
Abstract
Aims/hypothesis
Diabetes interferes with bone formation and impairs fracture healing, an important complication in humans and animal models. The aim of this study was to examine the impact of diabetes on mesenchymal stem cells (MSCs) during fracture repair.
Methods
Fracture of the long bones was induced in a streptozotocin-induced type 1 diabetic mouse model with or without insulin or a specific TNFα inhibitor, pegsunercept. MSCs were detected with cluster designation-271 (also known as p75 neurotrophin receptor) or stem cell antigen-1 (Sca-1) antibodies in areas of new endochondral bone formation in the calluses. MSC apoptosis was measured by TUNEL assay and proliferation was measured by Ki67 antibody. In vitro apoptosis and proliferation were examined in C3H10T1/2 and human-bone-marrow-derived MSCs following transfection with FOXO1 small interfering (si)RNA.
Results
Diabetes significantly increased TNFα levels and reduced MSC numbers in new bone area. MSC numbers were restored to normal levels with insulin or pegsunercept treatment. Inhibition of TNFα significantly reduced MSC loss by increasing MSC proliferation and decreasing MSC apoptosis in diabetic animals, but had no effect on MSCs in normoglycaemic animals. In vitro experiments established that TNFα alone was sufficient to induce apoptosis and inhibit proliferation of MSCs. Furthermore, silencing forkhead box protein O1 (FOXO1) prevented TNFα-induced MSC apoptosis and reduced proliferation by regulating apoptotic and cell cycle genes.
Conclusions/interpretation
Diabetes-enhanced TNFα significantly reduced MSC numbers in new bone areas during fracture healing. Mechanistically, diabetes-enhanced TNFα reduced MSC proliferation and increased MSC apoptosis. Reducing the activity of TNFα in vivo may help to preserve endogenous MSCs and maximise regenerative potential in diabetic patients.
Similar content being viewed by others

Introduction
Diabetes mellitus is one of the most common metabolic diseases, with 439 million people estimated to be affected worldwide by 2030 [1]. Diabetic patients have a greater risk of fracture and an increased risk of delayed union and non-union fracture healing [2–4]. Though the aetiology of diabetic complications is multifactorial, chronic inflammation is thought to play a critical role in several diabetic complications [5, 6]. It has been implicated in diabetic osteopaenia and reduced bone formation associated with impaired fracture repair in humans and in animal models of diabetes [7–9].
Fracture healing is a regenerative process that involves the coordinated activity of inflammatory cells, endothelial cells, stem cells, chondrocytes, osteoblasts and other cell types [10]. In an experimental animal model of fracture healing, mice with streptozotocin (STZ)-induced diabetes exhibited increased inflammation, enhanced osteoclastogenesis, loss of cartilage in the callus and reduced bone formation [7, 11, 12].
An inflammatory response is indispensable for proper fracture healing. Mice deficient in TNFα receptor or IL-6 show delayed fracture healing [13, 14]. However, animals with diabetes or rheumatoid arthritis exhibit elevated TNFα levels and display impaired fracture healing [7, 15]. TNFα inhibition increases fracture callus size in mouse models of diabetes [16].
Bone-marrow-derived MSCs differentiate to chondrocytes and osteoblasts, which play essential roles in endochondral ossification of fracture calluses. MSCs also function as trophic mediators that promote angiogenesis, have anti-apoptotic effects and reduce inflammation [17]. Therapeutic use of MSCs improves outcomes in the healing response to stroke, myocardial infarction and long bone fracture in animal models [18–20], and in the treatment of inflammatory diseases in phase 1 and 2 human clinical trials [21, 22]. Although MSCs are important in the repair of many different tissues, relatively little is known about the impact of diabetes on MSCs in fracture repair. Here, we determined that diabetes affects the number of MSCs in areas of new endochondral ossification in fracture healing in mice. Diabetes interferes with MSC proliferation and promotes MSC apoptosis in vivo through a mechanism that involves diabetes-enhanced TNFα levels. Thus, in fracture healing, diabetes-induced inflammation creates a suboptimal environment for MSC survival and ability to proliferate. Given the critical role that MSCs are thought to play in fracture healing, this deficit may significantly contribute to the negative effect of diabetes on fracture repair.
Methods
Animals
The research was carried out using 8-week-old male CD-1 mice purchased from Charles River Laboratories (Wilmington, MA, USA), conforming to a protocol approved by the American Association For Laboratory Animal Science (IACUC). The mouse model of type 1 diabetes induced by STZ has been previously described [11, 16]. Briefly, multiple injections of low-dose STZ (40 mg/kg, Sigma-Aldrich, St. Louis, MO, USA) were administered to mice daily for 5 days. Mice were considered to be diabetic when blood glucose levels exceeded 13.78 mmol/l (250 mg/dl). Mice were diabetic for at least 3 weeks prior to starting experiments (see electronic supplementary material [ESM] Table 1 for blood glucose levels). A closed–transverse fracture of the mouse tibia or femur was carried out as previously described [23, 24]. In one set of experiments, mice were treated with slow-release insulin as previously described [11]. In another set of experiments, pegsunercept was administered by intraperitoneal injection (4 mg/kg, Amgen, Thousand Oaks, CA, USA) 10 days post-fracture and every 3 days thereafter. The animals were killed by cervical dislocation and cardiac puncture under deep sedation with ketamine/xylazine/acepromazine. The samples of long bone were collected by carefully trimming the surrounding muscles.
Paraffin sections and histomorphometric analysis
Specimens were fixed, decalcified and embedded in paraffin as previously described [11]. Haematoxylin and eosin (H&E) staining was performed to determine the centre of the callus, where the callus was widest. All histology was performed in sections taken close to the centre of the callus. The amount of bone formation was determined by measuring the bone area within the callus in tissue sections. Data are presented as mm2 bone per callus and percentage of bone area per total callus area. Osteoblasts were identified as cuboidal cells in clusters of three or more cells in contact with the bone surface in Toluidine-Blue-stained sections. For each specimen, five to ten random fields throughout the areas of new endochondral ossification were examined under ×400 magnification.
Immunohistochemistry/fluorescence
Antigen retrieval was induced by pressure heating (>120°C) paraffin slides in 10 mmol/l sodium citrate (pH 6.0) using a 2100-Retriever (Aptum, Southampton, UK). A list of antibodies and reagents used is provided in ESM Table 2. For immunohistochemistry, the primary antibody was localised by incubation with a biotinylated secondary antibody and then avidin–horseradish peroxidase. For some analyses, a scale of immunopositive index was used as has been previously described [12]: 0 (no immunostaining); 1 (<10% lightly immunostained cells); 2 (<10% moderate-to-darkly immunostained cells); 3 (10–25% moderate-to-darkly immunostained cells); 4 (25–40% moderate-to-darkly immunostained cells). For immunofluorescence, primary antibody was localised by a biotinylated secondary antibody. To enhance the signal, avidin–biotin peroxidase enzyme complex and tyramide signal amplification was used. Visualisation was achieved using Alexa 546-conjugated streptavidin or fluorescein-conjugated avidin. For double immunofluorescence, residual biotin sites and peroxidase activity associated with the first primary antibody were blocked with avidin/biotin-blocking reagent and 3% (vol./vol.) hydrogen peroxide. IgG control experiments were carried out for each primary antibody, and images were acquired using the same exposure time. All histological images were analysed by two independent examiners under blinded conditions.
In vitro experiments
In vitro experiments were carried out with a mouse MSC line (C3T10H1/2) and primary human bone-marrow-derived MSCs (hBMSCs), which were isolated and characterised as previously described [25]. Briefly, isolation of hBMSCs was achieved with plastic adherence, and hBMSCs were characterised as CD73+CD90+CD105+CD45− cells that were capable of differentiating into osteoblasts and adipocytes. MSCs were grown and maintained in α-MEM + 10% (vol./vol.) FBS and 1% (vol./vol.) antibiotic/antimycotic mix. For all in vitro experiments, early-passage cells were used as previously determined by population doubling [25, 26].
For the apoptosis assay, the cells were allowed to reach 70% confluence and were then incubated with TNFα (10 ng/ml) in α-MEM + 0.5% FBS and 1% antibiotic/antimycotic mix. Apoptotic cells were labelled with annexin V–biotin or cleaved caspase-3 antibody. Five to eight random images were taken per well for the analysis. For the proliferation assay, 10% FBS medium was used to stimulate proliferation for 12 h. The BrdU Assay Kit (Cell Signaling, Danvers, MA, USA) was used according to the manufacturer’s protocol. BrdU incorporation was quantified by using a 96 well plate reader at λ = 450 nm (Infinite M200 PRO, Tecan, Morrisville, NC, USA).
Transfection experiments were carried out using Lipofectamine 2000 reagent and on-target plus smartpool human forkhead box protein O1 small interfering (si)RNA (10 nmol/l) in reduced-serum Opti-MEM for 6 h. The extraction of mRNA from each treatment group was carried out using an RNAeasy kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s protocol.
Statistical analysis
All statistical analyses were carried out with a software program (Minitab ver.16, www.minitab.com/en-us/). For insulin treatment and in vitro experiments, one-way ANOVA was used to determine statistical significance (p < 0.05). For pegsunercept-treatment experiments and appropriate in vitro experiments, two-way ANOVA followed by Tukey’s post-hoc test was used to denote significance. All data represent means ± SEM.
Results
Diabetes decreases CD271+ and Sca-1+ cells during endochondral ossification in healing fracture calluses
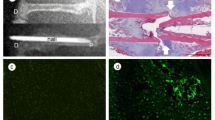
Active osteogenesis takes place in areas of endochondral bone formation during fracture healing, as shown in Fig. 1a. Diabetic mice exhibited a 53% decrease in new bone area in healing calluses compared with normoglycaemic mice (p < 0.05) (Fig. 1b). When bone area was normalised to the total callus area in groups, diabetic mice had 33% less bone formation (Fig. 1c). Diabetic mice also had reduced osteoblast numbers (Fig. 1d). We determined whether diabetes also led to a reduction in MSC numbers by immunohistochemistry using cluster designation-271 (also known as p75 neurotrophin receptor) and stem cell antigen 1 (Sca-1)-specific antibodies, which are reliable in vivo MSC markers in non-haematopoietic connective tissue [27, 28]. The numbers of CD271+ and Sca-1+ cells were significantly reduced in diabetic compared with normoglycaemic mice (p < 0.05) (Fig. 1e, f). The reduction in CD271+ and Sca-1+ MSCs was reversed when compared with diabetic mice with insulin treatment (p < 0.05), indicating that the decrease in MSCs was directly related to diabetic status. When examined by immunofluorescence, 21% of the total cell number were CD271+ in normoglycaemic mice and 13% were CD271+ in new bone in diabetic mice (p < 0.05) (Fig. 1g). Similar results were obtained when MSCs were examined as total CD271+ cells per mm2 (Fig. 1h). Even though there was relatively little haematopoietic tissue, we also examined MSCs as CD271+CD45− to rule out the possibility that cells were of haematopoietic origin. Almost all single CD271+ cells were also CD45−, confirming that the CD271+ cells detected were not haematopoietic (Fig. 1i). Interestingly, leucocyte numbers (CD45+ cells) were 1.5-fold higher in mice with diabetes compared with normoglycaemic mice (p < 0.05). This diabetic increase was restored to normal levels in diabetic mice that received insulin treatment (p < 0.05) (Fig. 1j), consistent with reports that diabetes enhances the inflammatory response during fracture healing [12].

Diabetes decreases the number of MSCs in areas of endochondral bone formation in healing fractures. (a) Left, H&E-stained section of a mouse tibial fracture callus on day 16 (original magnification ×40). Right, area of new endochondral bone formation (original magnification ×200). The dashed line demarcates the border between new bone and cartilage. (b) The area of new bone was quantified from H&E-stained sections of normoglycaemic mice, diabetic and diabetic + insulin-treated mice. (c) Bone formation normalised to total callus area among different groups. (d) Osteoblast counts were expressed as the number per mm of bone length in Toluidine Blue-stained sections. (e, f) CD271+ and Sca-1+ cells were quantified using an index ranging from 0 to 4 as described in Methods. (g–j) Images were analysed by immunofluorescence in normoglycaemic, diabetic and diabetic + insulin mice on day 16 after fracture. (g) CD271+ cells per total cell number determined by DAPI staining; (h) CD271+ cells per mm2; (i) CD271+CD45− cells per total cell number; (j) CD45+ cells per total cell number. n = 5 each. *p < 0.05 vs normoglycaemic or diabetic + insulin-treated mice. Diab, diabetic; Ins, insulin; NG, normoglycaemic; Ob, osteoblasts
The diabetes-induced decrease in MSC number is due to enhanced TNFα levels
Enhanced TNFα level is a key diabetic feature that contributes to diabetes-impaired soft and hard tissue wound healing [7, 29, 30]. Indeed, TNFα levels were 3.2-fold higher in diabetic compared with normoglycaemic mice, and pegsunercept treatment restored TNFα to normal levels (p > 0.05) (Fig. 2a). Moreover, diabetic mice had reduced bone formation and decreased numbers of osteoblasts, which were restored to normal levels by TNFα inhibition (p < 0.05) (Fig. 2b–d). The CD271+ MSC number decreased by almost 40% in diabetic mice (p < 0.05), and TNFα inhibition blocked the loss of MSCs caused by diabetes (p < 0.05) (Fig. 2e). Sca-1+ cells were also reduced by 50% in a TNFα-dependent manner in diabetic mice (Fig. 2f). These cells were not of haematopoietic origin as almost all Sca-1+ cells were CD45− (Fig. 2g). Moreover, pegsunercept treatment in diabetic mice decreased CD45+ cells to normoglycaemic levels (p < 0.05) (Fig. 2h). Thus, TNFα is largely responsible for the decrease in MSC number in diabetic fracture healing.

Inhibition of TNFα reduces the loss of MSC in diabetic but not normoglycaemic mice during fracture healing. Paraffin sections of 16 day femoral fracture calluses from the normoglycaemic mice, diabetic mice, normoglycaemic + pegsunercept and diabetic + pegsunercept were stained and analysed. (a) TNFα was measured as the mean fluorescent intensity in areas of newly formed bone in the fracture callus. (b) Bone area measured from the H&E-stained sections. (c) Bone formation normalised to total callus area in different groups. (d) Osteoblast counts were expressed as the number per mm bone. (e–h) Immunofluorescence analysis was carried out with CD271-, Sca-1- and CD45-specific antibodies. (e) CD271+ cells per total cell number; (f) Sca-1+ cells per total cell number; (g) Sca-1+CD45− cells per total cell number; (h) CD45+ cells per total cell number. n = 6 each. *p < 0.05 vs all groups. Diab, diabetic; NG, normoglycaemic; Ob, osteoblasts; PEG, pegsunercept
Diabetes increases apoptosis and decreases proliferation in MSCs
To identify potential mechanisms to explain the reduction in MSCs in fracture healing, we examined MSC apoptosis and proliferation. The number of TUNEL+ cells increased by 2.2-fold in diabetic fracture calluses compared with those in normoglycaemic mice (p < 0.05) (Fig. 3a). The number of apoptotic MSCs (double CD271+TUNEL+ cells) increased significantly, by 3.1-fold, in diabetic mice and insulin treatment blocked the MSC apoptosis (p < 0.05) (Fig. 3b). Immunofluorescence with a Ki67-specific antibody was used to detect actively proliferating cells. Diabetes reduced the number of Ki67+ cells by 54% (p < 0.05), and insulin treatment largely restored this (p < 0.05), so that it was similar to normoglycaemic levels (Fig. 3c). Diabetes reduced the number of proliferating MSCs (CD271+Ki67+) by 33% (p < 0.05), and insulin treatment in diabetic mice restored MSC proliferation (p < 0.05) similar to normal levels (Fig. 3d).

Diabetes increases apoptosis and decreases proliferation of MSCs in vivo. Fractures from 16 day specimens of normoglycaemic, diabetic and diabetic + insulin groups were examined for apoptotic and proliferating cells. Apoptotic cells were simultaneously identified with a fluorescent TUNEL assay or proliferating cells were identified by immunofluorescence with Ki67 antibody. Single- or double-positive cells were examined by multi-colour fluorescent microscopy. (a) TUNEL+ cells per total cell number; (b) CD271+TUNEL+ cells per total CD271+ cells; (c) Ki67+ cells per total cell number; (d) CD271+Ki67+ cells per total CD271+ cells. n = 5 each. *p < 0.05 vs normoglycaemic or diabetic + insulin. Diab, diabetic; Ins, insulin; NG, normoglycaemic
TNFα inhibition directly rescues diabetic effects in MSC apoptosis and proliferation
To determine how TNFα affects MSC apoptosis and proliferation, double immunofluorescence was performed on femoral fracture specimens treated with or without pegsunercept. Diabetic mice had a 3.6-fold increase in the total number of apoptotic cells compared with normoglycaemic mice (p < 0.05); this was reduced to normal values with pegsunercept treatment (p < 0.05) (Fig. 4a). Diabetic mice had a 3.9-fold increase in MSC apoptosis (p < 0.05), which was reversed with pegsunercept treatment (p < 0.05) (Fig. 4b). Approximately 13% of all cells in the new bone were proliferating as measured by Ki67 (Fig. 4c). In diabetic fractures, this number was lower at approximately 8%, and reverted to normal levels with pegsunercept treatment (p < 0.05) (Fig. 4c). When MSCs were examined, approximately 33% were proliferating; diabetes decreased this to about 21% (p < 0.05) (Fig. 4d). The reduction in MSC proliferation caused by diabetes was rescued by pegsunercept treatment (p < 0.05) (Fig. 4d). Taken together, our data suggest that elevated TNFα levels in diabetic fracture calluses induced higher levels of MSC apoptosis and reduced MSC proliferation.

Inhibition of TNFα rescues diabetes-induced apoptosis and anti-proliferation in MSCs. Femoral calluses 16 days after fracture in normoglycaemic and diabetic mouse groups treated with or without PEG were double-stained with CD271 antibody and apoptotic marker (TUNEL) or proliferation marker (Ki67). (a) TUNEL+ cells per total cell number; (b) CD271+TUNEL+ cells per total CD271+ cells; (c) Ki67+ cells per total cell number; (d) CD271+Ki67+ cells per total CD271+ cells. n = 6 each. *p < 0.05 vs normoglycaemic and diabetic + pegsunercept; † p < 0.05 vs normoglycaemic. Diab, diabetic; NG, normoglycaemic; PEG, pegsunercept
TNFα directly induces apoptosis and reduces proliferation in mouse MSCs and hBMSCs in vitro
To investigate whether TNFα alone is sufficient to induce diabetes-like effects in MSCs, in vitro experiments were carried out using the mouse MSC (mMSC) line, C3H10T1/2. Annexin V was used to detect apoptotic cells with membrane disruption. Annexin V+ mMSCs increased by 1.6-fold after 24 h of TNFα treatment (p > 0.05), and to 2.9-fold after 72 h (p < 0.05) (Fig. 5a). Similarly, TNFα increased cleaved caspase-3+ mMSCs, a downstream initiator of apoptosis, by 3.7-fold (p < 0.05) (Fig. 5b). Moreover, TNFα treatment decreased the mMSC proliferation rate after 24 h and 72 h by 36% and 73%, respectively (p < 0.05) (Fig. 5c). Primary hBMSCs were also examined. TNFα stimulated apoptosis in hBMSC, reflected by a 2.8-fold increase in annexin V+ cells and a 2.4-fold increase in cleaved caspase 3+ cells (p < 0.05) (Fig. 5d, e). The proliferation of hBMSCs decreased by 69% with TNFα stimulation (p < 0.05) (Fig. 5f). Thus, TNFα directly stimulates apoptosis and reduces proliferation in both mMSCs and hBMSCs, similar to the effect of diabetes shown in vivo.

TNFα induces apoptosis and reduces proliferation in mMSCs and hBMSCs. C3H10T1/2 cells (mMSC) and hBMSCs were treated with TNFα (10 ng/ml) for 24–72 h. Apoptosis was detected using annexin V and cleaved caspase-3 antibody, and proliferation was measured by the BrdU assay. (a, d) Annexin V+ cells per total cell number in mMSCs and hBMSCs, respectively. (b, e) Cleaved caspase-3+ cells per total cell number after 72 h of TNFα treatment in mMSCs and hBMSCs, respectively. (c, f) BrdU levels after serum-stimulated proliferation, measured at absorbance = 450 nm. *p < 0.05 vs 0 h treatment
Diabetes enhances forkhead box protein O1 nuclear localisation in MSCs in a TNFα-dependent manner
Forkhead box protein O1 (FOXO1), a transcription factor induced by TNFα, is implicated in the regulation of apoptosis and proliferation [31]. The percentage of all nucleated cells with FOXO1 localised to the nucleus increased by 5.8-fold in fractures in diabetic compared with normoglycaemic mice (p < 0.05) (Fig. 6a). TNFα inhibition significantly reduced FOXO1+ nuclear localisation (p < 0.05) (Fig. 6a). In contrast, diabetes had little effect on the overall level of FOXO1, as approximately 30% of nucleated cells were FOXO1+ in normoglycaemic and diabetic fracture calluses (p > 0.05) (Fig. 6b). When Sca-1+ MSCs were examined, more than 90% of Sca-1+ cells were FOXO1+, with little difference detected between groups (Fig. 6c). However, diabetes greatly increased the nuclear localisation of FOXO1+ in Sca-1+ MSC, and this was significantly reduced with TNFα inhibition (p < 0.05) (Fig. 6d). These data suggest that diabetes-induced TNFα stimulates nuclear localisation of FOXO1 in MSCs but does not affect its production.

TNFα inhibition reduces FOXO1 nuclear localisation in diabetic fractures in vivo. Immunofluorescence was carried out in femoral calluses 16 days after fracture in NG and diabetic mouse groups treated with or without PEG using Sca-1 and/or FOXO1 antibody. FOXO1+ and Sca-1+FOXO1+ cell numbers were examined. Nuclear localisation of FOXO1 was determined by the co-localisation of FOXO1 and DAPI stain. (a) Nuclear FOXO1+ cells per total cell number; (b) total FOXO1+ (cytoplasm or nucleus) per total cell number; (c) FOXO1+ cells per total Sca-1+ cells; (d) the ratio of Sca-1+nuclear FOXO1+ cells to Sca-1+total FOXO1+ cells. n = 6 each. *p < 0.05 vs all groups. Diab, diabetic; NG, normoglycaemic; PEG, pegsunercept
FOXO1 knockdown prevents TNFα-mediated MSC apoptosis and anti-proliferation
To establish that TNFα directly induces FOXO1 activity in MSCs, in vitro assays were carried out in hBMSCs following the approach described above. TNFα stimulated FOXO1 nuclear localisation after 24 h but did not alter overall FOXO1 levels (Fig. 7a, b). The effect of TNFα was examined in hBMSCs with FOXO1 knockdown. Specific siRNA reduced FOXO1 mRNA levels by approximately 80% and protein levels by 60% (Fig. 7c, d). TNFα-stimulated annexin V+ hBMSCs were reduced by 43%, and cleaved caspase 3+ hBMSCs were reduced by 33% with FOXO1 knockdown (p < 0.05) (Fig. 7e, f). Moreover, TNFα reduced serum-stimulated hBMSC proliferation by 45%, and FOXO1 silencing restored this inhibition by 90% of the decrease (p < 0.05) (Fig. 7g).

Silencing FOXO1 interferes with TNFα-mediated apoptosis and anti-proliferation. The hBMSCs were incubated in TNFα for 72 h, and immunofluorescence was carried out with FOXO1 antibody. For some experiments, hBMSCs were transfected with scrambled siRNA or FOXO1 siRNA (siFOXO1) for 6 h prior to TNFα incubation. (a) Nuclear FOXO1+ cells per total cell number; (b) FOXO1 levels measured by mean fluorescent intensity per cell. (c, d) Relative FOXO1 mRNA levels (c) and FOXO1 protein levels (d) after siFOXO1 silencing. (e) Annexin V+ cells per total cell number; (f) cleaved caspase-3+ cell per total cell number; (g) BrdU levels after serum-stimulated proliferation. * p < 0.05 vs control counterpart; † p < 0.05 vs siRNA controls. mfi, mean fluorescent intensity; siScr, scrambled siRNA
TNFα upregulates FOXO1-dependent apoptotic and cell cycle arrest genes in hBMSCs
To understand the molecular mechanism by which FOXO1 affects MSCs under inflammatory conditions, genes that regulate apoptosis and cell cycle were evaluated further. Among the apoptotic genes examined (BIM [also known as BCL2L11], Fas ligand [FASLG], caspase-3 [CASP3], caspase-8 [CASP8] and caspase-9 [CASP9]), caspase-8 and caspase-9 were significantly upregulated by TNFα. For both, silencing FOXO1 restored the mRNA levels to a normal value (Fig. 8a, b). When genes that regulate proliferation were examined (CDK1, cyclin D [CCND1], P21 [also known as CDKN1A] and P27 [also known as CDKN1B]), P21 and P27 mRNA levels were upregulated by TNFα, with the effect on both mRNAs blocked by FOXO1 knockdown (Fig. 8c, d).

TNFα-mediated apoptosis and cell cycle gene regulation is FOXO1 dependent. hBMSCs were transfected with control (scrambled siRNA) or FOXO1 siRNA, followed by TNFα treatment for 72 h. Quantitative RT-PCR was carried out and ribosomal protein L32 mRNA was used to normalise the mRNA values. (a–d) Quantification of relative mRNA fold changes for: (a) CASP8; (b) CASP9; (c) P21; and (d) P27. * p < 0.05 vs all groups
Discussion
A better understanding of how diabetes impairs fracture healing is important. Diabetes is an increasingly important healthcare issue and the disease has a significant impact on the skeleton and interferes with fracture repair [32, 33]. Here, we report the deleterious effects of diabetes on MSC viability during fracture healing. Diabetes reduced the number of endogenous MSCs in areas of endochondral bone formation, with the number restored to normal values by insulin treatment. To better understand mechanistically how diabetes affects MSCs, diabetic animals were treated with the TNFα inhibitor, pegsunercept.
Pegsunercept significantly increased the MSC number, demonstrating that diabetes-enhanced inflammation played a major role in reducing the MSC number. Furthermore, diabetes significantly increased MSC apoptosis and reduced proliferation in vivo. These negative effects were mediated by diabetes-enhanced inflammation. In vitro experiments demonstrated that TNFα directly increased MSC apoptosis and reduced DNA synthesis. The effect of TNFα was dependent on FOXO1 activity.
To quantify MSCs in vivo, specific markers were used, CD271 and Sca-1, which accurately identify MSC in areas with little haematopoietic tissue [28, 34]. Moreover, virtually all CD271+ and Sca-1+ cells were CD45−. CD271+ cells found in dermal and adipose tissue exhibit robust tri-lineage mesenchymal potential [34]. In addition, CD271+ cells administered in vivo home to fracture sites in mice [35]. Furthermore, Sca-1 is found on MSCs in murine synovial membrane and skeletal muscle that exhibits mesengenic properties [36, 37].
STZ-induced diabetes drastically reduced the number of MSCs in areas of new bone. Insulin treatment reversed serum glucose to normal levels in STZ-treated mice. It also restored the number of MSCs to normal levels in STZ-diabetic mice, together with the amount of new bone and osteoblast numbers. Thus, the reduction is not an untoward effect of STZ-induced diabetes in this model. Previous studies have demonstrated that bone marrow samples derived from diabetic mice have fewer MSCs compared with normoglycaemic animals [38, 39], and have significantly decreased MSC-colony-forming units ex vivo [40]. Our studies demonstrate for the first time that diabetic animals exhibit lower numbers of local endogenous MSCs in fracture callus, a mesenchymal tissue, in accordance with decreased bone formation in diabetic fracture healing.
Here, we have identified TNFα-mediated MSC apoptosis and anti-proliferation as the two mechanisms that explain the reduced MSC numbers in diabetic fracture healing. MSC apoptosis was returned to normal values when TNFα was inhibited in vivo, and TNFα sufficiently induced caspase-3 activation and apoptosis in hBMSCs in vitro. Previous in vitro studies have demonstrated that high glucose levels stimulate apoptosis and induce senescence in adipose-derived MSCs [41]. Consistent with these findings, a recent study showed that TNFα induced apoptosis in rat MSCs in vitro [42].
An essential component of an adequate healing process is the proliferative capacity of regenerative cells. MSC proliferation was reduced by 33% in diabetic fracture calluses, and TNFα inhibition significantly restored MSC proliferation. Moreover, TNFα induced upregulation of genes that interfere with progression through the cell cycle in a FOXO1-dependent manner. No previous in vivo studies have identified this mechanism for the effect of diabetes on MSC proliferation during fracture healing. Increased TNFα levels may delay the healing process in other mesenchymal tissues affected by diabetes.
FOXO1 is a transcription factor that regulates apoptosis and proliferation, and it is subject to activation by diabetic inflammatory stimuli in fibroblasts [29]. Diabetes increased FOXO1 nuclear localisation in MSCs in diabetic fracture approximately sixfold, and TNF inhibition significantly reduced this increase. FOXO1 knockdown in hBMSCs prevented the negative impact of TNFα on MSC apoptosis and proliferation. This is consistent with previous reports that FOXO1 plays an adverse role in fibroblasts [29], microvascular cells [43] and chondrocytes [7] in diabetic conditions. Thus, a high level of FOXO1 activation may mediate some of the negative effects of diabetes and contribute to diabetic complications.
Treatment of diabetic fractures with MSCs can promote healing [44]. Our studies suggest an alternative approach, which would be to enhance survival and support the proliferation of endogenous MSCs by reducing prolonged inflammation in diabetic fracture healing. This may be a simpler and more pragmatic approach than treatment of diabetic fractures with MSCs. Thus, targeting endogenous MSCs in order to maximise their regenerative potential using pharmacological intervention may ultimately be safer and more cost effective.
Abbreviations
- CD271:
-
Cluster designation-271 (also known as p75 neurotrophin receptor)
- FOXO1:
-
Forkhead box protein O1
- hBMSC:
-
Human bone-marrow-derived mesenchymal stem cell
- mMSC:
-
Mouse mesenchymal stem cell
- MSC:
-
Mesenchymal stem cell
- Sca-1:
-
Stem cell antigen-1
- si:
-
Small interfering
- STZ:
-
Streptozotocin
References
Shaw JE, Sicree RA, Zimmet PZ (2010) Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res Clin Pract 87:4–14
Khazai NB, Beck GR Jr, Umpierrez GE (2009) Diabetes and fractures: an overshadowed association. Curr Opin Endocrinol Diabetes Obes 16:435–445
Chaudhary SB, Liporace FA, Gandhi A, Donley BG, Pinzur MS, Lin SS (2008) Complications of ankle fracture in patients with diabetes. J Am Acad Orthop Surg 16:159–170
Adami S (2009) Bone health in diabetes: considerations for clinical management. Curr Med Res Opin 25:1057–1072
Graves DT, Kayal RA (2008) Diabetic complications and dysregulated innate immunity. Front Biosci 13:1227–1239
King GL (2008) The role of inflammatory cytokines in diabetes and its complications. J Periodontol 79:1527–1534
Kayal RA, Siqueira M, Alblowi J et al (2010) TNF-alpha mediates diabetes-enhanced chondrocyte apoptosis during fracture healing and stimulates chondrocyte apoptosis through FOXO1. J Bone Miner Res 25:1604–1615
Roszer T (2011) Inflammation as death or life signal in diabetic fracture healing. Inflamm Res 60:3–10
Moseley KF (2012) Type 2 diabetes and bone fractures. Curr Opin Endocrinol Diabetes Obes 19:128–135
Schindeler A, McDonald MM, Bokko P, Little DG (2008) Bone remodeling during fracture repair: the cellular picture. Semin Cell Dev Biol 19:459–466
Kayal RA, Alblowi J, McKenzie E et al (2009) Diabetes causes the accelerated loss of cartilage during fracture repair which is reversed by insulin treatment. Bone 44:357–363
Alblowi J, Tian C, Siqueira MF et al (2013) Chemokine expression is upregulated in chondrocytes in diabetic fracture healing. Bone 53:294–300
Gerstenfeld LC, Cho TJ, Kon T et al (2003) Impaired fracture healing in the absence of TNF-alpha signaling: the role of TNF-alpha in endochondral cartilage resorption. J Bone Miner Res 18:1584–1592
Yang X, Ricciardi BF, Hernandez-Soria A, Shi Y, Pleshko Camacho N, Bostrom MP (2007) Callus mineralization and maturation are delayed during fracture healing in interleukin-6 knockout mice. Bone 41:928–936
Diarra D, Stolina M, Polzer K et al (2007) Dickkopf-1 is a master regulator of joint remodeling. Nat Med 13:156–163
Alblowi J, Kayal RA, Siqueria M et al (2009) High levels of tumor necrosis factor-alpha contribute to accelerated loss of cartilage in diabetic fracture healing. Am J Pathol 175:1574–1585
Caplan AI, Dennis JE (2006) Mesenchymal stem cells as trophic mediators. J Cell Biochem 98:1076–1084
Chen J, Li Y, Katakowski M et al (2003) Intravenous bone marrow stromal cell therapy reduces apoptosis and promotes endogenous cell proliferation after stroke in female rat. J Neurosci Res 73:778–786
Min JY, Sullivan MF, Yang Y et al (2002) Significant improvement of heart function by cotransplantation of human mesenchymal stem cells and fetal cardiomyocytes in postinfarcted pigs. Ann Thorac Surg 74:1568–1575
Granero-Molto F, Weis JA, Miga MI et al (2009) Regenerative effects of transplanted mesenchymal stem cells in fracture healing. Stem Cells 27:1887–1898
Hare JM, Fishman JE, Gerstenblith G et al (2012) Comparison of allogeneic vs autologous bone marrow-derived mesenchymal stem cells delivered by transendocardial injection in patients with ischemic cardiomyopathy: the POSEIDON randomized trial. JAMA 308:2369–2379
Ciccocioppo R, Bernardo ME, Sgarella A et al (2011) Autologous bone marrow-derived mesenchymal stromal cells in the treatment of fistulising Crohn's disease. Gut 60:788–798
Gerstenfeld LC, Alkhiary YM, Krall EA et al (2006) Three-dimensional reconstruction of fracture callus morphogenesis. J Histochem Cytochem 54:1215–1228
Kayal RA, Tsatsas D, Bauer MA et al (2007) Diminished bone formation during diabetic fracture healing is related to the premature resorption of cartilage associated with increased osteoclast activity. J Bone Miner Res 22:560–568
Wang H, Brennan TA, Russell E et al (2013) R-Spondin 1 promotes vibration-induced bone formation in mouse models of osteoporosis. J Mol Med 91:1421–1429
Cristofalo VJ, Allen RG, Pignolo RJ, Martin BG, Beck JC (1998) Relationship between donor age and the replicative lifespan of human cells in culture: a reevaluation. Proc Natl Acad Sci U S A 95:10614–10619
Bernardo ME, Locatelli F, Fibbe WE (2009) Mesenchymal stromal cells. Ann N Y Acad Sci 1176:101–117
Xiao J, Yang X, Jing W et al (2013) Adipogenic and osteogenic differentiation of Lin(-)CD271(+)Sca-1(+) adipose-derived stem cells. Mol Cell Biochem 377:107–119
Siqueira MF, Li J, Chehab L et al (2010) Impaired wound healing in mouse models of diabetes is mediated by TNF-alpha dysregulation and associated with enhanced activation of forkhead box O1 (FOXO1). Diabetologia 53:378–388
Liu R, Bal HS, Desta T, Behl Y, Graves DT (2006) Tumor necrosis factor-alpha mediates diabetes-enhanced apoptosis of matrix-producing cells and impairs diabetic healing. Am J Pathol 168:757–764
Xu F, Zhang C, Graves DT (2013) Abnormal cell responses and role of TNF-alpha in impaired diabetic wound healing. Biomed Res Int 2013:754802
Clarke JL (2010) Building a coordinated care model for diabetes management. Popul Health Manag 13(Suppl 1):S3–S13
Saller A, Maggi S, Romanato G, Tonin P, Crepaldi G (2008) Diabetes and osteoporosis. Aging Clin Exp Res 20:280–289
Vaculik C, Schuster C, Bauer W et al (2012) Human dermis harbors distinct mesenchymal stromal cell subsets. J Invest Dermatol 132:563–574
Dreger T, Watson JT, Akers W et al (2014) Intravenous application of CD271-selected mesenchymal stem cells during fracture healing. J Orthop Trauma 28(Suppl 1):S15–S19
Kurth TB, Dell'accio F, Crouch V, Augello A, Sharpe PT, De Bari C (2011) Functional mesenchymal stem cell niches in adult mouse knee joint synovium in vivo. Arthritis Rheum 63:1289–1300
Valero MC, Huntsman HD, Liu J, Zou K, Boppart MD (2012) Eccentric exercise facilitates mesenchymal stem cell appearance in skeletal muscle. PLoS One 7:e29760
Shin L, Peterson DA (2012) Impaired therapeutic capacity of autologous stem cells in a model of type 2 diabetes. Stem Cells Transl Med 1:125–135
Gallagher KA, Liu ZJ, Xiao M et al (2007) Diabetic impairments in NO-mediated endothelial progenitor cell mobilization and homing are reversed by hyperoxia and SDF-1 alpha. J Clin Invest 117:1249–1259
Stolzing A, Sellers D, Llewelyn O, Scutt A (2010) Diabetes induced changes in rat mesenchymal stem cells. Cells Tissues Organs 191:453–465
Cramer C, Freisinger E, Jones RK et al (2010) Persistent high glucose concentrations alter the regenerative potential of mesenchymal stem cells. Stem Cells Dev 19:1875–1884
Mao J, Lv Z, Zhuang Y (2014) MicroRNA-23a is involved in tumor necrosis factor-alpha induced apoptosis in mesenchymal stem cells and myocardial infarction. Exp Mol Pathol 97:23–30
Behl Y, Krothapalli P, Desta T, Roy S, Graves DT (2009) FOXO1 plays an important role in enhanced microvascular cell apoptosis and microvascular cell loss in type 1 and type 2 diabetic rats. Diabetes 58:917–925
Breitbart EA, Meade S, Azad V et al (2010) Mesenchymal stem cells accelerate bone allograft incorporation in the presence of diabetes mellitus. J Orthop Res 28:942–949
Acknowledgements
We would like to thank M. Foroozia (University of Pennsylvania, School of Dental Medicine) for technical assistance with this manuscript.
Funding
This work was supported by National Institute of Health/National Institute of Arthritis and Muscular and Skin Diseases (NIH/NIAMS) grant AR-060055-03 (DTG) and R01AG028873 (RJP).
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
DTG, KIK and LSC contributed to the study conception and design. DTG is responsible for the integrity of the work as a whole. All authors contributed to the acquisition of data or analysis and interpretation of data, revised the manuscript and approved the final version of the manuscript to be published.
Author information
Authors and Affiliations
Corresponding author
Additional information
Kang I. Ko and Leila S. Coimbra contributed equally to this work
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Table 1
(PDF 7 kb)
ESM Table 2
(PDF 13 kb)
Rights and permissions
About this article

Cite this article
Ko, K.I., Coimbra, L.S., Tian, C. et al. Diabetes reduces mesenchymal stem cells in fracture healing through a TNFα-mediated mechanism. Diabetologia 58, 633–642 (2015). https://doi.org/10.1007/s00125-014-3470-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-014-3470-y