wu TC (2004) Comparison of HPV DNA vaccines employing intracellular targeting strategies. Gene Ther 11:1011–1018
Peng S, Ji H, Trimble C, He L, Tsai YC, Yeatermeyer J, Boyd DA, Hung CF, Wu TC (2004) Development of a DNA vaccine targeting human papillomavirus type 16 oncoprotein E6. J Virol 78:8468–8476
Kim TW, Lee JH, Hung CF, Peng S, Roden R, Wang MC, Viscidi R, Tsai YC, He L, Chen PJ, Boyd DA, Wu TC (2004) Generation and characterization of DNA vaccines targeting the nucleocapsid protein of severe acute respiratory syndrome coronavirus. J Virol 78:4638–4645
Spano JP, Bay JO, Blay JY, Rixe O (2005) Proteasome inhibition: a new approach for the treatment of malignancies. Bull Cancer 92:E61–E66 945–952
Adams J, Palombella VJ, Elliott PJ (2000) Proteasome inhibition: a new strategy in cancer treatment. Invest New Drugs 18:109–121
Min CK, Lee MJ, Eom KS, Lee S, Lee JW, Min WS, Kim CC, Kim M, Lim J, Kim Y, Han K (2007) Bortezomib in combination with conventional chemotherapeutic agents for multiple myeloma compared with bortezomib alone. Jpn J Clin Oncol 37:961–968
Uy GL, Trivedi R, Peles S, Fisher NM, Zhang QJ, Tomasson MH, DiPersio JF, Vij R (2007) Bortezomib inhibits osteoclast activity in patients with multiple myeloma. Clin Lymphoma Myeloma 7:587–589
Aghajanian C, Soignet S, Dizon DS, Pien CS, Adams J, Elliott PJ, Sabbatini P, Miller V, Hensley ML, Pezzulli S, Canales C, Daud A, Spriggs DR (2002) A phase I trial of the novel proteasome inhibitor PS341 in advanced solid tumor malignancies. Clin Cancer Res 8:2505–2511
Heider U, von Metzler I, Kaiser M, Rosche M, Sterz J, Rotzer S, Rademacher J, Jakob C, Fleissner C, Kuckelkorn U, Kloetzel PM, Sezer O (2008) Synergistic interaction of the histone deacetylase inhibitor SAHA with the proteasome inhibitor bortezomib in mantle cell lymphoma. Eur J Haematol 80:133–142
Faderl S, Rai K, Gribben J, Byrd JC, Flinn IW, O'Brien S, Sheng S, Esseltine DL, Keating MJ (2006) Phase II study of single-agent bortezomib for the treatment of patients with fludarabine-refractory B-cell chronic lymphocytic leukemia. Cancer 107:916–924
Mitsiades CS, McMillin D, Kotoula V, Poulaki V, McMullan C, Negri J, Fanourakis G, Tseleni-Balafouta S, Ain KB, Mitsiades N (2006) Antitumor effects of the proteasome inhibitor bortezomib in medullary and anaplastic thyroid carcinoma cells in vitro. J Clin Endocrinol Metab 91:4013–4021
Morris MJ, Kelly WK, Slovin S, Ryan C, Eicher C, Heller G, Scher HI (2007) A phase II trial of bortezomib and prednisone for castration resistant metastatic prostate cancer. J Urol 178:2378–2383 discussion 2383–2374
Schmid P, Kuhnhardt D, Kiewe P, Lehenbauer-Dehm S, Schippinger W, Greil R, Lange W, Preiss J, Niederle N, Brossart P, Freier W, Kummel S, Van de Velde H, Regierer A, Possinger K (2008) A phase I/II study of bortezomib and capecitabine in patients with metastatic breast cancer previously treated with taxanes and/or anthracyclines. Ann Oncol 19:871–876 [Epub ahead of print]
Lin KY, Guarnieri FG, Staveley-O'Carroll KF, Levitsky HI, August JT, Pardoll DM, Wu TC (1996) Treatment of established tumors with a novel vaccine that enhances major histocompatibility class II presentation of tumor antigen. Cancer Res 56:21–26
Huang B, Mao CP, Peng S, He L, Hung CF, Wu TC (2007) Intradermal administration of DNA vaccines combining a strategy to bypass antigen processing with a strategy to prolong dendritic cell survival enhances DNA vaccine potency. Vaccine 25:7824–7831
Chen CH, Wang TL, Hung CF, Yang Y, Young RA, Pardoll DM, Wu TC (2000) Enhancement of DNA vaccine potency by linkage of antigen gene to an HSP70 gene. Cancer Res 60:1035–1042
Wang TL, Ling M, Shih IM, Pham T, Pai SI, Lu Z, Kurman RJ, Pardoll DM, Wu TC (2000) Intramuscular administration of E7-transfected dendritic cells generates the most potent E7-specific anti-tumor immunity. Gene Ther 7:726–733
Hung CF, Tsai YC, He L, Wu TC (2007) Control of mesothelin-expressing ovarian cancer using adoptive transfer of mesothelin peptide-specific CD8+ T cells. Gene Ther 14:921–929
Brown CE, Wright CL, Naranjo A, Vishwanath RP, Chang WC, Olivares S, Wagner JR, Bruins L, Raubitschek A, Cooper LJ, Jensen MC (2005) Biophotonic cytotoxicity assay for high-throughput screening of cytolytic killing. J Immunol Methods 297:39–52
Walboomers JM, Jacobs MV, Manos MM, Bosch FX, Kummer JA, Shah KV, Snijders PJ, Peto J, Meijer CJ, Munoz N (1999) Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol 189:12–19
zur Hausen H (2002) Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer 2:342–350
Sun K, Welniak LA, Panoskaltsis-Mortari A, O'Shaughnessy MJ, Liu H, Barao I, Riordan W, Sitcheran R, Wysocki C, Serody JS, Blazar BR, Sayers TJ, Murphy WJ (2004) Inhibition of acute graft-versus-host disease with retention of graft-versus-tumor effects by the proteasome inhibitor bortezomib. Proc Natl Acad Sci U S A 101:8120–8125
Kang TH, Lee JH, Song CK, Han HD, Shin BC, Pai SI, Hung CF, Trimble C, Lim JS, Kim TW, Wu TC (2007) Epigallocatechin-3-gallate enhances CD8+ T cell-mediated antitumor immunity induced by DNA vaccination. Cancer Res 67:802–811
Lundqvist A, Abrams SI, Schrump DS, Alvarez G, Suffredini D, Berg M, Childs R (2006) Bortezomib and depsipeptide sensitize tumors to tumor necrosis factor-related apoptosis-inducing ligand: a novel method to potentiate natural killer cell tumor cytotoxicity. Cancer Res 66:7317–7325
Hallett WH, Ames E, Motarjemi M, Barao I, Shanker A, Tamang DL, Sayers TJ, Hudig D, Murphy WJ (2008) Sensitization of tumor cells to NK cell-mediated killing by proteasome inhibition. J Immunol 180:163–170
Cheng WF, Lee CN, Chang MC, Su YN, Chen CA, Hsieh CY (2005) Antigen-specific CD8+ T lymphocytes generated from a DNA vaccine control tumors through the Fas-FasL pathway. Mol Ther 12:960–968
Acknowledgements
This work was supported by the Flight Attendant Medical Research Institute and National Cancer Institute SPORE in Cervical Cancer P50 CA098252 and the 1 RO1 CA114425-01.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PPT 115 KB)
Rights and permissions
About this article
Cite this article
Tseng, CW., Monie, A., Wu, CY. et al. Treatment with proteasome inhibitor bortezomib enhances antigen-specific CD8+ T-cell-mediated antitumor immunity induced by DNA vaccination. J Mol Med 86, 899–908 (2008). https://doi.org/10.1007/s00109-008-0370-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-008-0370-y