
Abstract
Aims/hypothesis
Neural tube defects (NTDs) are a common malformation associated with diabetic embryopathy. Maternal hyperglycaemia-induced oxidative stress inhibits the expression of Pax3, a gene that is essential for neural tube closure, and increases the incidence of NTDs. Because oxidative stress can stimulate AMP-activated kinase (AMPK) activity, and AMPK can regulate gene transcription, we hypothesised that increased AMPK activity would mediate the adverse effects of maternal hyperglycaemia-induced oxidative stress on Pax3 expression and NTDs.
Methods
Pregnant mice were made transiently hyperglycaemic by glucose injection, or hypoxic by housing in a hypoxic chamber, or were treated with antimycin A to induce oxidative stress, and AMPK activity in the embryos was assayed. The effects of stimulating AMPK activity with 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside (AICAR) on Pax3 expression and NTDs were determined. Vitamin E or glutathione ethyl ester was used to reduce oxidative stress, and compound C was used to inhibit AMPK activation. Murine embryonic stem cells were employed as an in vitro model to study the effects of oxidative stress on AMPK activity and the effects of AMPK stimulation on Pax3 expression.
Results
Maternal hyperglycaemia stimulated AMPK activity, and stimulation of AMPK with AICAR inhibited Pax3 expression (in vivo and in vitro) and increased NTDs (in vivo). Stimulation of AMPK by hyperglycaemia, hypoxia or antimycin A was inhibited by antioxidants. The AMPK inhibitor compound C blocked the effects of hyperglycaemia or AA on Pax3 expression and NTDs.
Conclusions/interpretation
Stimulation of AMPK in embryos during a diabetic pregnancy mediates the effects of hyperglycaemia-induced oxidative stress to disturb the expression of the critical Pax3 gene, thereby causing NTDs.
Similar content being viewed by others


Introduction
Pregestational maternal diabetes increases the risk of congenital malformations three- to fivefold [1]. In human embryos, the malformations are induced within the first 6 weeks of gestation, coincident with the initiation of organogenesis. Most developing organ systems can be affected by maternal diabetes, but defects of the neural tube and heart are among the most common [1].
Studies using rodent embryos have implicated reactive oxygen species (ROS) in the adverse effects of diabetic pregnancy on congenital defects [2–8]. Increased glucose delivery to the embryo is responsible for increased oxidative stress [9, 10]; this oxidative stress is generated through a complex network that includes decreased production of reduced glutathione (GSH) from oxidised glutathione (GSSG) as a result of increased flux through the hexosamine biosynthetic pathway [11], and decreased glutathione synthesis [4]. Oxidative stress appears to disturb development, not by a direct genotoxic stress, but by inhibiting the expression of critical developmental control genes [10]. In particular, expression of Pax3, a gene encoding a transcription factor that is required for neural tube, neural crest and somitic mesoderm development, is inhibited by excess glucose and oxidative stress, and this is associated with an increased incidence of neural tube defects (NTDs) [9, 10], as well as cardiac outflow tract defects [12].
During normal development, the early post-implantation mammalian embryo is physiologically hypoxic, with an oxygen concentration of 2–8% [13]. We previously showed that maternal hyperglycaemia increases glucose metabolism by the embryo, and this further depletes intra-embryonic oxygen, causing excessive hypoxia in the embryo [14]. Hypoxia can stimulate the mitochondrial production of superoxide (O2•−) by a process that is dependent on mitochondrial complexes I, II and III (whereas the production of O2•− during normoxia is dependent only on complexes I and III) [15–17]. Thus, we hypothesised that inducing embryo hypoxia would recapitulate the effects of maternal hyperglycaemia on ROS production, gene expression and NTDs, and that inducing hyperoxia in these embryos would prevent these adverse effects during the diabetic pregnancy.
Indeed, housing pregnant mice in 12% oxygen (compared with approximately 20% oxygen in room air) increased embryonic hydrogen peroxide (H2O2), inhibited Pax3 expression and induced NTDs. Conversely, inducing hyperoxia by housing pregnant mice in by 30% oxygen blocked the effects of maternal diabetes in increasing embryonic H2O2, inhibiting Pax3 expression and inducing NTDs. Furthermore, the antioxidants vitamin E and glutathione ethyl ester (GSH-EE) blocked the effects of maternal hypoxia or hyperglycaemia on H2O2 production, Pax3 expression and NTDs [14]. These results indicate that, during diabetic pregnancy, excess glucose metabolism by embryos depletes intra-embryonic oxygen, which increases O2•− production. Increased O2•− production, along with decreased GSH production, causes oxidative stress, which leads to impaired expression of Pax3, thereby disturbing neural tube closure. How oxidative stress leads to impaired Pax3 expression was not, however, understood.
AMP-activated protein kinase (AMPK) is a heterotrimeric enzyme consisting of a catalytic α subunit and regulatory β and γ subunits. It is activated under conditions in which the ratio of AMP to ATP is increased, such as during exercise and in hypoxia [18, 19]. The mitochondrial generation of H2O2 can also stimulate AMPK through the production of the hydroxyl radical (OH•) [20]. Activated AMPK can translocate to the nucleus [21] and regulate gene expression by phosphorylating nuclear proteins such as transcriptional coactivator 300 (p300), hepatocyte nuclear factor 4α (HNF4α), thyroid receptor interacting protein 6 (TRIP6), nuclear respiratory factor 1 (NRF-1), protein tyrosine phosphatase nuclear 45 kDa form (TC45), peroxisome proliferator-activated receptor gamma coactivator 1α (PGC-1α), histone deacetylase 5 and hypoxia inducible factor 1α (HIF-1α) [22–29]. Thus, AMPK could mediate the effects of oxidative stress resulting from maternal hyperglycaemia in altering the expression of specific developmental control genes, and therefore lead to malformations.
This study was based upon the following chain of events that had previously been shown: maternal hyperglycaemia induces embryo hypoxic stress [14]; embryo hypoxic stress induces oxidative stress [14]; hypoxic and oxidative stress stimulate AMPK activity [18–20]; oxidative stress inhibits Pax3 expression [10, 11]; and insufficient Pax3 expression induces NTDs [9, 30, 31]. It has not, however, been determined how the oxidative stress signal affects the inhibition of Pax3 expression. Here we tested the hypothesis that increased AMPK activity mediates the effects of embryonic oxidative stress in inhibiting Pax3 expression and inducing NTDs. To do this, we investigated whether maternal hyperglycaemia stimulated AMPK activity in embryos, whether stimulation of AMPK by hyperglycaemia was mediated by oxidative stress, and whether stimulation of AMPK activity inhibited Pax3 expression and induced NTDs. These experiments were performed in vivo, using embryos from a pregnant mouse model, and in vitro, using murine embryonic stem cells (mESCs) as an in vitro model of differentiating mouse neuroepithelium.
Methods
Animal procedures
Female ICR mice (Taconic, Germantown, NY, USA) aged 6–8 weeks were placed in cages with ICR males and checked every morning for the presence of a copulation plug. Noon on the day a copulation plug was found was determined to be embryonic day (E) 0.5. For induction of transient hyperglycaemia, pregnant mice were injected s.c. on E7.5 with 2 ml 12.5% glucose in PBS at approximately hourly intervals between 09:00 and 17:00 hours to maintain blood glucose ≥ 17 mmol/l as described [9]. For assay of AMPK activity in response to maternal hyperglycaemia, pregnant mice were given a single injection of glucose at 12:00 hours on E7.5.
Transient hypoxia was induced by housing pregnant mice in chambers containing 12% oxygen as described [14], beginning at 12:00 hours on E7.5. Antimycin A (AA) 3 mg/kg (Sigma, St Louis, MO, USA) dissolved in 25% propylene glycol [10], or 50 mg/kg 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside (AICAR) (Sigma) dissolved in PBS, was administered s.c. at 12:00 hours on E7.5. GSH-EE (Sigma) dissolved in PBS was administered at 1 mmol/kg [11] as a single injection at the same time as the injection of AA or induction of hypoxia. Vitamin E-treated mice were fed Lab Diet 5020 (Purina, St Louis, MO, USA) that had been supplemented with 0.125% (wt/wt) (+)-α-tocopherol succinate (Sigma) beginning on E0.5 as described [10]. Compound C (20 mg/kg; Calbiochem, La Jolla, CA, USA) dissolved in vehicle (2% NaOAc neutralised with NaOH) was administered at 09:00, 13:00 and 17:00 hours on E7.5.
Pregnant mice were killed at times indicated on E7.5 for assay of embryonic AMPK activity, on E8.5 for assay of Pax3 mRNA, or on E10.5 for scoring of NTDs. Embryos were dissected from the uterus in cold PBS and immediately frozen in liquid nitrogen, except for E10.5 embryos, which were examined immediately. All procedures using animals followed the Principles of Laboratory Animal Care (National Institutes of Health publication no. 85–23, revised 1985; http://grants1.nih.gov/grants/olaw/references/phspol.htm) and were approved by the Institutional Animal Care and Use Committee of the Joslin Diabetes Center.
Embryonic stem cell culture
D3 mESCs (ATCC) were cultured on 0.1% gelatin-coated tissue culture dishes in DMEM (4,500 mg/l glucose; Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal calf serum (Atlanta Biologicals, Atlanta, GA, USA), 110 μmol/l β-mercaptoethanol (Sigma), 1X penicillin/streptomycin (Invitrogen), 1,000 units/ml of leucocyte inhibitory factor, 2 mmol/l glutamine, 1X nucleosides and 1X non-essential amino acids (all from Millipore, Billerica, MA, USA). Cultures were induced to form neuronal precursors as described [32], except that 0.5 μmol/l retinoic acid (Sigma) was added to the embryoid body cultures. Cultures were terminated after 2 days of selection of neuronal precursors from embryoid bodies. The following were added on day 1 of selection of neuronal precursors from embryoid bodies: AA 10 μmol/l in ethanol, 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside (AICAR) 1 μmol/l, compound C 20 μmol/l, GSH-EE 250 μmol/l and dl-α-tocopherol acetate (Fluka, Buchs, Switzerland) 1 μg/ml in 40% propylene glycol.
Real-time RT-PCR
Total RNA was extracted using Ultraspec reagent (Biotecx Laboratories, Friendswood, TX, USA). Real-time RT-PCR was performed as described using rRNA as the normalisation control as described [10].
AMPK assays
AMPK activity was assayed either by enzyme activity assay or by a measure of activated AMPK relative to total AMPK, as indicated. AMPK enzyme activity was assayed by transfer of 32P from [32P]ATP (2.22 × 1011 Bq/mmol) to AMARA peptide (AMARAASAAASARRR) as described [33] using embryos from two pooled pregnancies per data point. PO4-AMPK (activated) and total AMPK were assayed by immunoblot using pooled embryos from individual pregnancies as described [34]. The band intensity of autoradiographs exposed in a linear range was determined using Adobe Photoshop (Adobe, San Jose, CA, USA). Antibodies against AMPK (pan-α) and the α1 and αs catalytic subunits were obtained from Upstate Biotechnology (Waltham, MA, USA), and the antibody against PO4-AMPK was obtained from Cell Signaling Technology (Beverly, MA, USA).
Statistical analyses
Except where noted, all experiments were performed in triplicate, using three separate pregnancies or three cell cultures. Data were analysed by one-way ANOVA followed by Neuman–Keuls post test using GraphPad Prism software v. 4.0 (San Diego, CA, USA).
Results
Maternal hyperglycaemia stimulates AMPK activity in embryos
To test whether maternal hyperglycaemia stimulates AMPK activity in embryos, transient hyperglycaemia (≥16.65 mmol/l) was induced in pregnant mice by s.c. glucose injection on E7.5. Glucose injection significantly increased maternal blood glucose concentrations by 15 min (Fig. 1a) and increased the phosphorylated (active) form of AMPK by 30 min (Fig. 1b and c). AMPK enzyme activity was stimulated with the same time course as AMPK phosphorylation (Fig. 1d), confirming that increased AMPK phosphorylation was due to increased AMPK enzyme activity. Of note, although maternal blood glucose levels were significantly elevated by 15 min after glucose injection, stimulation of AMPK activity in embryos was not observed until after 15 min, but AMPK activity remained elevated for at least 30 min after maternal glycaemia was no longer significantly elevated.

a Maternal blood glucose levels before and up to 90 min after induction of transient hyperglycaemia on E7.5 of pregnancy by s.c. glucose administration. b Immunoblot of PO4-AMPK and total AMPK from whole embryos before and up to 90 min after induction of maternal hyperglycaemia. c Quantitation of scanned bands of PO4 (P)-AMPK/total AMPK from (b). Band intensity is expressed as arbitrary units (AU) relative to time 0 (t0). d AMPK enzyme activity (pmol/mg protein × min) in embryos before or up to 90 min following induction of maternal hyperglycaemia. AMPK activity 90 min after administration of AICAR (50 mg/kg) served as a positive control. Because assay of PO4-AMPK/AMPK by immunoblot is more sensitive than assay of AMPK enzyme activity, immunoblot of PO4-AMPK/AMPK was used to measure the stimulation of AMPK activity in subsequent experiments. All panels: *p < 0.05 vs t0; **p < 0.01 vs t0. n = pooled embryos from three pregnancies per time point, except in (d), where six pregnancies per time point were used, and embryos from two pregnancies were pooled for triplicate reactions per time point
Stimulation of AMPK activity inhibits Pax3 expression and induces NTD
To test whether stimulating AMPK could inhibit Pax3 expression and induce NTDs, pregnant mice were given a single injection of AICAR (50 mg/kg) at 12:00 hours on E7.5. This concentration of AICAR significantly stimulates AMPK activity (Fig. 1d) but did not induce hypoglycaemia (blood glucose was 7.1 ± 0.28 mmol/l in AICAR-treated animals, vs 8.43 ± 0.25 mmol/l in controls). AICAR inhibited Pax3 expression on E8.5 and increased NTDs that were detectable on E10.5 (Fig. 2a,b). The effects of AICAR on Pax3 expression and NTDs were comparable to the effects of maternal hyperglycaemia induced transiently during E7.5.

a Real-time RT-PCR analysis of Pax3 mRNA from E8.5 embryos of mice injected with saline or glucose during E7.5, or given a single injection of AICAR on E7.5. Pax3 was normalised to rRNA and expressed relative to that of control embryos from saline-treated pregnancies. b Per cent NTD in E10.5 embryos from pregnancies treated as in a. n = three pregnancies per treatment group. c AMPK activity (PO4 [P]-AMPK/total AMPK) in differentiating neuronal precursors derived from mESCs to that had been cultured without or with 1 mmol/l AICAR for times indicated beginning on day 4 of the culture of neuronal precursors. d Real-time RT-PCR of Pax3 in undifferentiated (UD) mESCs, differentiating neuronal precursors derived from mESCs (D) or differentiating neuronal precursors treated with 1 mmol/l AICAR for 48 h (D + AICAR). Pax3 was normalised to rRNA and expressed relative to that of differentiating neuronal precursors. ***p < 0.001 vs control (embryos of saline-treated pregnancies or untreated cells). *p < 0.05 vs differentiating mESCs
As an alternative approach, we studied the role of stimulating AMPK activity on embryo gene expression in vitro, using mESCs as a model of differentiating neuroepithelium. mESCs can be induced to differentiate into a neural lineage [32], and mESC-derived neuronal precursors exhibit a gene expression profile similar to that of neuroepithelium, including Pax3 [35–37]. AICAR stimulated AMPK activity by differentiating mESC-derived neuronal precursors as early as 2 h after its addition to the culture media, and AMPK activity remained elevated for at least 24 h (Fig. 2c). AICAR inhibited Pax3 expression by differentiating mESCs to a level that was not significantly different from that of undifferentiated mESC (Fig. 2d).
ROS mediate the effects of hyperglycaemia-induced hypoxia to stimulate AMPK
Hyperglycaemia and hypoxia stimulate the production of ROS (O2•−, H2O2 and OH•) [10, 14–17]. During hypoxia, increasing AMP:ATP ratios stimulate AMPK activity [18, 19], but AMPK can also be simulated by ROS [20]. To test whether ROS could be responsible for stimulating embryonic AMPK activity following hyperglycaemia-induced hypoxia, we tested the effects of the antioxidants GSH-EE and vitamin E in inhibiting AMPK activity in the embryos of pregnant mice that were housed in 12% oxygen on E7.5. AMPK activity was stimulated 30, 60 and 90 min after inducing hypoxia, and the stimulation of AMPK was completely blocked by GSH-EE or vitamin E (Fig. 3a). This indicates that the effects of hypoxia to stimulate AMPK activity in embryos are solely due to increased levels of ROS.

a AMPK activity in E7.5 embryos recovered at time 0 (t0) or 30–90 min after housing pregnant mice in 12% oxygen chambers. GSH-EE was administered by i.p. injection at t0. Vitamin E (vit E) was administered by supplementing the chow with vitamin E succinate beginning on E0.5 of pregnancy. t0 controls consisted of three groups each of three pregnancies that were untreated (hypoxia), were injected with GSH-EE and immediately recovered, or had only been treated with vitamin E since E0.5. ***p < 0.001 vs hypoxia. Hypoxia alone, black bars; hypoxia + GSH-EE, grey bars; hypoxia + vitamin E, hatched bars. b AMPK activity in E7.5 embryos recovered at time 0 or 30–90 min after administration of 3 mg/kg AA to pregnant mothers. Embryos recovered 90 min after glucose injection ± GSH-EE or vitamin E were used as hyperglycaemia controls. t0 controls are as described in a, and GSH-EE and vitamin E were administered as in a. † p < 0.001 vs AA; ***p < 0.001 vs glucose. AA (or glucose) alone, black bars; AA (or glucose) + GSH-EE, grey bars; AA (or glucose) + vitamin E, hatched bars. c AMPK activity in mESCs at the times indicated after the addition of 10 mmol/l AA to cultures. AICAR, which served as a positive control, was added for 90 min. *p < 0.05, **p < 0.01, ***p < 0.001 vs t0. d AMPK activity in mESCs treated or not treated with AA, GSH-EE or vitamin E for 60 min. ***p < 0.001 vs all other groups. e Real-time RT-PCR of Pax3 mRNA normalised to rRNA in mESCs. mESCs were undifferentiated (UD) or differentiated to neuronal precursors (D) that were treated or not treated with AA, GSH-EE or vitamin E. **p < 0.01 vs D. f Percentage of NTDs in E10.5 embryos from pregnancies treated on E7.5 as in panel b. **p < 0.01 vs C; ***p < 0.001 vs C
To test whether induction of ROS is sufficient to stimulate AMPK, AA was administered to pregnant mice on E7.5 to stimulate mitochondrial O2•− production [38]. We previously showed that administering AA to pregnant mice increases embryonic ROS, inhibits Pax3 expression and increases NTDs [10, 14]. AA also inhibits Pax3 expression in differentiating P19 embryonal carcinoma cells [10]. As shown in Fig. 3b, AA stimulated AMPK activity in embryos with a time course similar to that stimulated by maternal hyperglycaemia, seen in Fig. 1c. The effect of AA in stimulating AMPK activity was suppressed by vitamin E or GSH-EE (Fig. 3b). Stimulation of AMPK activity by hyperglycaemia was also suppressed by vitamin E or GSH-EE (Fig. 3b). These results indicate that ROS are both necessary and sufficient to stimulate AMPK activity in response to hyperglycaemia.
D3 mESCs, which were isolated in high-glucose DMEM, are insensitive to glucose-induced ROS production, but do produce ROS in response to AA. As shown in Fig. 3c, AA stimulated AMPK activity in differentiating mESCs, and as shown in Fig. 3d, stimulation of AMPK by AA was suppressed by vitamin E and GSH-EE. Just as in the embryo [10, 14], AA inhibited Pax3 expression in differentiating mESCs (Fig. 3e). GSH-EE and vitamin E suppressed the inhibition of Pax3 expression caused by AA, but not that caused by AICAR. Furthermore, GSH-EE and vitamin E suppressed NTDs in the embryos of mice that had been treated with AA or glucose, as previously observed [10, 14], but GSH-EE and vitamin E did not suppress NTDs in the embryos of mice that had been treated with AICAR (Fig. 3f). Taken together, these results suggest that stimulation of AMPK activity by oxidative stress inhibits Pax3 expression and induces NTDs, and that antioxidants prevent the stimulation of AMPK activity, inhibition of Pax3 expression and induction of NTD by oxidative stress.
Inhibiting AMPK activation suppresses the adverse effects of hyperglycaemia or oxidative stress on Pax3 expression and NTDs
The catalytic α subunit of AMPK has two isoforms: α1 and α2 [39]. Both isoforms are expressed by mESCs and mouse embryos (Fig. 4a) and both isoforms are activated by AICAR and AA (Fig. 4b,c). Activation of both the α1 and α2 isoforms is inhibited by the selective inhibitor of AMPK kinase compound C [40]. Compound C significantly inhibited the stimulation of AMPK by AA, as well as by AICAR (Fig. 4d). Compound C also prevented inhibition of Pax3 expression by AA and AICAR (Fig. 4e). Notably, compound C prevented NTDs caused by maternal hyperglycaemia (Fig. 4f).

a Immunoblot of total AMPK following immunoprecipitation using antibodies for the α1 or α2 subunits of AMPK from three cultures of differentiating mESCs or from three separate litters of E7.5 or E10.5 mouse embryos, or from the skeletal muscle (S), liver (L) or heart (H) of adult animals. b AMPK activity assays were performed as in Fig. 1 following immunoprecipitation of AMPKα1 from differentiating mESCs treated with AICAR or AA, or from the heart of an adult mouse treated with AICAR. ***p < 0.001 vs control; **p < 0.01 vs control. c As in b, except that the activity of α2-immunoprecipitated AMPK was assayed. d AMPK activity in differentiating neuronal precursors derived from mESCs that had been cultured without or with AICAR, AA or compound C for 90 min. ***p < 0.001 vs control, compound C; **p < 0.01 vs AA; *p < 0.05 vs AICAR. e Real-time RT-PCR of Pax3 mRNA normalised to rRNA from undifferentiated (UD) mESCs, or differentiating neuronal precursors (D) that were untreated, or treated with compound C, AICAR, AICAR + compound C, AA or AA + compound C. ***p < 0.001 vs D, compound C, AA + compound C, AICAR + compound C. f NTDs in embryos recovered on E10.5 from mothers treated or not treated with glucose or compound C on E7.5. Pregnant mice were made hyperglycaemic as in Fig. 1. Compound C (20 mg/kg) was administered at 09:00, 13:00 and 15:00 hours. **p < 0.01 vs all other treatment groups
Discussion
The data reported here demonstrate that ROS stimulate AMPK activity, and that AMPK inhibits the expression of the essential embryonic gene Pax3, thereby causing NTDs. Moreover, because the effects of hypoxia on AMPK activation were prevented by antioxidants, this suggests that the effects of hyperglycaemia-induced hypoxia in stimulating AMPK are solely due to the production of ROS. This is the first evidence that an enzyme that is directly regulated by ROS, and which can regulate gene expression, regulates the expression of an embryonic gene that is associated with diabetic embryopathy. A schematic diagram of how hyperglycaemia leads to AMPK stimulation and Pax3 inhibition, and how AMPK was experimentally stimulated and inhibited in this study, is shown in Fig. 5.

Proposed model by which stimulation of AMPK in embryos resulting from maternal hyperglycaemia causes NTDs associated with diabetic embryopathy. The mechanisms by which AMPK inhibits Pax3 expression, indicated by ‘?’, have yet to be determined
Involvement of an enzymatic sensor of metabolic energy that has the potential to regulate gene transcription explains how oxidative stress, which would be expected to occur throughout the embryo during diabetic pregnancy, can cause specific structural defects rather than ubiquitous cellular demise. We previously showed that AA can block Pax3 expression without causing DNA damage [10]. Thus, altered Pax3 expression appears to occur by a regulated process, as genotoxicity would be non-specific and would cause gross malformation throughout the embryo. Moreover, expression of the constitutively expressed genes fibronectin and 36b4 (also known as Rplp0) is not affected by maternal diabetes [30], indicating that only some genes are regulated by processes that are susceptible to cellular redox status. It is also possible that the conformation or intra- or intermolecular disulfide bonds of proteins involved in Pax3 transcription regulation might be altered in the presence of increased ROS levels. However, the data presented here, in particular the effects of compound C on Pax3 expression and NTDs, suggest that stimulation of AMPK activity is sufficient to explain the adverse effects of oxidative stress on Pax3 expression and NTDs.
We considered that the transcription factor HIF-1α, which senses hypoxia and ROS and elicits transcriptional responses [17, 27], might be involved in mediating the effects of hyperglycaemia-induced oxidative stress on Pax3 expression. If HIF-1α is involved, it would probably be acting as a downstream target of AMPK [27]. It is unlikely that HIF-1α is acting upstream of AMPK, because the time course to stimulate AMPK activity is too rapid to allow for the transcription and translation of HIF-1α targets. It is unlikely that HIF-1α is acting independently of AMPK, because the effects of oxidative stress or hyperglycaemia on Pax3 expression or NTDs were completely blocked by compound C. Furthermore, HIF-1α-null embryos develop NTDs resembling those caused by maternal diabetes [41], which may be secondary to defective vascularisation. This indicates that inactivation of HIF-1α-dependent transcription does not rescue Pax3 expression during embryonic hypoxia and argues against the involvement of HIF-1α in mediating the effects of oxidative stress on embryonic gene expression and NTD.
Many of the experiments were performed using chemical activation or inhibition of oxidative stress and AMPK pathways. Therefore, it is important to consider the specificity of chemical effects. For example, AA is a specific inhibitor of mitochondrial complex III, which generates O2•− production [42]. However, the antioxidants vitamin E and GSH-EE could have effects other than in restoring GSH levels, for example, in normalising diacylglycerol (DAG) and activity of protein kinase C (PKC), or in providing substrate for glutathiolation reactions [43, 44]. However, the fact that the effects of AA were blocked by two different antioxidants argues that the effects of AA are caused by the production of ROS, and that the effects of vitamin E and GSH-EE are due to their activities as antioxidants. Similarly, AICAR or compound C might have effects on signalling pathways other than AMPK; however, there are no known enzymatic pathways that are both stimulated by AICAR and inhibited by compound C [45], suggesting that the effects of AICAR or compound C were due to modulation of AMPK activity.
These experiments explain how antioxidants have been effective in animal models of diabetic embryopathy. GSH-EE, or N-acetylcysteine (NAC), which is converted to GSH, would compensate for the decreased synthesis of GSH or generation from GSSG during diabetic pregnancy [4, 11] and decrease the accumulation of H2O2, which stimulates AMPK activity. Vitamin E (α-tocopherol) functions in the glutathione peroxidase pathway and can maintain normal GSH levels [46], as well as scavenging ROS at membranes [5, 6, 10, 47]. Vitamin C [7] may serve as an electron donor for endogenous vitamin E, thereby restoring its antioxidant capacity.
These studies also provide a new understanding of how other enzymatic pathways that have been implicated in diabetic embryopathy may function upstream of AMPK activation by decreasing the cellular supply of GSH. Expression of the gene encoding γ-glutamylcysteine synthetase is inhibited by maternal diabetes, and this leads to decreased GSH synthesis [4]. Increased production of DAG and activity of PKC have been observed in the embryos and decidua of diabetic mice or rats [48, 49]. DAG-sensitive PKC can stimulate NADPH oxidase [50], thereby decreasing the availability of NADPH, which is necessary for the reduction of GSSG to GSH. However, increased DAG and PKC were studied using embryos after malformations had already occurred, so it could not be determined whether increased PKC activity had occurred prior to the onset of gene expression that led to defective organogenesis.
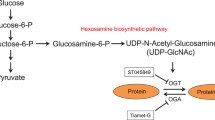
Finally, we have shown that there is increased activity of the hexosamine synthetic pathway in response to maternal hyperglycaemia [11]. This appears to result from an increased provision of substrate from glycolysis. Glucosamine 6-phosphate, which is produced by glutamine/fructose-6-phosphate amidotransferase, the rate-limiting enzyme for hexosamine synthesis, inhibits glucose-6-phosphate dehydrogenase (G6PD), the rate-limiting enzyme of the pentose shunt pathway. Since G6PD activity is coupled to the reduction of NADP+ to NADPH, inhibition of G6PD activity leads to a decreased ration of GSH to GSSG [11]. The embryopathic effects of glucosamine can be blocked by GSH-EE [11], indicating that the embryopathic effects of increased flux through the hexosamine synthetic pathway are due to decreased antioxidative capacity of the embryo.
In our studies, transient induction of maternal hyperglycaemia on E7.5 was used to replicate the effects of maternal diabetes, as we previously showed that hyperglycaemia on E7.5 is necessary and sufficient to cause decreased Pax3 expression and increased NTDs associated with diabetic embryopathy [9]. Furthermore, maternal hyperglycaemia induces hypoxic and oxidative stress, and antioxidants or hyperoxia block the effects of diabetic pregnancy on oxidative stress, Pax3 expression and NTDs [10, 11, 14]. Thus, while the effects of diabetic pregnancy on AMPK activity were not directly examined in this study, it would be expected that AMPK activity would be increased in the embryos of diabetic mice, and that embryos of diabetic mice with corrected hypoxia or oxidative stress would display normalised AMPK activity.
Although we tested the effects of stimulating AMPK activity only on E7.5, the stage of development at which the induction of Pax3 expression (which starts on E8.5) is susceptible to maternal hyperglycaemia [30], and AMPK activity remained elevated even after maternal blood glucose levels were returning to normal, we did not determine how long after the hyperglycaemic episode AMPK remained active. Because blood glucose levels fluctuate during diabetes, it is expected that episodes of increased AMPK activity parallel hyperglycaemic excursions. Further investigation is necessary in order to map the molecular consequences of AMPK stimulation at specific stages of development.
It should be noted that stimulation of AMPK by ROS inhibited Pax3 expression both in mouse embryos and in mESC-derived neuronal precursors. Thus, mESC-derived neuronal precursors, which can provide large quantities of cellular material for biochemical and molecular procedures, provide a valuable in vitro model to complement whole animal experiments. The use of mESCs will be instrumental in future studies to identify AMPK targets and to further elucidate how Pax3 gene transcription is regulated by oxidative stress.
Abbreviations
- AA:
-
Antimycin A
- AICAR:
-
5-Aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside
- AMPK:
-
AMP-activated protein kinase
- DAG:
-
Diacylglycerol
- GSH:
-
Reduced glutathione
- GSH-EE:
-
Glutathione ethyl ester
- GSSG:
-
Oxidised glutathione
- mESC:
-
Murine embryonic stem cells
- NAC:
-
N-Acetylcysteine
- NTD:
-
Neural tube defect
- PKC:
-
Protein kinase C
- ROS:
-
Reactive oxygen species
References
Zabihi S, Loeken MR (2010) Understanding diabetic teratogenesis: where are we now and where are we going? Birth Defects Res A Clin Mol Teratol 88:779–790
Eriksson UJ, Borg LAH (1991) Protection by free oxygen radical scavenging enzymes against glucose-induced embryonic malformations in vitro. Diabetologia 34:325–331
Hagay ZJ, Weiss Y, Zusman I et al (1995) Prevention of diabetes-associated embryopathy by overexpression of the free radical scavenger copper zinc superoxide dismutase in transgenic mouse embryos. Am J Obstet Gynecol 173:1036–1041
Trocino RA, Akazawa S, Ishibashi M et al (1995) Significance of glutathione depletion and oxidative stress in early embryogenesis in glucose-induced rat embryo culture. Diabetes 44:992–998
Viana M, Herrera E, Bonet B (1996) Teratogenic effects of diabetes mellitus in the rat. Prevention by vitamin E. Diabetologia 39:1041–1046
Siman CM, Eriksson UJ (1997) Vitamin E decreases the occurrence of malformations in the offspring of diabetic rats. Diabetes 46:1054–1061
Siman CM, Eriksson UJ (1997) Vitamin C supplementation of the maternal diet reduces the rate of malformation in the offspring of diabetic rats. Diabetologia 40:1416–1424
Sivan E, Lee YC, Wu YK, Reece EA (1997) Free radical scavenging enzymes in fetal dysmorphogenesis among offspring of diabetic rats. Teratology 56:343–349
Fine E, Horal M, Chang T, Fortin G, Loeken M (1999) Evidence that hyperglycemia causes altered gene expression, apoptosis, and neural tube defects in a mouse model of diabetic pregnancy. Diabetes 48:2454–2462
Chang TI, Horal M, Jain S, Wang F, Patel R, Loeken MR (2003) Oxidant regulation of gene expression and neural tube development: Insights gained from diabetic pregnancy on molecular causes of neural tube defects. Diabetologia 46:538–545
Horal M, Zhang Z, Virkamaki A, Stanton R, Loeken MR (2004) Activation of the hexosamine pathway causes oxidative stress and abnormal embryo gene expression: involvement in diabetic teratogenesis. Birth Defects Res Part A Clin Mol Teratol 70:519–527
Morgan SC, Relaix F, Sandell LL, Loeken MR (2008) Oxidative stress during diabetic pregnancy disrupts cardiac neural crest migration and causes outflow tract defects. Birth Defects Res A Clin Mol Teratol 82:453–463
Fischer B, Bavister BD (1993) Oxygen tension in the oviduct and uterus of rhesus monkeys, hamsters and rabbits. J Reprod Fertil 99:673–679
Li R, Chase M, Jung SK, Smith PJS, Loeken MR (2005) Hypoxic stress in diabetic pregnancy contributes to impaired embryo gene expression and defective development by inducing oxidative stress. Am J Physiol Endocrinol Metab 289:E591–E599
Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT (1998) Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci U S A 95:11715–11720
Paddenberg R, Ishaq B, Goldenberg A et al (2003) Essential role of complex II of the respiratory chain in hypoxia-induced ROS generation in the pulmonary vasculature. Am J Physiol Lung Cell Mol Physiol 284:L710–L719
Chandel NS, McClintock DS, Feliciano CE et al (2000) Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem 275:25130–25138
Hardie DG, Hawley SA (2001) AMP-activated protein kinase: the energy charge hypothesis revisited. Bioessays 23:1112–1119
Hayashi T, Hirshman MF, Fujii N, Habinowski SA, Witters LA, Goodyear LJ (2000) Metabolic stress and altered glucose transport: activation of AMP-activated protein kinase as a unifying coupling mechanism. Diabetes 49:527–531
Choi SL, Kim SJ, Lee KT et al (2001) The regulation of AMP-activated protein kinase by H(2)O(2). Biochem Biophys Res Commun 287:92–97
McGee SL, Howlett KF, Starkie RL, Cameron-Smith D, Kemp BE, Hargreaves M (2003) Exercise increases nuclear AMPK alpha2 in human skeletal muscle. Diabetes 52:926–928
Yang W, Hong YH, Shen XQ, Frankowski C, Camp HS, Leff T (2001) Regulation of transcription by AMP-activated protein kinase: phosphorylation of p300 blocks its interaction with nuclear receptors. J Biol Chem 276:38341–38344
Hong YH, Varanasi US, Yang W, Leff T (2003) AMP-activated protein kinase regulates HNF4alpha transcriptional activity by inhibiting dimer formation and decreasing protein stability. J Biol Chem 278:27495–27501
Solaz-Fuster MC, Gimeno-Alcaniz JV, Casado M, Sanz P (2006) TRIP6 transcriptional co-activator is a novel substrate of AMP-activated protein kinase. Cell Signal 18:1702–1712
Bergeron R, Ren JM, Cadman KS et al (2001) Chronic activation of AMP kinase results in NRF-1 activation and mitochondrial biogenesis. Am J Physiol Endocrinol Metab 281:E1340–E1346
Lam MH, Michell BJ, Fodero-Tavoletti MT, Kemp BE, Tonks NK, Tiganis T (2001) Cellular stress regulates the nucleocytoplasmic distribution of the protein-tyrosine phosphatase TCPTP. J Biol Chem 276:37700–37707
Lee M, Hwang JT, Lee HJ et al (2003) AMP-activated protein kinase activity is critical for hypoxia-inducible factor-1 transcriptional activity and its target gene expression under hypoxic conditions in DU145 cells. J Biol Chem 278:39653–39661
McGee SL, van Denderen BJ, Howlett KF et al (2008) AMP-activated protein kinase regulates GLUT4 transcription by phosphorylating histone deacetylase 5. Diabetes 57:860–867
Jager S, Handschin C, St-Pierre J, Spiegelman BM (2007) AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci U S A 104:12017–12022
Phelan SA, Ito M, Loeken MR (1997) Neural tube defects in embryos of diabetic mice: role of the Pax-3 gene and apoptosis. Diabetes 46:1189–1197
Auerbach R (1954) Analysis of the developmental effects of a lethal mutation in the house mouse. J Exp Zool 127:305–329
Lee SH, Lumelsky N, Studer L, Auerbach JM, McKay RD (2000) Efficient generation of midbrain and hindbrain neurons from mouse embryonic stem cells. Nat Biotechnol 18:675–679
Downs SM, Hudson ER, Hardie DG (2002) A potential role for AMP-activated protein kinase in meiotic induction in mouse oocytes. Dev Biol 245:200–212
Musi N, Fujii N, Hirshman MF et al (2001) AMP-activated protein kinase (AMPK) is activated in muscle of subjects with type 2 diabetes during exercise. Diabetes 50:921–927
Yamada G, Kioussi C, Schubert FR et al (1994) Regulated expression of Brachyury (T), NKX1.1 and Pax genes in embryoid bodies. Biochem Biophys Res Commun 199:552–563
Yan Y, Yang D, Zarnowska ED et al (2005) Directed differentiation of dopaminergic neuronal subtypes from human embryonic stem cells. Stem Cells 23:781–790
Perry P, Sauer S, Billon N et al (2004) A dynamic switch in the replication timing of key regulator genes in embryonic stem cells upon neural induction. Cell Cycle 3:1645–1650
Boveris A (1977) Mitochondrial production of superoxide radical and hydrogen peroxide. Adv Exp Med Biol 78:67–82
Hardie DG (1999) Roles of the AMP-activated/SNF1 protein kinase family in the response to cellular stress. Biochem Soc Symp 64:13–27
Zhou G, Myers R, Li Y et al (2001) Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 108:1167–1174
Kotch LE, Iyer NV, Laughner E, Semenza GL (1999) Defective vascularization of HIF-1alpha-null embryos is not associated with VEGF deficiency but with mesenchymal cell death. Dev Biol 209:254–267
Turrens JF, Alexandre A, Lehninger AL (1985) Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch Biochem Biophys 237:408–414
Sullivan DM, Wehr NB, Fergusson MM, Levine RL, Finkel T (2000) Identification of oxidant-sensitive proteins: TNF-alpha induces protein glutathiolation. Biochemistry 39:11121–11128
Kunisaki M, Fumio U, Nawata H, King GL (1996) Vitamin E normalizes diacylglycerol-protein kinase C activation induced by hyperglycemia in rat vascular tissues. Diabetes 45:S(3)117–S(3)119
Bradley EA, Eringa EC, Stehouwer CD et al (2010) Activation of AMP-activated protein kinase by 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside in the muscle microcirculation increases nitric oxide synthesis and microvascular perfusion. Arterioscler Thromb Vasc Biol 30:1137–1142
Jain SK, McVie R, Smith R (2000) Vitamin E supplementation restores glutathione and malondialdehyde to normal concentrations in erythrocytes of type-1 diabetic patients. Diabetes Care 23:1389–1394
Sivan E, Reece EA, Wu Y-K, Homko CJ, Polansky M, Borenstein M (1996) Dietary vitamin E prophylaxis and diabetic embryopathy: morphologic and biochemical analysis. Am J Obstet Gynecol 175:793–799
Hiramatsu Y, Sekiguchi N, Hayashi M et al (2002) Diacylglycerol production and protein kinase C activity are increased in a mouse model of diabetic embryopathy. Diabetes 51:2804–2810
Gareskog M, Wentzel P (2004) Altered protein kinase C activation associated with rat embryonic dysmorphogenesis. Pediatr Res 56:849–857
Hua H, Munk S, Goldberg H, Fantus IG, Whiteside CI (2003) High glucose-suppressed endothelin-1 Ca2+ signaling via NADPH oxidase and diacylglycerol-sensitive protein kinase C isozymes in mesangial cells. J Biol Chem 278:33951–33962
Acknowledgements
We are grateful for assistance and advice on the AMPK assay from members of the Goodyear Laboratory, and for culture of mESCs from F. Wang. M. Horal and J. Blanchard provided technical support. This project was supported by National Institutes of Health grant RO1DK58300 (to MRL) and was assisted by core facilities supported by a Diabetes Endocrine Research Center grant, P30DK036836, from the National Institute of Diabetes and Digestive and Kidney Diseases.
Contribution statement
YW, MV and ST contributed to the conception and design of the experiments, execution of the experiments, analysis and interpretation of the data, and revision of the manuscript and gave final approval of the version to be published. MR contributed to the conception and design of the experiments, execution of the experiments, analysis and interpretation of the data, and drafting and revision of the manuscript for important intellectual content, and gave final approval of the version to be published.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wu, Y., Viana, M., Thirumangalathu, S. et al. AMP-activated protein kinase mediates effects of oxidative stress on embryo gene expression in a mouse model of diabetic embryopathy. Diabetologia 55, 245–254 (2012). https://doi.org/10.1007/s00125-011-2326-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-011-2326-y