
Abstract
The renal collecting system serves the fine-tuning of renal acid–base secretion. Acid-secretory type-A intercalated cells secrete protons via a luminally expressed V-type H+-ATPase and generate new bicarbonate released by basolateral chloride/bicarbonate exchangers including the AE1 anion exchanger. Efficient proton secretion depends both on the presence of titratable acids (mainly phosphate) and the concomitant secretion of ammonia being titrated to ammonium. Collecting duct ammonium excretion requires the Rhesus protein RhCG as indicated by recent KO studies. Urinary acid secretion by type-A intercalated cells is strongly regulated by various factors among them acid–base status, angiotensin II and aldosterone, and the Calcium-sensing receptor. Moreover, urinary acidification by H+-ATPases is modulated indirectly by the activity of the epithelial sodium channel ENaC. Bicarbonate secretion is achieved by non-type-A intercalated cells characterized by the luminal expression of the chloride/bicarbonate exchanger pendrin. Pendrin activity is driven by H+-ATPases and may serve both bicarbonate excretion and chloride reabsorption. The activity and expression of pendrin is regulated by different factors including acid–base status, chloride delivery, and angiotensin II and may play a role in NaCl retention and blood pressure regulation. Finally, the relative abundance of type-A and non-type-A intercalated cells may be tightly regulated. Dysregulation of intercalated cell function or abundance causes various syndromes of distal renal tubular acidosis underlining the importance of these processes for acid–base homeostasis.
Similar content being viewed by others


Introduction
Extracellular pH and systemic acid–base status are critical for normal organ and cellular function. Deranged acid–base status is associated with higher morbidity and mortality in patients with chronic kidney disease [15]. Extracellular pH affects bone density and stability, and mild chronic metabolic acidosis has been suspected to contribute to osteopenia and osteoporosis [10, 77]. Rickets and osteomalacia are often observed in patients with inborn syndromes of renal tubular acidosis (see below). Extracellular acidosis affects skeletal muscle metabolism and induces a catabolic state [15]. In the setting of chronic kidney disease, metabolic acidosis contributes to peripheral insulin resistance and lower leptin secretion [46, 109, 186]. Thus, extracellular pH has to be tightly kept in the normal physiologic range of pH 7.36–7.44 to maintain normal organ and body function.
Acid–base status is influenced and regulated by the activity of many organs including skeletal muscle (i.e., exercise), intestine (i.e., loss of acid or bicarbonate), bone (i.e., incorporation or release of carbonate and phosphate), and by dietary intake or physical activity. Kidney and respiration play a central role in both controlling and maintaining systemic acid–base status by affecting the levels of pCO2, bicarbonate, and nonvolatile acids.
The kidney controls and maintains systemic acid–base status by three intricately linked mechanisms: the reabsorption of filtered bicarbonate, the excretion of acids (or, if necessary, of alkali), and the de novo generation of ammonium and bicarbonate. The latter process allows the excretion of acids and the replenishing of bicarbonate used to buffer acids. The reabsorption of filtered bicarbonate is mostly achieved by the proximal tubule and to a lesser extent by the thick ascending limb and the distal convoluted tubule (for review, [68]). Urine entering the connecting tubule contains only minute amounts of bicarbonate [68].
Ammoniagenesis occurs only in the proximal tubule; the mechanisms of ammonium excretion will be discussed in more detail below. The ultimate fine-tuning of renal acid or base excretion takes place in the various segments of the collecting system involving various cell types and distinct transport proteins and is subject to tight regulation. The importance of collecting duct acid–base excretion to overall systemic acid–base balance is highlighted by several rare inherited disorders affecting collecting duct acid–base transport proteins or their regulation.
Classic work performed by R.F. Pitts, G. Giebisch, G. Malnic, M.L. Halperin, R. Richterich, R.C. Morris, A. Sebastian, N.E. Madias, H.N. Hulter, W.B. Schwartz, P.R.Steinmetz, R.J. Alpern, Q. Al-Awqati, and many others has elucidated and described the fundamental processes that contribute to urinary acidification using a variety of techniques and animal models. These data are the basis for the more recent molecular approaches dissecting the mechanisms of urinary acidification.
Extensive in vivo experiments in various species including dog, guinea pig, rabbit, mouse, rat, and humans indicate that distinct differences exist regarding the basal rates of urinary acidification, the extent of adaptive responses to alkali or acid loading, and the morphological characteristics of the collecting duct (i.e., relative number of different intercalated cell subtypes). Among other reasons, the specific dietary requirements of these species (relative dietary alkali or acid load, electrolyte content) have been discussed as potential explanations. However, it is beyond the scope of this review to discuss these differences.
The purpose of this article is to give a short overview of the mechanisms of acid–base excretion along the collecting system and its regulation on various levels and to discuss briefly dysregulation and inherited disorders of these mechanisms leading to distal renal tubular acidosis (dRTA). This review will discuss mainly more recent data coming from molecular and functional studies in mouse and rats or from genetic studies in humans.
Various segments and cells along the collecting duct contribute to renal acid–base regulation
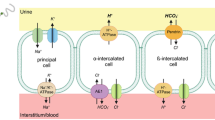
Cells contributing to final urinary acidification are distributed over several segments along the nephron and collecting system. The first intercalated cells, characterized by the expression of luminal H+-ATPases and the anion exchangers AE1 (Anion exchanger 1) or pendrin occur in the late distal convoluted tubule (DCT2) [25, 91, 105, 184]. The subsequent segments of the collecting system, namely the connecting tubule (CNT), the cortical collecting duct (CCD), the outer medullary collecting duct (OMCD), and the initial third of the inner medullary collecting duct (iIMCD) contain various subtypes of intercalated cells. At least two subtypes of intercalated cells can be distinguished based on the expression of specific proteins: type-A intercalated cells and non-type-A intercalated cells (Fig. 1). Type-A intercalated cells are characterized by the presence of luminal H+-ATPases and a basolateral anion exchanger, AE1. In contrast, non-type-A intercalated cells express the anion exchanger pendrin on the luminal pole. H+-ATPase expression in non-type-A intercalated cells may be luminal, basolateral, or both membranes [91, 184, 202]. However, some authors have further subclassified non-type-A intercalated cells based on subcellular H+-ATPase distribution [91, 184]. Non-type-A intercalated cells also express the NHE regulatory factor 1 (NHERF1) [32]. All types of intercalated cells express carbonic anhydrase II and the transcription factor Foxi1 (forkhead box I1) as additional cell-specific markers [27]. Importantly, type-A intercalated cells are dispersed from the late distal convoluted tubule to the initial inner medullary collecting duct. In contrast, non-type-A intercalated cells are mostly expressed in the DCT2 and connecting tubule and less in the cortical collecting duct. Only a few non-type-A intercalated cells are found in the outer stripe of the outer medulla in adult kidney [92, 170, 207]. Pendrin-positive cells are found also in the inner medulla and inner stripe of the outer medulla during nephrogenesis but disappear during the first postnatal days [29, 170].

Two types of intercalated cells. Intercalated cells are expressed from the late distal convoluted tubule to the initial third of the inner medullary collecting duct (red shaded). Left cell model of non-type-A intercalated cell. These cells express on the luminal membrane the chloride/bicarbonate exchanger pendrin mediating bicarbonate excretion and chloride absorption. Bicarbonate is produced from CO2 and H2O catalyzed by carbonic anhydrase II (CAII). Non-type-A intercalated cells express also V-type H+-ATPases which can be found on the basolateral and/or luminal membrane and which may drive pendrin transport activity. Chloride is released across the basolateral membrane through chloride channels that consist of ClC-kb and Barttin subunits. Right cell model of type-A intercalated cell. Bicarbonate and proton generation is catalyzed by CAII providing protons for luminal V-type H+-ATPases and bicarbonate for basolateral chloride/bicarbonate exchangers including AE1. Type-A intercalated cells also express basolateral KCC4 KCl-cotransporters that may function in maintaining in low intracellular chloride. Type-A intercalated cells express also on their luminal membrane H+/K+-ATPases that are not further discussed in this review and serve mostly preservation of potassium during potassium deficiency. Moreover, both type-A and non-type-A intercalated cells participate in ammonium excretion as further detailed in Fig. 3
The classic view states that type-A intercalated cells secrete acid whereas non-type-A intercalated cells are responsible for bicarbonate excretion. This simple classification is challenged by several recent findings. The luminal expression of several subunits of intercalated cell specific H+-ATPase has been detected in the principal cells of the connecting tubule (Wagner, Loffing, unpublished observations). Moreover, intercalated cells may serve not only acid–base transport but also the regulation of electrolyte homeostasis. Pendrin function, as discussed below, may contribute importantly to chloride reabsorption along the CNT and CCD. Genetic ablation of the B1 H+-ATPase subunit in mice causes a syndrome of massive salt loss (Chambrey, Loffing, Wagner, Eladari, unpublished data) which has also been described in patients with classic forms of dRTA [160]. Moreover, expression of flow-activated potassium channels (maxiK) has been described in intercalated cells, thereby contributing to potassium homeostasis [128, 129]. However, it is beyond the scope of this review to discuss the potential role of intercalated cells in electrolyte homeostasis.
The relative contribution of these subsegments to acid–base excretion and final urinary acidification has been difficult to establish. The importance of the segments between the distal tubule and inner medullary collecting duct for urinary acidification has been recognized a long time ago by Ullrich et al. and Gottschalk et al. using microcatheters and microperfusion [63, 191] and the kinetics of acidification were determined under conditions of acidosis and alkalosis by Malnic and Giebisch [61, 199]. These data demonstrated that urinary pH is approximately 0.4 units pH lower than plasma pH in the distal convoluted tubule and acidifies by as much as 2 units pH reaching values of approximately pH 5.5 at the medullary tip. However, the exact quantitative contribution of single subsegments of the collecting duct system has remained unclear since these segments are not accessible to micropuncture. Studies in mice lacking either the B1 H+-ATPase subunit (ATP6V1B1) or the alpha subunit of the epithelial Na+ channel (ENaC) have shed some light on the importance of the connecting tubule as a major segment [97]. Application of loop diuretics increases electrogenic urinary acidification, an effect abolished by inhibitors of ENaC function such as amiloride or triamterene [73]. In mice lacking the B1 H+-ATPase subunit, specifically expressed in intercalated cells, urinary pH is more alkaline at baseline and does not acidify upon furosemide application. In contrast, in mice that lack αENaC expression in all segments of the collecting duct but with preserved αENaC expression in the connecting tubule, urinary pH is acidified normally after furosemide treatment. These data indicate that the connecting tubule is sufficient to maintain normal electrogenic urinary acidification [97].
The major transport proteins
H-ATPases
V-type H+-ATPases are multisubunit protein complex consisting of at least 14 subunits in humans (for a detailed review on H+-ATPase structure, function, and regulation, see recent reviews: [54, 55, 119, 122, 202]). Most subunits occur in different isoforms that may be specific to organs, cell types, or subcellular organelles. In general, H+-ATPases are organized in two domains, a cytosolic domain (V1) binding and hydrolyzing ATP and a membrane bound domain (V0) mediating proton translocation (Fig. 2). Both domains are connected through a stalk-like structure. Activity of H+-ATPases may be regulated by trafficking, domain assembly/disassembly, and changes in the ratio of ATP hydrolysis/H+-pumping as well as by other means [55, 122, 202]. H+-ATPases couple the hydrolysis of ATP to the movement of protons across membranes and are found not only in the plasma membrane but are also mostly expressed in many intracellular organelles. Subcellular localization, regulation, and function of different H+-ATPase populations may at least in part be regulated by the presence of specific subunit isoforms. The B1, a4, and d2 isoforms have been labeled as intercalated cell specific. However, these isoforms are expressed also in other organs such as epididymis or inner ear and are found also in the thick ascending limb of the loop of Henle (B1) or the proximal tubule (a4) [53, 173]. Localization and expression of the d2 subunit has not been reported in full detail to date [166, 168].

Model of the structure of vacuolar H+-ATPases. H+-ATPases are multisubunit membrane-bound enzymes consisting of two major subunits, a cytosolic V1 domain (shaded in orange-red) and the membrane associated V0 domain (blue colored). Both domains are connected by a stalk that mediates the energy from ATP hydrolysis to H+-transfer. Mutations in the B1 isoform of the B subunit (shaded in red) cause distal renal tubular acidosis with sensorineural deafness. Moreover, mutations have been found in the a2, a3, and a4 isoforms of the a subunit (shaded in dark blue) in patients with cutis laxa (a2), osteopetrosis (a3), or distal renal tubular acidosis (a4). Figure adapted from references [55, 202]
Moreover, staining for several H+-ATPase subunits including B1 and a4 has been observed also in cells in the connecting tubule that express ENaC or AQP2 (Aquaporin 2 water channel), typical markers of principal cells. The function of these subunits or full pumps in principal cells has not been investigated in detail. Whether pumps in principal cells of the CNT contribute to urinary acidification is unknown. H+-ATPase staining of principal cells is not detected in the CCD and later segments.
Expression of H+-ATPases along the nephron during embryonic development in mouse kidney occurs apparently differentially in the different segments of the nephron in a cell-type-specific coordinated manner [79]. Also after birth, H+-ATPase expression and abundance increases and reaches adult levels only after about 18–20 days postnatally coinciding with weaning and full urinary acidification [29].
Anion exchangers: AE1 and pendrin
The collecting duct expresses a variety of anion exchangers including members of the SLC4 transporter family ((Anion exchanger isoforms 1–4) AE1, AE2, AE3, AE4, NBCn1 (electroneutral sodium-bicarbonate cotransporter 1)) and the SLC26 transporter family (pendrin, SLC26A7). The role and regulation of AE2, AE3, AE4, NBCn1, and SLC26A7 in the collecting duct are mostly unknown; phenotypes of the respective KO mouse models have not been reported. Thus, this section of the review will focus only on the function and regulation of AE1 and pendrin in the collecting duct.
AE1
AE1 belongs to a subfamily of electroneutral anion exchangers of the SLC4 family of bicarbonate transporters (for review, [7, 38, 138, 144]). The AE1 isoform of the kidney is an N-terminally truncated version of the red blood cell band3/AE1 protein due to alternative splicing of the first exon. Kidney AE1 (kAE1) lacks the first 65 amino acids in humans [8, 96]. AE1 expression is basolateral and its presence characterizes type-A intercalated cells in the collecting system [9]. AE1 mediates basolateral release of intracellular bicarbonate against extracellular chloride, thereby secreting newly generated bicarbonate into the interstitial space/blood. AE1 may form a transport metabolon together with carbonic anhydrase II which is bound to a C-terminal stretch of amino acids and enhances AE1 transport activity [176]. Similarly, AE1 may also interact with the extracellular carbonic anhydrase isoform IV (CA IV) [175].
The importance of AE1 for normal acid–base status is underlined by the fact that mutations in AE1 cause distal renal tubular acidosis as discussed below. A mouse model lacking AE1 in red blood cells and kidney demonstrated massive hyperchloremic metabolic acidosis [174]. In freshly isolated OMCD type-A intercalated cells, the specific AE1 inhibitor diBA(5)C4 reduced total chloride/bicarbonate exchange activity only by about 50% and had no effect in OMCDs from AE1 deficient mice. More surprisingly, total chloride/bicarbonate exchange activity was only reduced by 30% in type-A intercalated cells from AE1 KO mice [174]. Thus, type-A intercalated cells express several basolateral chloride/bicarbonate exchangers, but the contribution of AE1 is critical to normal function, which other anion exchanger(s) mediate basolateral chloride/bicarbonate exchange has not been elucidated to date. Due to the lack of specific inhibitors, it has remained difficult to distinguish different anion exchangers.
Little is known about the regulation of AE1 abundance, polarized expression, and activity. Williamson et al. have shown that trafficking of kAE1may be regulated by the phosphorylation status of two tyrosine residues (Y359 and Y904) which may affect polarization as well as recycling of AE1 [215]. Whether phosphorylation of these residues occurs in vivo and by which stimuli is unknown. Trafficking of AE1 from the Golgi to the plasma membrane may also be regulated through interactions with an integrin-linked kinase [84].
AE1 mRNA and protein expression is increased in rat kidney during metabolic and respiratory acidosis [43, 74, 151]. Interestingly, in mouse kidney, only AE1 protein but not mRNA are enhanced (Mohebbi, Van der Wijst, Perna, Capasso, Wagner, unpublished results). Aldosterone has also been reported to stimulate basolateral anion exchange activity in isolated perfused OMCDs from normal or adrenalectomized rabbit [70]. Presently, it is unknown if this anion exchanger activity reflects AE1 transport activity or alternative transporters. Of note, in mouse kidney, the aldosterone analog deoxycorticosterone acetate (DOCA) increases AE1 mRNA but not protein expression (Mohebbi, Van der Wijst, Perna, Capasso, Wagner, unpublished results).
Pendrin
The anion exchanger pendrin (PDS, SLC26A4) was initially identified as being mutated in patients suffering from Pendred syndrome (OMIM #274600) characterized by hypothyroidism, goiter, and sensorineural deafness [50, 159]. In the kidney, pendrin is specifically expressed on the luminal membrane of non-type-A intercalated cells [92, 149]. There pendrin may mediate chloride/bicarbonate exchange releasing bicarbonate into urine and reabsorbing chloride. Indeed, both in vivo and in vitro experiments using Pds knockout mice indicate that pendrin is critical for bicarbonate secretion during metabolic alkalosis [149]. Accordingly, downregulation of protein expression during NH4Cl, (NH4)2SO4 or acetazolamide induced metabolic acidosis has been described [57, 67, 203]. During bicarbonate-loading, increased pendrin abundance was observed, whereas potassium depletion caused reduced protein levels, an effect that may further enhance metabolic alkalosis during K+ restriction [57, 203].
Moreover, pendrin may also be important for transcellular chloride reabsorption in the connecting tubule and cortical collecting duct. Increased chloride delivery to the distal nephron and collecting duct is associated with reduced pendrin expression levels [139, 192]. Apparently, pendrin expression is sensitive to luminal chloride concentration but less to systemic chloride status. A role of pendrin in collecting duct chloride reabsorption is further supported by the findings that pendrin expression is regulated during chloride depletion [197, 208], Pds KO mice are resistant to DOCA and NaCl induced hypertension [196], and angiotensin II stimulates chloride reabsorption in isolated CCDs from wild-type but not from pendrin-deficient mice [132]. However, it is not entirely clear if chloride depletion alone or the accompanying metabolic alkalosis increased pendrin expression. Moreover, the influence of aldosterone or the aldosterone analog DOCA on pendrin expression is controversial. Verlander et al. reported increased pendrin expression in DOCA-treated mice [196], whereas we failed to detect changes in pendrin expression in DOCA-treated mice (Mohebbi, Wagner, unpublished data). Moreover, functional analysis of the pendrin promoter in various cell lines showed in the presence of aldosterone reduced promoter activity when transfected in HEK cells but no effects in other cell lines [1].
Pendrin activity may be controlled on at least four different levels, namely mRNA and protein expression as well as subcellular localization. Enhanced luminal pendrin localization was observed in animals loaded with bicarbonate [203], given DOCA [196], or during chloride depletion [197], whereas several treatments altered total pendrin abundance in the kidney [57, 67, 139, 192, 203]. Studies in various cell lines provided evidence for direct regulatory domains in the promoter region sensitive to intracellular pH and possibly to aldosterone [1]. A fourth and indirect means of regulation may be changes in the number of pendrin expressing non-type-A intercalated cells as observed in states of chronic metabolic acidosis or altered distal chloride delivery associated with a changed relative abundance of pendrin positive cells [67, 192, 203].
The exact role of pendrin in humans has not been established to date. A recent case report described development of massive hypochloremic metabolic alkalosis in a patient with Pendred syndrome upon treatment with thiazide diuretics [134] which might indicate that pendrin may indeed be necessary to defend against or compensate for metabolic alkalosis as in the pendrin deficient mouse model.
The KCl-cotransporter KCC4
Genetic ablation of the KCl-cotransporter KCC4 was reported by Boettger et al. to cause distal renal tubular acidosis [28]. KCC4 was found to be expressed basolaterally in type-A intercalated cells, Kcc4 deficient mice excreted more alkaline urine and had lower arterial base excess values indicative for renal tubular acidosis. However, it is not clear if this acidosis is of the distal type since KCC4 is also abundantly expressed in the proximal tubule and urinary bicarbonate levels were not reported. Intracellular chloride concentrations were measured in single intercalated cells using energy dispersive X-ray microanalysis and found to be elevated. The authors proposed that KCC4 serves in type-A intercalated cells to release chloride back into interstitium and thereby maintain AE1 activity [28]. However, type-A intercalated cells appear to express also the ClC-Kb/Barttin chloride channel on the same membrane which may serve a similar function. Thus, the role of chloride transporting proteins in the basolateral membrane of (type A) intercalated cells remains to be fully elucidated.
Ammonium excretion: Crucial role of RhCG
The process of renal ammonia/ammonium excretion is complex, involves several nephron segments with distinct mechanisms and is only partly understood (for review, [62, 95, 213, 214]). In the proximal tubule, ammonium is formed from the metabolism of glutamine, a process stimulated by a variety of factors including metabolic acidosis, glucocorticoids, or potassium depletion. Import of glutamine in the proximal tubule cells occurs most likely through the basolateral SNAT3 glutamine transporter regulated by acid–base status and glucocorticoids [65, 83, 115, 123]. The ammoniagenic pathway produces NH3 and bicarbonate and is also highly regulated during metabolic acidosis [41, 42, 75, 123]. Ammonium is excreted in the proximal tubule either into venous blood or into urine via an apical sodium/proton exchanger (NHE), most likely involving the NHE3 isoform which accepts NH +4 instead of H+ [68]. However, other isoforms, such as NHE8 [64], may also participate as the Nhe3 deficient mouse model with transgenic overexpression of NHE3 in the intestine shows no major derangement of systemic acid–base status as indicated by normal blood bicarbonate levels [217]. In the thick ascending limb of the loop of Henle, ammonium is actively reabsorbed via luminal Na+/K+/2Cl− (NKCC2) cotransporters where ammonium is accepted instead of potassium. Additional NH +4 may enter cells through luminal potassium channels, possibly renal outer medullary K+ channels. There is also a component of “passive” transport via the paracellular route driven by the lumen-positive potential [62]. Ammonium is released via the basolateral membrane into interstitium by not completely characterized and understood mechanisms. A role for the electroneutral sodium-bicarbonate cotransporter NBCn1 (Slc4a7) has been proposed [76, 126]. Release via isoforms of the KCl cotransporter subfamily might be another possibility; however, the exact localization of the KCC1-4 isoforms along the nephron has not been reported in detail. This step of massive reabsorption by the thick ascending limb epithelium accumulates high concentrations of ammonia/ammonium in the interstitium, thereby providing the driving force for the uptake of interstitial ammonia/ammonium by the adjacent collecting duct cells.
The final step of ammonia/ammonium excretion is mediated by the collecting duct. The major site of ammonia/ammonium excretion is the outer medullary collecting duct. However, during metabolic acidosis, a strong increase in ammonia/ammonium excretion is also found in the connecting tubule, cortical collecting duct, and inner medullary collecting duct [95]. Ammonium accumulates in the medullary interstitium to high concentrations due to the reabsorption in the thick ascending limb. It is thought that this high ammonium concentration provides at least part of the driving force for ammonium excretion into urine. Ammonium secretion results most likely from the trapping of ammonia in the tubular lumen as ammonium after being titrated by protons stemming from active H+ secretion (possibly driven by V-type H+-ATPases). Thus, luminal acidification by the H+-ATPases contributes to the driving force for ammonium secretion. In the inner medullary collecting duct, H+ secretion maintains acidic urinary pH and, thereby, stabilizes NH3 secretion (as a result of a larger NH3 gradient between interstitium and acidic urine (low NH3 but high NH +4 concentrations)). Thus, evidence from functional experiments indicates that ammonium secretion along the collecting duct requires a large NH3 permeability and active H+ secretion. During metabolic acidosis, ammoniagenesis increases; ammonium reabsorption in the thick ascending limb is stimulated (partly by increasing NKCC2 expression [12]), ammonium accumulation in the medullary interstitium is enhanced, and H+ secretion along the collecting duct is increased (to a large extent due to increased insertion of functional H+-ATPases into the luminal membrane of type-A intercalated cells). Exocytosis of H+-ATPase containing vesicles may be directly stimulated by ammonium via a v-SNARE dependent mechanism [56]. Taken together, these factors favor largely increased ammonium excretion along the collecting duct.
Ammonium excretion in the collecting duct requires at least two transport steps, basolateral uptake, and luminal excretion. These mechanisms may be functionally and molecularly distinct. Detailed studies by Susan Wall and colleagues demonstrated several uptake mechanisms for ammonium on the basolateral side, namely Na+/K+/2Cl− cotransport, possibly NKKC1, and Na+/K+-ATPase [204, 206]. In both cases, NH +4 can be transported instead of potassium. Little evidence has been obtained for a specific ammonia transport pathway. The role of NKCC1 in basolateral NH +4 uptake may be rather small since pharmacological blockade or genetic deletion of NKCC1 has little effect on ammonium excretion [205, 209].
In contrast, the luminal membrane has a high permeability for NH3 [222]. H+/K+-ATPases are expressed on the luminal membrane and in the case of the so-called colonic isoform, the possibility of active transport of ammonium has been demonstrated [36, 40, 180]. Whether this is relevant in vivo is unknown at present.
The prevailing hypothesis was that ammonium excretion in the collecting duct occurs through nonionic diffusion in the form of NH3, subsequent protonation, and trapping of NH +4 in the lumen. This process would not require any specific transport proteins and be regulated only by intracellular NH3 concentrations and luminal acidification.
In 2000, Marini and colleagues provided evidence for a role of Rhesus-like proteins in ammonium/ammonia transport using yeast complementation studies. Three mammalian proteins, RhAG, RhBG, and RhCG were identified [111]. RhBG and RhCG were found to the expressed in mammalian kidney where RhBG is expressed in the basolateral membrane and RhCG on the luminal membrane of all cells along the connecting tubule and intercalated cells only in the cortical and medullary collecting duct [49, 140, 198]. The subcellular localization of RhCG has remained, however, controversial. Eladari et al. described only luminal staining for RhCG in rat kidney [49, 140], whereas the laboratory of D. Weiner has reported both luminal and basolateral staining for RhCG in human [69] and rat kidney [88, 162] and also in mouse kidney [89, 198].
Several studies addressed the possibility that RhBG and RhCG may underlie NH +4 /NH3 transport in the collecting duct by studying their transport properties in various cell models [16, 117, 224], their regulation during metabolic acidosis [162, 163], or by generating Rhbg deficient mice [34]. Taken together, these studies indicate that RhBG and RhCG can mediate transport of NH +4 and/or NH3 but the stoichiometry, the species of transported ions, and transport mode have remained controversial. Additional insights into the transport mechanism and species of transported ions/molecules mediated by Rh proteins came from genetic ablation of Rh proteins in green alga and from crystallization of the bacterial homologue AmtB. Evidence from green alga suggests that the Rh1 protein may be linked to a bidirectional CO2 gas channel [171]. The crystal structure of the AmtB demonstrates a binding site for NH +4 which led to the hypothesis that NH +4 is deprotonated to NH3 which then may permeate the transporter/channel as uncharged gas [85, 94, 223]. At the intracellular face of the protein, NH3 may be protonated again. Thus, Rh proteins may form part of an ammonia permeable gas channel and may be involved in the transport of NH +4 or NH3 in the kidney collecting duct (Fig. 3).

Ammonium exretion in the collecting duct. Ammonium is excreted into urine by an active, regulated, and at least two-step process. First, ammonium is taken up into collecting duct cells (mostly intercalated cells) from interstitium. This step may be mediated by several transport proteins localized in the basolateral membrane that are able to accept NH +4 instead of K+ ions: the Na+/K+-ATPase and the NKKC1 Na+/K+/2Cl− cotransporter. The existence of basolateral RhCG proteins is controversial. On the luminal membrane, RhCG is expressed and is involved in the net flux of NH3. The exact transport mechanism, however, remains to be established. Parallel secretion of protons, mostly by H+-ATPases, and to a lesser extent by H+/K+-ATPases acidifies urine and traps NH +4 in the lumen, thereby leading to its final excretion
Two recent studies directly addressed the role of Rhbg and Rhcg in the mammalian kidney performing genetic deletion of these genes. Surprisingly, Rhbg-deficient mice showed no defect in renal ammonium excretion in vivo and ammonium or ammonia permeabilities in the isolated perfused collecting duct showed no difference [34]. In contrast, deletion of Rhcg causes incomplete distal renal tubular acidosis with more alkaline urine, metabolic acidosis, and impaired renal ammonium excretion [26]. Under basal conditions, only a mild reduction in urinary ammonium excretion was found in Rhcg KO mice and no evidence detected for metabolic acidosis. Oral acid challenges caused a much more pronounced metabolic acidosis due to a massive reduction in urinary ammonium excretion whereas titratable acids were normally excreted. The renal defect was further analyzed on the cellular level using in vitro intracellular pH measurements of microperfused CCD and OMCD demonstrating a decrease in the alkalinization rate during luminal application of NH4Cl suggesting reduced net NH3 permeability by almost 70%. Similarly, assessment of transepithelial NH3 permeability in microperfused CCDs showed massively impaired NH3 fluxes. Thus, Rhcg is required for renal ammonium excretion and may be involved in mediating luminal net NH3 efflux [26]. This study, hence, established a new paradigm of ammonium transport in the collecting duct requiring the presence of RhCG and indicating that ammonia excretion does not occur by simple nonionic diffusion.
A few studies have also addressed the regulation of RhBG and RhCG during states of increased or altered urinary ammonium excretion. In rodent models of metabolic acidosis, reduced renal mass, or cyclosporine-induced acidosis, no evidence for altered Rhbg abundance or localization was obtained. In contrast, the group of D. Weiner has reported that Rhcg protein abundance is strongly enhanced and that Rhcg staining becomes more luminal under these circumstances [162, 163]. In contrast, we found in several mouse models of increased ammonium excretion reduced Rhcg mRNA levels (Devuyst, Wagner, unpublished results). Clearly, further work is required to address this discrepancy and to understand acute and chronic regulation of these interesting transport proteins.
Regulation
Acute regulation by hormones and other factors: angiotensin II and aldosterone, CaSR
Acid excretion along the collecting duct is tightly regulated by both systemic as well as local factors. The renin–angiotensin II–aldosterone system (RAAS) appears to be a major stimulator. RAAS activation occurs during metabolic acidosis [13, 66, 80, 153] and blockade of the RAAS impairs renal acid excretion during acidosis [72, 161]. Moreover, deficiency of aldosterone secretion or signaling causes hyperkalemic distal renal tubular acidosis [60, 104, 161]. Similarly, animal studies indicate that angiotensin II and aldosterone are important stimulators of collecting duct acid excretion [19, 100, 101, 110]. On a cellular level, both angiotensin II and aldosterone appear to be strong stimuli for H+-ATPase activity in type-A intercalated cells. Type-A intercalated cells express angiotensin receptors type 1 (AT1R) , where angiotensin II increases intracellular Ca2+ and stimulates H+-ATPase activity in a protein kinase C dependent manner [133, 148]. Also, aldosterone has direct stimulatory effects on type-A intercalated cell function. Hayes demonstrated that adrenalectomy decreased and aldosterone supplementation restored or even increased luminal H+-ATPase activity and basolateral chloride/bicarbonate exchanger activity in the rabbit outer medullary collecting duct [70]. Interestingly, also acute stimulatory aldosterone effects could be observed within a few minutes after application to isolated mouse outer medullary collecting ducts. Aldosterone stimulated H+-ATPase activity within minutes, an effect not affected by inhibition of the mineralocorticoid receptor or inhibitors of transcription and translation [216]. The nongenomic effect of aldosterone appears also to be mediated by increased intracellular calcium and protein kinase C (Winter, Velic, Kampik, Wagner, unpublished results).
Local factors that seem to regulate intercalated cell function include extracellular Calcium, CO2, and pH. Increased pCO2 stimulates exocytosis of H+-ATPase containing vesicles in the outer medullary collecting duct [156]. Moreover, incubation of collecting ducts in vitro at acidic pH enhances type-A intercalated cell function [190]. How type-A intercalated cells sense extracellular pH or changes in pCO2 is unknown to date.
Hypercalcemia and subsequent hypercalciuria often causes increased diuresis and stronger urinary acidification. In patients with recurrent calcium containing kidney stones, defective urinary acidification has been detected and is thought to promote crystal formation and stone development [185]. Principal and intercalated cells along the medullary collecting duct express on the luminal membrane the calcium-sensing receptor (CaSR) [152]. Acute activation of the CaSR has been shown to blunt vasopressin stimulated water reabsorption [152]. Renkema et al. could recently demonstrate that the CaSR plays also a major role in urinary acidification and prevention of kidney stones [142]. They used the Trpv5 −/− hypercalciuric mouse model that presents with massive increased urine flow and acidic urine and showed that generation of double KO mice lacking also the B1 H+-ATPase subunit prevented urinary acidification. Hypercalciuric mice with defective urinary acidification developed massive nephrocalcinosis and hydronephrosis and died of renal failure at the age of 8–12 weeks. Moreover, it could be shown that Calcium or the CaSR agonist neomycin stimulated H+-ATPase activity in the in vitro microdissected medullary collecting duct from wildtype or Trpv5 −/− mice but not from B1 KO mice. Thus, activation of the CaSR by high luminal calcium concentrations may trigger a compensatory or preventive process leading to reduced water reabsorption and increased proton secretion thereby reducing the risk of calcium precipitations and kidney-stone formation.
Interestingly, most factors known to stimulate H+-ATPase activity in type-A intercalated cells appear to stimulate H+-ATPase activity by trafficking of H+-ATPases or accessory/stimulatory proteins into the luminal membrane [133, 148, 156, 216]. In the mIMCD3 type-A intercalated cell model, the trafficking of H+-ATPase containing vesicles to the luminal membrane involves a protein complex containing Munc-18-2, syntaxin 1-A, SNAP23, and VAMP forming a SNARE complex [5, 17, 18, 102, 121]. Similarly, cleavage of cellubrevin with tetanus toxin prevents stimulation of H+-ATPase activity in the outer medullary collecting duct and epididymis [31, 148]. The B1 isoform of the H+-ATPase complex appears to play a critical role in the stimulation of H+-ATPase activity by trafficking. In the absence of the B1 isoform, type-A intercalated cells express more luminal B2 isoform which may help to sustain basal H+-ATPase activity [131]. However, in the absence of a functional B1 isoform, H+-ATPases fail to respond to stimulatory factors such as metabolic acidosis, angiotensin II, or CaSR activation [131, 148, 221], thereby explaining at least in part the phenotype of incomplete dRTA in mice lacking the B1 subunit [52].
Regulation through altered protein expression
Acid–base status, electrolytes, as well as some hormones affect abundance of some proteins involved in acid–base transport along the collecting duct. However, it is not clear in all cases if changes in total mRNA or protein abundance are due to true alterations in protein abundance. In some instances, these changes may rather reflect differences in cell number or cannot be clearly distinguished from changes in mRNA or protein expression in total kidney for proteins expressed not exclusively along the collecting duct. Only few reports have been able to overcome the technical difficulties of collecting enough material from dissected and isolated subsegments to examine mRNA or protein levels [35, 123].
Acidosis is associated with increased expression of components of the acid-extruding machinery such as AE1 mRNA and protein as discussed above. In contrast, proteins expressed specifically in non-type-A intercalated cells such as pendrin appear to be downregulated during acidosis, an effect probably due to both reduced number of non-type-A intercalated cells and less transporter/cell as reflected also in greatly diminished immunostaining intensities. Regulation of transcripts has been investigated systematically by serial analysis of gene expression (SAGE) technology using isolated outer medullary collecting ducts from mice subjected to ammonium chloride or potassium depletion-induced urinary acidification. A large number of transcripts encoding for acid–base transporters, regulatory enzymes and kinases, as well as for ion channels was detected [35]. However, it is not clear if these altered transcript levels reflect only changes in transcription or mRNA stability or not also tubular hypertrophy after prolonged treatments. Nevertheless, these data may provide evidence for coordinated regulation of a set of genes in the collecting duct. New technologies such as fluorescence based sorting of specific tubule segments combined with lower requirements for total mRNA for microarray hybridization or more sensitive second-generation sequencing and proteome technologies may offer great avenues for identifying regulated proteins as well as for novel regulators.
Chronic regulation by remodeling
The relative abundance of the different cell types varies along the collecting duct and changes also with electrolyte and acid–base status, a process termed remodeling [3, 14, 158, 203]. During chronic metabolic acidosis, the increase in the relative abundance of type-A intercalated cells has been reported most likely at the expense of non-type-A intercalated cells. Likewise, chronic inhibition of carbonic anhydrase activity or genetic ablation of carbonic anhydrase II causes remodeling of the collecting duct [14, 30]. Apoptotic removal of non-type-A intercalated cells from the medulla during development has been well documented [90, 108, 170]. If this occurs also during adaptation to metabolic acidosis or other electrolyte disturbances has not been reported.
The molecular mechanisms initiating and controlling collecting duct remodeling are not elucidated to date. Reversal of polarity has been discussed based on the observation that H+-ATPases change subcellular localization from basolateral to apical membranes during acidosis and that cells with apical bicarbonate secretion acquire basolateral bicarbonate excretion [21, 157, 158, 173]. Cell culture experiments with primary cells from rabbit collecting duct cells suggested that non-type-A intercalated cells may differentiate into type-A intercalated and principal cells [51]. However, uncertainty about the purity of preparations and the lack of good markers for different cell types leave open questions. Interestingly, a protein, hensin (also known as DMBT1 or Muclin), was identified that induced differentiation of cells with characteristics of non-type-A intercalated cells into type-A intercalated like cells in cell culture. The production and secretion of hensin is stimulated during metabolic acidosis and in isolated perfused cortical collecting ducts application of anti-hensin antibodies prevented the reversal of functional polarity suggesting that hensin may play an important role in the acute adaptive remodeling of the collecting duct [2–4, 182, 201]. More recently, three different mouse models deficient for hensin/DMBT1 were reported. Two of these mouse models were apparently viable [44, 143] whereas the third model was lethal at a very early stage [200]. The reason for this discrepancy is unknown at this moment. Polymerization of hensin is required for its effects on epithelia. This complex process may involve interactions with different members of the integrin family and be coordinated by cyclophilin [135, 200].
More recently, the forkhead transcription factor Foxi1 was identified to be highly expressed in intercalated cells. Its genetic ablation causes distal renal tubular acidosis in Foxi1 KO mice due to loss of differentiation of collecting duct cells [27]. It was also shown that the intercalated cell specific AE1, Pendrin, and AE4 transporters may be transcriptionally regulated by Foxi1 [27, 98]. If Foxi1 is regulated during development or by factors that induce collecting duct remodeling is presently unknown. Induction of Foxi1 during nephrogenesis occurs at a time point when first intercalated cells and intercalated cell-specific transport proteins appear further highlighting that Foxi1 plays an important role in intercalated cell differentiation [79].
Proliferation of intercalated cells may significantly contribute to collecting duct remodeling during metabolic acidosis. In mouse kidney, intercalated cells appear to proliferate at a low rate under basal conditions [212]. In contrast, in rat kidney, we failed to detect evidence for significant basal proliferation of intercalated cells (Bacic, Nowik, Kaissling, Wagner, unpublished data). However, appearance of cells positive for the proliferation markers BrdU, PCNA, or Ki67 occurs as early as 12 h after induction of metabolic acidosis in rats. Interestingly, these proliferating cells appear to be terminally differentiated since cells form part of the tubular lumen and stain positively for the type-A intercalated cell-specific AE1 anion exchanger (Bacic, Nowik, Kaissling, Wagner, unpublished results). Doucet and colleagues had performed SAGE analysis on OMCDs from acidotic mice and detected among other regulated transcripts also GDF15 (growth differentiation factor 15) to be highly upregulated [35]. Using markers of proliferation, these authors report two distinct phases of proliferation in mouse kidney: an early phase around 3 days after induction of metabolic acidosis which may be characterized by axial growth and was abolished in GDF15 deficient mice or when PI3-kinase or mTOR activity were blocked with rapamycin [193]. Consequently, GDF15-deficient mice developed more pronounced metabolic acidosis. The later phase of proliferation occurring 1 week after acidosis induction was not dependent on GFD15 and may be primarily associated with transversal proliferation [193]. Thus, type-A intercalated cell proliferation plays clearly a role in the kidneys adaptation to an acid-load and may consist of distinct phases that are differentially regulated. The fact that the number and relative abundance of different subtypes of intercalated cells may change during metabolic acidosis should be kept in mind in interpreting measurements of total protein abundance of intercalated cell-specific proteins since changes may reflect either altered cell numbers and/or altered protein expression per cell.
Inborn errors of transport: mutations in AE1, B1, a4, and CAII
Rare-inherited familiar forms of dRTA have greatly enhanced our understanding how the collecting duct secretes acid. Syndromes of dRTA are characterized by the inability of the kidney to produce acidic urine (pH < 5.5) in the face of metabolic acidosis. dRTA is often associated with disorders of potassium homeostasis (hypokalemic in type I and hyperkalemic in type IV dRTA). Moreover, metabolic acidosis and alkaline urine alone or in combination may promote hypercalciuria and subsequent formation of kidney stones and/or nephrocalcinosis. To date, three different genes have been identified that cause classic type I dRTA when mutated: the SLC4A1 (AE1) anion exchanger [33, 82], ATP6V1B1, and ATP6V0A4 encoding the B1 and a4 subunits of the V-type H+-ATPase [81, 169]. Mutations in various components of the aldosterone synthesis or signaling network cause forms of apparent or pseudohypoaldosteronism associated with renal tubular acidosis (so-called type IV dRTA) [104]. Moreover, mutations in carbonic anhydrase II (CAII), expressed in the proximal tubule cells and intercalated cells along the collecting duct, cause a mixed type of dRTA with a combination of proximal and distal features, i.e., bicarbonate wasting and acidification defect [147, 165].
Mutations in either the ATP6V1B1 (B1 subunit) or ATP6V0A4 (a4 subunit) genes cause autosomal recessive forms of distal renal tubular acidosis. B1 and a4 form part of renal H+-ATPase pumps [53, 120, 167, 173]. Expression of the B1 isoform is not restricted to intercalated cells but occurs also at lower levels in the thick ascending limb of the loop of Henle. In contrast, the a4 subunit isoform is found in the cells lining the proximal tubule, the thick ascending limb of the loop of Henle, and all subtypes of intercalated cells. Both subunits are also expressed in extrarenal tissues, mainly the epididymis and in cells of the stria vascularis of the inner ear. Differences in male fertility have not been reported to date, whereas patients with ATP6V1B1 mutations suffer from sensorineural deafness early in childhood which is resistant to alkali therapy [81]. Similarly, patients with ATP6V0A4 mutations develop sensorineural deafness which, however, may occur later in life than in ATP6V1B1 patients [177, 194].
Most mutations in the B1 subunit studied to date appear to affect either assembly and/or function of the H+-ATPase complex. Experiments in IMCD3 or HEK cells transfected with various B1 mutants as well as yeast complementation assays with wild-type and B1 mutants found similar defects affecting pump assembly [59, 221]. Interestingly, a B1 variant, considered as rather common polymorphism showed a major defect in the yeast complementation assay suggesting that it may affect pump activity in vivo [59]. Moreover, experiments using mice lacking the B1 subunit demonstrated that the B1 subunit is required for maximal urinary acidification [52]. Under basal conditions, B1 KO mice produced more alkaline urine but had otherwise normal systemic acid–base parameters. However, an oral acid challenge with NH4Cl caused severe metabolic hyperchloremic acidosis and decompensation [52]. Along the same line, H+-ATPase activity in isolated OMCDs was normal under basal conditions but did not increase in OMCDs from acid-loaded B1 KO mice [131] or upon stimulation with angiotensin II [148]. This may be explained by the inability of proton pumps lacking the B1 subunit to traffic to the luminal membrane in response to various stimuli. Indeed, immunohistochemistry showed membrane-associated staining for proton pumps in the OMCD and IMCD of KO mice under basal conditions with enhanced B2 staining [130, 131]. In acid-loaded wild-type mice, proton pump staining in the luminal membrane of type-A intercalated cells increased whereas no increase was detected in KO mice. Collectively, these data suggest that the B1 subunit appears to confer the ability to H+-ATPases to increase their membrane associated activity in intercalated cells in response to external stimuli.
Much less is known about the role of the a4 isoform and the pathomechanisms leading to dRTA in patients. Despite the fact that the a4 subunit is detected along the entire human, rat, and mouse nephron with intense staining in the brush border of the proximal tubule as well as luminal and basolateral localization in all subtypes of intercalated cells [155, 173], no clinical symptoms have been reported from patients indicating reduced function of the proximal tubule. This may be due to the fact that the kidney expresses also the a1, a2, and a3 isoforms of this pump subunit and that expression patterns with a4 widely overlap [155]. The phenotype of an a4 deficient mouse model has not been reported to date. Some mutations in the a4 isoform have been investigated in more detail in vitro. Complementation assays in yeast lacking the homologous Vph1p gene demonstrated defective acidification and growth. One a4 mutant (W520L) demonstrated an interesting phenotype reducing expression of other subunits of the pump suggesting a dominant negative effect [125]. Similarly, the R807Q mutant expresses only low amount of proteins consistent with less stable mutant protein. The a4 isoform apparently interacts with the glycolytic enzyme phosphofructokinase 1 which may link pump function to energy supply [179]. This interaction is disrupted in the G820R a4 mutant leading to decreased pump activity despite an only mildly impaired ability to hydrolyze ATP [178].
Mutations in the AE1 anion exchanger underlie recessive and dominant cases of dRTA and can lead to two distinct phenotypes: red blood cell deformities or distal renal tubular acidosis. Interestingly, in some but not all patients, these two phenotypes do occur simultaneously, but certain mutations are always associated with only one or another phenotype [6, 7]. The literature on AE1 mutations and underlying molecular mechanisms of disease has been summarized in some recent excellent reviews [6, 7, 58, 99, 187, 218]. The most common recessive dRTA-causing mutation, G701D, interacts with the chaperonine glycophorin which appears to rescue the mutant protein in red blood cells. Glycophorin is absent from intercalated cells possibly explaining the cell-specific phenotype of this particular mutation [183]. A number of other recessive mutations have been described and partially characterized. These mutations are relatively common in Southeast Asia and are often associated with red blood phenotypes [86, 87].
The autosomal recessive pattern of mutations such as G701D, S773P, or the deletion mutant ∆400–408, may be explained by the retention of mutant protein intracellularly whereas enough normal proteins reach the membrane in heterozygous patients [39, 93, 183, 195].
In contrast, dominant dRTA-causing AE1 mutations are rarely associated with red blood cell phenotypes and occur more often in Caucasian patients. Mechanistically, these mutations may affect the polarized localization of AE1 at the basolateral membrane of type-A intercalated cells, remain intracellular, or lose activity [6, 7, 39, 47, 93, 141, 150, 188, 189]. The autosomal dominant pattern of inheritance in certain mutants is possibly due to the fact that the transporter dimerizes or that partial rerouting of mutant AE1 to the luminal membrane of type-A intercalated cells may shunt normal acid secretion.
It should be noted that the impact of AE1 mutations were all studied in various in vitro cell line models relying on stable or transient transfections of mutant AE1. Renal biopsy material from patients with AE1 mutations is rare, and only two cases have been reported in the literature. The S613F mutant is predicted from cell culture models to lead to partially misrouted apical AE1 expression [188]. In contrast, no luminal expression of AE1 was detected in the kidney from a patient carrying the S613F mutation [210]. However, some intracellular AE1 staining was detected; AE1 was absent from the basolateral side. Interestingly, the number of type-A intercalated cells appeared to be greatly reduced in this particular kidney biopsy, and remaining type-A intercalated cells appeared small and abnormal in shape. In the kidney from a patient with the dominant R589H mutation, no AE1 staining was detected in intercalated cells which were reduced in number [164]. Similarly, in mice lacking total AE1 expression, we detected a reduced number of intercalated cells, which might indicate that functional AE1 is required for normal type-A intercalated cell proliferation, differentiation, or survival (unpublished results).
Mutations in CAII are associated with a severe disease characterized by the occurrence of osteopetrosis, (distal) renal tubular acidosis, and cerebral calcifications due to the expression of CAII in all these tissues [147]. In the kidney, CAII is localized in the cytosol of proximal tubular cells as well as in all subtypes of intercalated cells and plays an important role in the intracellular hydration of CO2 for the generation of bicarbonate and protons [137]. Moreover, CAII is (directly) interacting with several acid–base transporters such as AE1 (see above). Impaired CAII function, thus, leads to reduced generation of transport substrates of bicarbonate and proton translocating pumps and carriers with subsequent loss of bicarbonate reabsorption in the proximal tubule, bicarbonate generation in the collecting duct, and reduced proton secretion by intercalated cells. Studies in rats with chronic pharmacological carbonic anhydrase inhibition with acetazolamide and mice with genetic ablation of carbonic anhydrase revealed both effects on the remodeling of the collecting duct [14, 30]. Chronic carbonic anhydrase inhibition increased the relative number of type-A intercalated cells at the expense of non-type-A intercalated cells in the CCD, whereas in the OMCD the number of intercalated cells increased and principal cells were reduced [14]. In contrast, genetic deletion of carbonic anhydrase II in mice causes an overall depletion of intercalated cells in all regions of the collecting duct [30]. Thus, chronic impairment of intercalated cell function may impact on differentiation or survival of these cells and lead to replacement by principal cells. Whether this is also the case in human kidney has not been examined to date.
Dent’s disease: defective ClC5 chloride/proton exchangers
Dent’s disease is primarily a proximal tubule disorder characterized by low-molecular-weight proteinuria that may be associated with hypercalciuria, nephrocalcinosis, and renal failure. It is caused by inactivating mutations of the renal chloride-proton exchanger ClC-5, which colocalizes with the vacuolar H+-ATPase in proximal tubule cells and type-A intercalated cells [48, 107, 124]. Investigations of renal biopsies of patients with inactivating mutations of ClC-5 revealed that apical H+-ATPase expression was absent in type-A intercalated cells, whereas the polarity of H+-ATPase was modified in proximal tubule cells. The significance of these abnormal H+-ATPase localizations will need further studies in patients to understand the defect in tubular acidification that is reported in a subset of patients with Dent’s disease [116, 154].
Acquired problems of collecting duct acid–base transport
A number of acquired states are associated with dysregulation of collecting duct acid–base transport and can be caused by a variety of diseases such autoimmune disease (Sjögren’s disease, autoimmune hypothyrodism), isolated hypothyroidism [114, 127, 219], or hypoaldosteronism. Moreover, a number of drugs may impair the collecting ducts ability to excrete acid or to adapt appropriately to altered systemic acid–base status. These drugs include lithium [11, 22, 113, 118, 146], the immunosuppressants cyclosporine [172] and FK506 [71, 220], amphotericin [112, 145], or toxins such as toluene [24, 181].
Sjögren’s disease affects kidney function in about one third of all cases and may cause dRTA [136]. Autoantibodies isolated from patients react with different structures of the kidney including intercalated cells, but the exact antigen(s) have not been further identified. Kidney biopsies have been investigated of few patients and reduced or absent staining for the E and B1 H+-ATPase subunit [20, 37, 45, 78, 210] or the AE1 exchanger reported [210]. In a recent series of five patients, we confirmed reduced H+-ATPase expression (a4 and B1 subunits) in intercalated cells, complete absence of AE1 immunoreactivity, and a reduction in the total number of intercalated cells (Mohebbi, Lemaire, Devuyst, Wagner, unpublished results). Thus, Sjögren’s disease may cause dRTA due to loss of important collecting duct acid–base transport proteins and less intercalated cells. The order of these events as well as the primary immunologic insult in the kidney need to be further clarified.
The use of the calcineurin inhibitors cyclosporine and FK506 is often associated with dRTA in the setting of patients receiving kidney or other organ transplants [71, 220]. In a rat model, cyclosporine has been shown to cause dRTA [23]. Interestingly, Watanabe et al. demonstrated in the CCD of cyclosporine-treated rats that after an acute acid exposure for 3 h, the adaptive downregulation of bicarbonate secretion by non-type-A intercalated cells was abolished [211]. This effect was secondary to inhibition of the cyclophilin activity by cyclosporine since inhibition of calcineurin alone (by FK506) did not affect the adaptive response in the CCD [211]. Moreover, cyclophilin appears critical for hensin polymerization required to induce terminal differentiation and plasticity of intercalated cells [135].
Application of FK506 to rats did not alter acid–base status or renal acid excretion but transiently caused more pronounced metabolic acidosis upon acid-loading. Detailed analysis of acid–base transport protein expression in the kidney revealed inappropriately high pendrin expression and late reduction in non-type-A intercalated cell numbers during acid-loading (Mohebbi, Wagner, manuscript in revision). These data point again to delayed adaption of the collecting duct due to calcineurin inhibition. However, at least in the case of cyclosporine, dRTA develops independent from calcineurin function suggesting different mechanisms of action of cyclosporine and FK506 [211]. Thus, cyclophilin and hensin may be required for the rapid response of the collecting duct to acid-loads within hours whereas calcineurin may modulate more chronically the expression of pendrin and subsequently the number of non-type-A intercalated cells. A role of calcineurin in the collecting duct is further supported by the fact that calcineurin may be involved in aquaporin-2 water channel regulation and trafficking [103].
Summary and future perspectives
Research over the past 60 years has uncovered major mechanisms of renal acid–base handling, the critical role of the collecting duct in excretion of acids or bicarbonate, identified many molecules involved in these transport processes and their regulation. Transgenic mouse models and rare inherited human diseases have highlighted the importance of some mechanisms described and have allowed to start dissecting molecular pathways.
Despite this tremendous progress, we still lack insights in major components of the function and regulation of the collecting duct in renal fine-tuning of acid–base homeostasis. The precise mechanism(s) mediating ammonium excretion remained to be uncovered and the role of the Rhesus proteins RhBG and RhCG investigated. Also the role of other transport proteins such as the AE4 anion exchanger, the eletroneutral NBCn1 (SLC4A7) transporter, or the K+/Cl− cotransporter KCC4 needs to be clarified. Regulation of the H+-ATPase, its exact subunit composition in various subdomains of intercalated cells, and its (physical and functional) interaction with other intercalated cell proteins is only poorly understood.
Regulation of collecting duct acid–base handling occurs on various levels ranging from cell proliferation on the one side to acute regulation of transport processes on the other side. We are only starting to understand that and how cell proliferation may contribute to collecting duct acid–base control. Obviously, cell proliferation and differentiation must be controlled and regulated tightly. Moreover, the developmental origin and differentiation of the various cell types making up the collecting duct is only partially elucidated. The role of segment or cell-specific transcription factors such as Foxi1 will be important to understand normal development of the collecting duct as well as regeneration of cells and nephrons. Another major open question is how are remodeling of the collecting duct or transport processes acutely and chronically adapted to the systemic and local acid–base status of the body. Proton activated G protein coupled receptors OGR1 and GPR4 have been identified [106] and are expressed also in the kidney. If these receptors, however, contribute to the control of collecting duct acid–base handling has not been reported. Several hormones have been shown to be increased during metabolic acidosis such as endothelin or the angiotensin II-aldosterone axis. In vivo and in vitro evidence demonstrated their importance. How these and possibly other hormones respond to acid–base status and how they regulate collecting duct function needs to be further examined.
References
Adler L, Efrati E, Zelikovic I (2008) Molecular mechanisms of epithelial cell-specific expression and regulation of the human anion exchanger (pendrin) gene. Am J Physiol Cell Physiol 294:C1261–C1276
Al-Awqati Q (1996) Plasticity in epithelial polarity of renal intercalated cells: targeting of the H+-ATPase and band 3. Am J Physiol 270:C1571–C1580
Al-Awqati Q (2003) Terminal differentation of intercalated cells: The role of Hensin. Annu Rev Physiol 65:567–583
Al-Awqati Q, Vijayakumar S, Takito J, Hikita C, Yan L, Wiederholt T (1999) Terminal differentiation in epithelia: the Hensin pathway in intercalated cells. Semin Nephrol 19:415–420
Alexander EA, Shih T, Schwartz JH (1997) H+ secretion is inhibited by clostridial toxins in an inner medullary collecting duct cell line. Am J Physiol 273:F1054–F1057
Alper SL (2002) Genetic diseases of acid–base transporters. Annu Rev Physiol 64:899–923
Alper SL (2003) Diseases of mutations in the SLC4A1/AE1 (band 3) Cl-/HCO -3 exchanger. In: Broer S, Wagner CA (eds) Membrane transporter diseases. Kluwer Academic/ Plenum Publishers, New York, pp 39–63
Alper SL, Kopito RR, Libresco SM, Lodish HF (1988) Cloning and characterization of a murine band 3-related cDNA from kidney and from a lymphoid cell line. J Biol Chem 263:17092–17099
Alper SL, Natale J, Gluck S, Lodish HF, Brown D (1989) Subtypes of intercalated cells in rat kidney collecting duct defined by antibodies against erythroid band 3 and renal vacuolar H+-ATPase. Proc Natl Acad Sci U S A 86:5429–5433
Arnett TR (2008) Extracellular pH regulates bone cell function. J Nutr 138:415S–418S
Arruda JA, Dytko G, Mola R, Kurtzman NA (1980) On the mechanism of lithium-induced renal tubular acidosis: studies in the turtle bladder. Kidney Int 17:196–204
Attmane-Elakeb A, Mount DB, Sibella V, Vernimmen C, Hebert SC, Bichara M (1998) Stimulation by in vivo and in vitro metabolic acidosis of expression of rBSC-1, the Na+-K+(NH +4 )-2Cl- cotransporter of the rat medullary thick ascending limb. J Biol Chem 273:33681–33691
Augustinsson O, Johansson K (1986) Ammonium chloride induced acidosis and aldosterone secretion in the goat. Acta Physiol Scand 128:535–540
Bagnis C, Marshansky V, Breton S, Brown D (2001) Remodeling the cellular profile of collecting ducts by chronic carbonic anhydrase inhibition. Am J Physiol Renal Physiol 280:F437–F448
Bailey JL (2005) Metabolic acidosis: an unrecognized cause of morbidity in the patient with chronic kidney disease. Kidney Int Suppl 96:S15–S23
Bakouh N, Benjelloun F, Hulin P, Brouillard F, Edelman A, Cherif-Zahar B, Planelles G (2004) NH3 is involved in the NH4+ transport induced by the functional expression of the human Rh C glycoprotein. J Biol Chem 279:15975–15983
Banerjee A, Li G, Alexander EA, Schwartz JH (2001) Role of SNAP-23 in trafficking of H+-ATPase in cultured inner medullary collecting duct cells. Am J Physiol Cell Physiol 280:775–781
Banerjee A, Shih T, Alexander EA, Schwartz JH (1999) SNARE proteins regulate H+-ATPase redistribution to the apical membrane in rat renal inner medullary collecting duct cells. J Biol Chem 274:26518–26522
Barreto-Chaves ML, Mello-Aires M (1996) Effect of luminal angiotensin II and ANP on early and late cortical distal tubule HCO -3 reabsorption. Am J Physiol 271:F977–F984
Bastani B, Haragsim L, Gluck SL, Siamopoulos KC (1995) Lack of H+-ATPase in distal nephron causing hypokalemia distal RTA in a patient with Sjögren's syndrome. Nephrol Dial Transplant 10:908–913
Bastani B, Purcell H, Hemken P, Trigg D, Gluck S (1991) Expression and distribution of renal vacuolar proton-translocating adenosine triphosphatase in response to chronic acid and alkali loads in the rat. J Clin Invest 88:126–136
Batlle D, Gaviria M, Grupp M, Arruda JA, Wynn J, Kurtzman NA (1982) Distal nephron function in patients receiving chronic lithium therapy. Kidney Int 21:477–485
Batlle DC, Gutterman C, Tarka J, Prasad R (1986) Effect of short-term cyclosporine A administration on urinary acidification. Clinical nephrology 25(Suppl 1):S62–S69
Batlle DC, Sabatini S, Kurtzman NA (1988) On the mechanism of toluene-induced renal tubular acidosis. Nephron 49:210–218
Biner HL, Arpin-Bott MP, Loffing J, Wang X, Knepper M, Hebert SC, Kaissling B (2002) Human cortical distal nephron: distribution of electrolyte and water transport pathways. J Am Soc Nephrol 13:836–847
Biver S, Belge H, Bourgeois S, Van Vooren P, Nowik M, Scohy S, Houillier P, Szpirer J, Szpirer C, Wagner CA, Devuyst O, Marini AM (2008) A role for Rhesus factor Rhcg in renal ammonium excretion and male fertility. Nature 456:339–343
Blomqvist SR, Vidarsson H, Fitzgerald S, Johansson BR, Ollerstam A, Brown R, Persson AE, Bergstrom GG, Enerback S (2004) Distal renal tubular acidosis in mice that lack the forkhead transcription factor Foxi1. J Clin Invest 113:1560–1570
Boettger T, Hubner CA, Maier H, Rust MB, Beck FX, Jentsch TJ (2002) Deafness and renal tubular acidosis in mice lacking the K-Cl co-transporter Kcc4. Nature 416:874–878
Bonnici B, Wagner CA (2004) Postnatal expression of transport proteins involved in acid–base transport in mouse kidney. Pflugers Arch 448:16–28
Breton S, Alper SL, Gluck SL, Sly WS, Barker JE, Brown D (1995) Depletion of intercalated cells from collecting ducts of carbonic anhydrase II-deficient (CAR2 null) mice. Am J Physiol 269:F761–F774
Breton S, Nsumu NN, Galli T, Sabolic I, Smith PJ, Brown D (2000) Tetanus toxin-mediated cleavage of cellubrevin inhibits proton secretion in the male reproductive tract. Am J Physiol Renal Physiol 278:F717–F725
Breton S, Wiederhold T, Marshansky V, Nsumu NN, Ramesh V, Brown D (2000) The B1 subunit of the H+ATPase is a PDZ domain-binding protein. Colocalization with NHE-RF in renal B-intercalated cells. J Biol Chem 275:18219–18224
Bruce LJ, Cope DL, Jones GK, Schofield AE, Burley M, Povey S, Unwin RJ, Wrong O, Tanner MJ (1997) Familial distal renal tubular acidosis is associated with mutations in the red cell anion exchanger (Band 3, AE1) gene. J Clin Invest 100:1693–1707
Chambrey R, Goossens D, Bourgeois S, Picard N, Bloch-Faure M, Leviel F, Geoffroy V, Cambillau M, Colin Y, Paillard M, Houillier P, Cartron JP, Eladari D (2005) Genetic ablation of Rhbg in the mouse does not impair renal ammonium excretion. Am J Physiol Renal Physiol 289:F1281–F1290
Cheval L, Morla L, Elalouf JM, Doucet A (2006) Kidney collecting duct acid–base "regulon". Physiol Genomics 27:271–281
Codina J, Pressley TA, DuBose TD Jr (1999) The colonic H+, K+-ATPase functions as a Na+-dependent K+(NH4+)-ATPase in apical membranes from rat distal colon. J Biol Chem 274:19693–19698
Cohen EP, Bastani B, Cohen MR, Kolner S, Hemken P, Gluck SL (1992) Absence of H+-ATPase in cortical collecting tubules of a patient with Sjogren's syndrome and distal renal tubular acidosis. J Am Soc Nephrol 3:264–271
Cordat E, Casey JR (2009) Bicarbonate transport in cell physiology and disease. Biochem J 417:423–439
Cordat E, Kittanakom S, Yenchitsomanus PT, Li J, Du K, Lukacs GL, Reithmeier RA (2006) Dominant and recessive distal renal tubular acidosis mutations of kidney anion exchanger 1 induce distinct trafficking defects in MDCK cells. Traffic 7:117–128
Cougnon M, Bouyer P, Jaisser F, Edelman A, Planelles G (1999) Ammonium transport by the colonic H+-K+-ATPase expressed in Xenopus oocytes. Am J Physiol 277:C280–C287
Curthoys NP, Gstraunthaler G (2001) Mechanism of increased renal gene expression during metabolic acidosis. Am J Physiol Renal Physiol 281:F381–F390
Curthoys NP, Taylor L, Hoffert JD, Knepper MA (2007) Proteomic analysis of the adaptive response of rat renal proximal tubules to metabolic acidosis. Am J Physiol Renal Physiol 292:F140–F147
Da Silva JC Jr, Perrone RD, Johns CA, Madias NE (1991) Rat kidney band 3 mRNA modulation in chronic respiratory acidosis. Am J Physiol 260:F204–F209
De Lisle RC, Xu W, Roe BA, Ziemer D (2008) Effects of Muclin (Dmbt1) deficiency on the gastrointestinal system. Am J Physiol Gastrointest Liver Physiol 294:G717–G727
DeFranco PE, Haragsim L, Schmitz PG, Bastani B (1995) Absence of vacuolar H+-ATPase pump in the collecting duct of a patient with hypokalemic distal renal tubular acidosis and Sjogren's syndrome. J Am Soc Nephrol 6:295–301
DeFronzo RA, Beckles AD (1979) Glucose intolerance following chronic metabolic acidosis in man. Am J Physiol 236:E328–E334
Devonald MA, Smith AN, Poon JP, Ihrke G, Karet FE (2003) Non-polarized targeting of AE1 causes autosomal dominant distal renal tubular acidosis. Nat Genet 33:125–127
Devuyst O, Christie PT, Courtoy PJ, Beauwens R, Thakker RV (1999) Intra-renal and subcellular distribution of the human chloride channel, CLC-5, reveals a pathophysiological basis for Dent's disease. Hum Mol Genet 8:247–257
Eladari D, Cheval L, Quentin F, Bertrand O, Mouro I, Cherif-Zahar B, Cartron JP, Paillard M, Doucet A, Chambrey R (2002) Expression of RhCG, a New Putative NH3/NH +4 Transporter, along the Rat Nephron. J Am Soc Nephrol 13:1999–2008
Everett LA, Glaser B, Beck JC, Idol JR, Buchs A, Heyman M, Adawi F, Hazani E, Nassir E, Baxevanis AD, Sheffield VC, Green ED (1997) Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS). Nat Genet 17:411–422
Fejes-Toth G, Naray-Fejes-Toth A (1992) Differentiation of renal beta-intercalated cells to alpha-intercalated and principal cells in culture. Proc Natl Acad Sci U S A 89:5487–5491
Finberg KE, Wagner CA, Bailey MA, Paunescu TG, Breton S, Brown D, Giebisch G, Geibel JP, Lifton RP (2005) The B1 subunit of the H+ATPase is required for maximal urinary acidification. Proc Nat Acad Sci USA 102:13616–13621
Finberg KE, Wagner CA, Stehberger PA, Geibel JP, Lifton RP (2003) Molecular Cloning and Characterization of Atp6v1b1, the Murine Vacuolar H+-ATPase B1-Subunit. Gene 318:25–34
Forgac M (1999) Structure and properties of the vacuolar (H+)-ATPases. J Biol Chem 274:12951–12954
Forgac M (2007) Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat Rev Mol Cell Biol 8:917–929
Frank AE, Wingo CS, Andrews PM, Ageloff S, Knepper MA, Weiner ID (2002) Mechanisms through which ammonia regulates cortical collecting duct net proton secretion. Am J Physiol Renal Physiol 282:F1120–F1128
Frische S, Kwon TH, Frokiaer J, Madsen KM, Nielsen S (2003) Regulated expression of pendrin in rat kidney in response to chronic NH4Cl or NaHCO3 loading. Am J Physiol Renal Physiol 284:F584–F593
Fry AC, Karet FE (2007) Inherited renal acidoses. Physiology (Bethesda) 22:202–211
Fuster DG, Zhang J, Xie XS, Moe OW (2008) The vacuolar-ATPase B1 subunit in distal tubular acidosis: novel mutations and mechanisms for dysfunction. Kidney Int 73:1151–1158
Geller DS, Rodriguez-Soriano J, Vallo Boado A, Schifter S, Bayer M, Chang SS, Lifton RP (1998) Mutations in the mineralocorticoid receptor gene cause autosomal dominant pseudohypoaldosteronism type I. Nat Genet 19:279–281
Giebisch G, Malnic G, De Mello GB, De Mello Aires M (1977) Kinetics of luminal acidification in cortical tubules of the rat kidney. J Physiol 267:571–599
Good DW (1994) Ammonium transport by the thick ascending limb of Henle's loop. Annu Rev Physiol 56:623–647
Gottschalk CW, Lassiter WE, Mylle M (1960) Localization of urine acidification in the mammalian kidney. Am J Physiol 198:581–585
Goyal S, Mentone S, Aronson PS (2005) Immunolocalization of NHE8 in rat kidney. Am J Physiol Renal Physiol 288:F530–F538
Gu S, Villegas CJ, Jiang JX (2005) Differential regulation of amino acid transporter SNAT3 by insulin in hepatocytes. J Biol Chem 280:26055–26062
Gyorke ZS, Sulyok E, Guignard JP (1991) Ammonium chloride metabolic acidosis and the activity of renin-angiotensin-aldosterone system in children. Eur J Pediatr 150:547–549
Hafner P, Grimaldi R, Capuano P, Capasso G, Wagner CA (2008) Pendrin in the mouse kidney is primarily regulated by Cl- excretion but also by systemic metabolic acidosis. Am J Physiol Cell Physiol 295:C1658–C1667
Hamm LL, Alpern RJ, Preisig PA (2008) Cellular mechanisms of renal tubular acidification. In: Alpern RJ, Hebert SC (eds) Seldin and Giebisch's The Kidney Physiology and Pathophysiology. Elsevier, Amsterdam, pp 1539–1585
Han KH, Croker BP, Clapp WL, Werner D, Sahni M, Kim J, Kim HY, Handlogten ME, Weiner ID (2006) Expression of the ammonia transporter, rh C glycoprotein, in normal and neoplastic human kidney. J Am Soc Nephrol 17:2670–2679
Hays SR (1992) Mineralocorticoid modulation of apical and basolateral membrane H+/OH-/HCO -3 transport processes in the rabbit inner stripe of outer medullary collecting duct. J Clin Invest 90:180–187
Heering P, Ivens K, Aker S, Grabensee B (1998) Distal tubular acidosis induced by FK506. Clin Transplant 12:465–471
Henger A, Tutt P, Riesen WF, Hulter HN, Krapf R (2000) Acid–base and endocrine effects of aldosterone and angiotensin II inhibition in metabolic acidosis in human patients. J Lab Clin Med 136:379–389
Hropot M, Fowler N, Karlmark B, Giebisch G (1985) Tubular action of diuretics: distal effects on electrolyte transport and acidification. Kidney Int 28:477–489
Huber S, Asan E, Jons T, Kerscher C, Puschel B, Drenckhahn D (1999) Expression of rat kidney anion exchanger 1 in type-A intercalated cells in metabolic acidosis and alkalosis. Am J Physiol 277:F841–F849
Ibrahim H, Lee YJ, Curthoys NP (2008) Renal response to metabolic acidosis: role of mRNA stabilization. Kidney Int 73:11–18
Jakobsen JK, Odgaard E, Wang W, Elkjaer ML, Nielsen S, Aalkjaer C, Leipziger J (2004) Functional up-regulation of basolateral Na+-dependent HCO3- transporter NBCn1 in medullary thick ascending limb of K+-depleted rats. Pflugers Arch 448:571–578
Jehle S, Zanetti A, Muser J, Hulter HN, Krapf R (2006) Partial neutralization of the acidogenic Western diet with potassium citrate increases bone mass in postmenopausal women with osteopenia. J Am Soc Nephrol 17:3213–3222
Joo KW, Jeon US, Han JS, Ahn C, Kim S, Lee JS, Kim GH, Cho YS, Kim YH, Kim J (1998) Absence of H+-ATPase in the intercalated cells of renal tissues in classic distal renal tubular acidosis. Clinical nephrology 49:226–231
Jouret F, Auzanneau C, Debaix H, Wada GH, Pretto C, Marbaix E, Karet FE, Courtoy PJ, Devuyst O (2005) Ubiquitous and kidney-specific subunits of vacuolar H+-ATPase are differentially expressed during nephrogenesis. J Am Soc Nephrol 16:3235–3246
Kalhoff H, Manz F (2001) Nutrition, acid–base status and growth in early childhood. Eur J Nutr 40:221–230
Karet FE, Finberg KE, Nelson RD, Nayir A, Mocan H, Sanjad SA, Rodriguez SJ, Santos F, Cremers CW, Di Pietro A, Hoffbrand BI, Winiarski J, Bakkaloglu A, Ozen S, Dusunsel R, Goodyer P, Hulton SA, Wu DK, Skvorak AB, Morton CC, Cunningham MJ, Jha V, Lifton RP (1999) Mutations in the gene encoding B1 subunit of H+-ATPase cause renal tubular acidosis with sensorineural deafness. Nat Genet 21:84–90
Karet FE, Gainza FJ, Gyory AZ, Unwin RJ, Wrong O, Tanner MJ, Nayir A, Alpay H, Santos F, Hulton SA, Bakkaloglu A, Ozen S, Cunningham MJ, di Pietro A, Walker WG, Lifton RP (1998) Mutations in the chloride-bicarbonate exchanger gene AE1 cause autosomal dominant but not autosomal recessive distal renal tubular acidosis. Proc Natl Acad Sci U S A 95:6337–6342
Karinch AM, Lin CM, Meng Q, Pan M, Souba WW (2007) Glucocorticoids have a role in renal cortical expression of the SNAT3 glutamine transporter during chronic metabolic acidosis. Am J Physiol Renal Physiol 292:F448–F455
Keskanokwong T, Shandro HJ, Johnson DE, Kittanakom S, Vilas GL, Thorner P, Reithmeier RA, Akkarapatumwong V, Yenchitsomanus PT, Casey JR (2007) Interaction of integrin-linked kinase with the kidney chloride/bicarbonate exchanger, kAE1. J Biol Chem 282:23205–23218
Khademi S, O'Connell J 3rd, Remis J, Robles-Colmenares Y, Miercke LJ, Stroud RM (2004) Mechanism of ammonia transport by Amt/MEP/Rh: structure of AmtB at 1.35 A. Science 305:1587–1594
Khositseth S, Sirikanaerat A, Khoprasert S, Opastirakul S, Kingwatanakul P, Thongnoppakhun W, Yenchitsomanus PT (2008) Hematological abnormalities in patients with distal renal tubular acidosis and hemoglobinopathies. Am J Hematol 83:465–471
Khositseth S, Sirikanerat A, Wongbenjarat K, Opastirakul S, Khoprasert S, Peuksungnern R, Wattanasirichaigoon D, Thongnoppakhun W, Viprakasit V, Yenchitsomanus PT (2007) Distal renal tubular acidosis associated with anion exchanger 1 mutations in children in Thailand. Am J Kidney Dis 49(841):850
Kim HY, Baylis C, Verlander JW, Han KH, Reungjui S, Handlogten ME, Weiner ID (2007) Effect of reduced renal mass on renal ammonia transporter family, Rh C glycoprotein and Rh B glycoprotein, expression. Am J Physiol Renal Physiol 293:F1238–F1247
Kim HY, Verlander JW, Bishop JM, Cain BD, Han KH, Igarashi P, Lee HW, Handlogten ME, Weiner ID (2009) Basolateral Expression of the ammonia transporter family member, Rh C Glycoprotein, in the Mouse Kidney. Am J Physiol Renal Physiol 296(3):F543–F555
Kim J, Cha JH, Tisher CC, Madsen KM (1996) Role of apoptotic and nonapoptotic cell death in removal of intercalated cells from developing rat kidney. Am J Physiol 270:F575–F592
Kim J, Kim YH, Cha JH, Tisher CC, Madsen KM (1999) Intercalated cell subtypes in connecting tubule and cortical collecting duct of rat and mouse. J Am Soc Nephrol 10:1–12
Kim YH, Kwon TH, Frische S, Kim J, Tisher CC, Madsen KM, Nielsen S (2002) Immunocytochemical localization of pendrin in intercalated cell subtypes in rat and mouse kidney. Am J Physiol Renal Physiol 283:F744–F754
Kittanakom S, Cordat E, Akkarapatumwong V, Yenchitsomanus PT, Reithmeier RA (2004) Trafficking defects of a novel autosomal recessive distal renal tubular acidosis mutant (S773P) of the human kidney anion exchanger (kAE1). J Biol Chem 279:40960–40971
Knepper MA, Agre P (2004) Structural biology. The atomic architecture of a gas channel. Science 305:1573–1574
Knepper MA, Packer R, Good DW (1989) Ammonium transport in the kidney. Physiol Rev 69:179–249
Kollert-Jons A, Wagner S, Hubner S, Appelhans H, Drenckhahn D (1993) Anion exchanger 1 in human kidney and oncocytoma differs from erythroid AE1 in its NH2 terminus. Am J Physiol 265:F813–F821
Kovacikova J, Winter C, Loffing-Cueni D, Loffing J, Finberg KE, Lifton RP, Hummler E, Rossier B, Wagner CA (2006) The connecting tubule is the main site of the furosemide-induced urinary acidification by the vacuolar H+-ATPase. Kidney Int 70:1706–1716
Kurth I, Hentschke M, Hentschke S, Borgmeyer U, Gal A, Hubner CA (2006) The forkhead transcription factor Foxi1 directly activates the AE4 promoter. Biochem J 393:277–283
Laing CM, Toye AM, Capasso G, Unwin RJ (2005) Renal tubular acidosis: developments in our understanding of the molecular basis. Int J Biochem Cell Biol 37:1151–1161
Levine DZ, Iacovitti M, Buckman S, Burns KD (1996) Role of angiotensin II in dietary modulation of rat late distal tubule bicarbonate flux in vivo. J Clin Invest 97:120–125
Levine DZ, Iacovitti M, Buckman S, Hincke MT, Luck B, Fryer JN (1997) ANG II-dependent HCO -3 reabsorption in surviving rat distal tubules: expression/activation of H+-ATPase. Am J Physiol 272:F799–F808
Li G, Alexander EA, Schwartz JH (2003) Syntaxin isoform specificity in the regulation of renal H+-ATPase exocytosis. J Biol Chem 278:19791–19797
Li SZ, McDill BW, Kovach PA, Ding L, Go WY, Ho SN, Chen F (2007) Calcineurin-NFATc signaling pathway regulates AQP2 expression in response to calcium signals and osmotic stress. Am J Physiol Cell Physiol 292:C1606–C1616
Lifton RP, Gharavi AG, Geller DS (2001) Molecular mechanisms of human hypertension. Cell 104:545–556
Loffing J, Loffing-Cueni D, Valderrabano V, Klausli L, Hebert SC, Rossier BC, Hoenderop JG, Bindels RJ, Kaissling B (2001) Distribution of transcellular calcium and sodium transport pathways along mouse distal nephron. Am J Physiol Renal Physiol 281:F1021–F1027
Ludwig MG, Vanek M, Guerini D, Gasser JA, Jones CE, Junker U, Hofstetter H, Wolf RM, Seuwen K (2003) Proton-sensing G-protein-coupled receptors. Nature 425:93–98
Luyckx VA, Goda FO, Mount DB, Nishio T, Hall A, Hebert SC, Hammond TG, Yu AS (1998) Intrarenal and subcellular localization of rat CLC5. Am J Physiol 275:F761–F769
Madsen KM, Verlander JW, Kim J, Tisher CC (1991) Morphological adaption of the collecting duct to acid–base disturbances. Kidney Int Suppl 33:S57–S63
Mak RH (1998) Effect of metabolic acidosis on insulin action and secretion in uremia. Kidney Int 54:603–607
Makhanova N, Lee G, Takahashi N, Sequeira Lopez ML, Gomez RA, Kim HS, Smithies O (2006) Kidney function in mice lacking aldosterone. Am J Physiol Renal Physiol 290:F61–F69
Marini AM, Matassi G, Raynal V, Andre B, Cartron JP, Cherif-Zahar B (2000) The human Rhesus-associated RhAG protein and a kidney homologue promote ammonium transport in yeast. Nat Genet 26:341–344
McCurdy DK, Frederic M, Elkinton JR (1968) Renal tubular acidosis due to amphotericin B. New Eng J Med 278:124–131
Mehta PK, Sodhi B, Arruda JA, Kurtzman NA (1979) Interaction of amiloride and lithium on distal urinary acidification. J Lab Clin Med 93:983–994
Michael UF, Chavez R, Cookson SL, Vaamonde CA (1976) Impaired urinary acidification in the hypothyroid rat. Pflugers Arch 361:215–220
Moret C, Dave MH, Schulz N, Jiang JX, Verrey F, Wagner CA (2007) Regulation of renal amino acid transporters during metabolic acidosis. Am J Physiol Renal Physiol 292:F555–F566
Moulin P, Igarashi T, Van Der Smissen P, Cosyns JP, Verroust P, Thakker RV, Scheinman SJ, Courtoy PJ, Devuyst O (2003) Altered polarity and expression of H+-ATPase without ultrastructural changes in kidneys of Dent's disease patients. Kidney Int 63:1285–1295
Nakhoul NL, Hamm LL (2004) Non-erythroid Rh glycoproteins: a putative new family of mammalian ammonium transporters. Pflugers Arch 447:807–812
Nascimento L, Rademacher DR, Hamburger R, Arruda JA, Kurtzman A (1977) On the mechanism of lithium-induced renal tubular acidosis. J Lab Clin Med 89:455–462
Nelson N, Harvey WR (1999) Vacuolar and plasma membrane proton-adenosinetriphosphatases. Physiol Rev 79:361–385
Nelson RD, Guo XL, Masood K, Brown D, Kalkbrenner M, Gluck S (1992) Selectively amplified expression of an isoform of the vacuolar H+-ATPase 56-kilodalton subunit in renal intercalated cells. Proc Natl Acad Sci U S A 89:3541–3545
Nicoletta JA, Ross JJ, Li G, Cheng Q, Schwartz J, Alexander EA, Schwartz JH (2004) Munc-18–2 regulates exocytosis of H+-ATPase in rat inner medullary collecting duct cells. Am J Physiol Cell Physiol 287:C1366–C1374
Nishi T, Forgac M (2002) The vacuolar (H+)-ATPases–nature's most versatile proton pumps. Nat Rev Mol Cell Biol 3:94–103
Nowik M, Lecca MR, Velic A, Rehrauer H, Brandli AW, Wagner CA (2008) Genome-wide gene expression profiling reveals renal genes regulated during metabolic acidosis. Physiol Genomics 32:322–334
Obermuller N, Gretz N, Kriz W, Reilly RF, Witzgall R (1998) The swelling-activated chloride channel ClC-2, the chloride channel ClC-3, and ClC-5, a chloride channel mutated in kidney stone disease, are expressed in distinct subpopulations of renal epithelial cells. J Clin Invest 101:635–642
Ochotny N, Van Vliet A, Chan N, Yao Y, Morel M, Kartner N, von Schroeder HP, Heersche JN, Manolson MF (2006) Effects of human a3 and a4 mutations that result in osteopetrosis and distal renal tubular acidosis on yeast V-ATPase expression and activity. J Biol Chem 281:26102–26111
Odgaard E, Jakobsen JK, Frische S, Praetorius J, Nielsen S, Aalkjaer C, Leipziger J (2004) Basolateral Na+-dependent HCO3- transporter NBCn1-mediated HCO3- influx in rat medullary thick ascending limb. J Physiol 555:205–218
Oster JR, Michael UF, Perez GO, Sonneborn RE, Vaamonde CA (1976) Renal acidification in hypothyroid man. Clinical nephrology 6:398–403
Pacha J, Frindt G, Sackin H, Palmer LG (1991) Apical maxi K channels in intercalated cells of CCT. Am J Physiol 261:F696–F705
Palmer LG, Frindt G (2007) High-conductance K channels in intercalated cells of the rat distal nephron. Am J Physiol Renal Physiol 292:F966–F973
Paunescu TG, Da Silva N, Marshansky V, McKee M, Breton S, Brown D (2004) Expression of the 56 kDa B2 subunit isoform of the vacuolar H+ATPase in proton secreting cells of the kidney and epididymis. Am J Physiol Cell Physiol 287:C149–C162
Paunescu TG, Russo LM, Da Silva N, Kovacikova J, Mohebbi N, Van Hoek AN, McKee M, Wagner CA, Breton S, Brown D (2007) Compensatory membrane expression of the V-ATPase B2 subunit isoform in renal medullary intercalated cells of B1-deficient mice. Am J Physiol Renal Physiol 293:F1915–F1926
Pech V, Kim YH, Weinstein AM, Everett LA, Pham TD, Wall SM (2006) Angiotensin II increases chloride absorption in the cortical collecting duct in mice through a pendrin-dependent mechanism. Am J Physiol Renal Physiol 292(3):F914–F920
Pech V, Zheng W, Pham TD, Verlander JW, Wall SM (2008) Angiotensin II activates H+-ATPase in type-A intercalated cells. J Am Soc Nephrol 19:84–91
Pela I, Bigozzi M, Bianchi B (2008) Profound hypokalemia and hypochloremic metabolic alkalosis during thiazide therapy in a child with Pendred syndrome. Clin Nephrol 69:450–453
Peng H, Vijayakumar S, Schiene-Fischer C, Li H, Purkerson JM, Malesevic M, Liebscher J, Al-Awqati Q, Schwartz GJ (2009) Secreted cyclophilin A, a peptidylprolyl cis-trans isomerase, mediates matrix assembly of hensin, a protein implicated in epithelial differentiation. J Biol Chem 284(10):6465–6475
Pertovaara M, Korpela M, Kouri T, Pasternack A (1999) The occurrence of renal involvement in primary Sjogren's syndrome: a study of 78 patients. Rheumatology (Oxford) 38:1113–1120
Purkerson JM, Schwartz GJ (2007) The role of carbonic anhydrases in renal physiology. Kidney Int 71:103–115
Pushkin A, Kurtz I (2006) SLC4 base (HCO3 -, CO3 2-) transporters: classification, function, structure, genetic diseases, and knockout models. Am J Physiol Renal Physiol 290:F580–F599
Quentin F, Chambrey R, Trinh-Trang-Tan MM, Fysekidis M, Cambillau M, Paillard M, Aronson PS, Eladari D (2004) The Cl-/HCO -3 exchanger pendrin in the rat kidney is regulated in response to chronic alterations in chloride balance. Am J Physiol Renal Physiol 287:F1179–F1188
Quentin F, Eladari D, Cheval L, Lopez C, Goossens D, Colin Y, Cartron JP, Paillard M, Chambrey R (2003) RhBG and RhCG, the Putative Ammonia Transporters, Are Expressed in the Same Cells in the Distal Nephron. J Am Soc Nephrol 14:545–554
Quilty JA, Li J, Reithmeier RA (2002) Impaired trafficking of distal renal tubular acidosis mutants of the human kidney anion exchanger kAE1. Am J Physiol Renal Physiol 282:F810–F820
Renkema KY, Velic A, Dijkman HB, Verkaart S, van der Kemp AW, Nowik M, Timemrmans K, Doucet A, Wagner CA, Bindels RJ, Hoenderop JG (2009) Calcium-sensing receptor-mediated urinary acidification prevents renal stone formation. J Amer Soc Nephrol in press
Renner M, Bergmann G, Krebs I, End C, Lyer S, Hilberg F, Helmke B, Gassler N, Autschbach F, Bikker F, Strobel-Freidekind O, Gronert-Sum S, Benner A, Blaich S, Wittig R, Hudler M, Ligtenberg AJ, Madsen J, Holmskov U, Annese V, Latiano A, Schirmacher P, Amerongen AV, D'Amato M, Kioschis P, Hafner M, Poustka A, Mollenhauer J (2007) DMBT1 confers mucosal protection in vivo and a deletion variant is associated with Crohn's disease. Gastroenterology 133:1499–1509
Romero MF, Fulton CM, Boron WF (2004) The SLC4 family of HCO 3 - transporters. Pflugers Arch 447:495–509
Roscoe JM, Goldstein MB, Halperin ML, Schloeder FX, Stinebaugh BJ (1977) Effect of amphotercin B on urine acidification in rats: implications for the pathogenesis of distal renal tubular acidosis. J Lab Clin Med 89:463–470
Roscoe JM, Goldstein MB, Halperin ML, Wilson DR, Stinebaugh BJ (1976) Lithium-induced impairment of urine acidification. Kidney Int 9:344–350
Roth DE, Venta PJ, Tashian RE, Sly WS (1992) Molecular basis of human carbonic anhydrase II deficiency. Proc Natl Acad Sci U S A 89:1804–1808
Rothenberger F, Velic A, Stehberger PA, Kovacikova J, Wagner CA (2007) Angiotensin II stimulates vacuolar H+-ATPase activity in renal acid-secretory intercalated cells from the outer medullary collecting duct. J Am Soc Nephrol 18:2085–2093
Royaux IE, Wall SM, Karniski LP, Everett LA, Suzuki K, Knepper MA, Green ED (2001) Pendrin, encoded by the Pendred syndrome gene, resides in the apical region of renal intercalated cells and mediates bicarbonate secretion. Proc Natl Acad Sci U S A 98:4221–4226
Rungroj N, Devonald MA, Cuthbert AW, Reimann F, Akkarapatumwong V, Yenchitsomanus PT, Bennett WM, Karet FE (2004) A novel missense mutation in AE1 causing autosomal dominant distal renal tubular acidosis retains normal transport function but is mistargeted in polarized epithelial cells. J Biol Chem 279:13833–13838
Sabolic I, Brown D, Gluck SL, Alper SL (1997) Regulation of AE1 anion exchanger and H+-ATPase in rat cortex by acute metabolic acidosis and alkalosis. Kidney Int 51:125–137
Sands JM, Naruse M, Baum M, Jo I, Hebert SC, Brown EM, Harris HW (1997) Apical extracellular calcium/polyvalent cation-sensing receptor regulates vasopressin-elicited water permeability in rat kidney inner medullary collecting duct. J Clin Invest 99:1399–1405
Schambelan M, Sebastian A, Katuna BA, Arteaga E (1987) Adrenocortical hormone secretory response to chronic NH4Cl-induced metabolic acidosis. Am J Physiol 252:E454–460
Scheinman SJ (1998) X-linked hypercalciuric nephrolithiasis: clinical syndromes and chloride channel mutations. Kidney Int 53:3–17
Schulz N, Dave MH, Stehberger PA, Chau T, Wagner CA (2007) Differential localization of vacuolar H+-ATPases containing a1, a2, a3, or a4 (ATP6V0A1–4) subunit isoforms along the nephron. Cell Physiol Biochem 20:109–120
Schwartz GJ, Al-Awqati Q (1985) Carbon dioxide causes exocytosis of vesicles containing H+ pumps in isolated perfused proximal and collecting tubules. J Clin Invest 75:1638–1644
Schwartz GJ, Barasch J, Al-Awqati Q (1985) Plasticity of functional epithelial polarity. Nature 318:368–371
Schwartz GJ, Tsuruoka S, Vijayakumar S, Petrovic S, Mian A, Al-Awqati Q (2002) Acid incubation reverses the polarity of intercalated cell transporters, an effect mediated by hensin. J Clin Invest 109:89–99
Scott DA, Wang R, Kreman TM, Sheffield VC, Karniski LP (1999) The Pendred syndrome gene encodes a chloride-iodide transport protein. Nat Genet 21:440–443
Sebastian A, McSherry E, Morris RC Jr (1976) Impaired renal conservation of sodium and chloride during sustained correction of systemic acidosis in patients with type 1, classic renal tubular acidosis. J Clin Invest 58:454–469
Sebastian A, Sutton JM, Hulter HN, Schambelan M, Poler SM (1980) Effect of mineralocorticoid replacement therapy on renal acid–base homeostasis in adrenalectomized patients. Kidney Int 18:762–773
Seshadri RM, Klein JD, Kozlowski S, Sands JM, Kim YH, Han KH, Handlogten ME, Verlander JW, Weiner ID (2006) Renal expression of the ammonia transporters, Rhbg and Rhcg, in response to chronic metabolic acidosis. Am J Physiol Renal Physiol 290:F397–F408
Seshadri RM, Klein JD, Smith T, Sands JM, Handlogten ME, Verlander JW, Weiner ID (2006) Changes in subcellular distribution of the ammonia transporter, Rhcg, in response to chronic metabolic acidosis. Am J Physiol Renal Physiol 290:F1443–F1452
Shayakul C, Jarolim P, Zachlederova M, Prabakaran D, Cortez-Campeao D, Kalabova D, Stuart-Tilley AK, Ideguchi H, Haller C, Alper SL (2004) Characterization of a highly polymorphic marker adjacent to the SLC4A1 gene and of kidney immunostaining in a family with distal renal tubular acidosis. Nephrol Dial Transplant 19:371–379
Sly WS, Shah GN (2001) The carbonic anhydrase II deficiency syndrome: osteopetrosis with renal tubular acidosis and cerebral calcification. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The metabolic and molecular bases of inherited disease. McGraw-Hill, New York, pp 5331–5343
Smith AN, Borthwick KJ, Karet FE (2002) Molecular cloning and characterization of novel tissue-specific isoforms of the human vacuolar H+-ATPase C, G and d subunits, and their evaluation in autosomal recessive distal renal tubular acidosis. Gene 297:169–177
Smith AN, Finberg KE, Wagner CA, Lifton RP, Devonald MA, Su Y, Karet FE (2001) Molecular cloning and characterization of Atp6n1b: a novel fourth murine vacuolar H+-ATPase a-subunit gene. J Biol Chem 276:42382–42388
Smith AN, Jouret F, Bord S, Borthwick KJ, Al-Lamki RS, Wagner CA, Ireland DC, Cormier-Daire V, Frattini A, Villa A, Kornak U, Devuyst O, Karet FE (2005) Vacuolar H+-ATPase d2 subunit: molecular characterization, developmental regulation, and localization to specialized proton pumps in kidney and bone. J Am Soc Nephrol 16:1245–1256
Smith AN, Skaug J, Choate KA, Nayir A, Bakkaloglu A, Ozen S, Hulton SA, Sanjad SA, Al-Sabban EA, Lifton RP, Scherer SW, Karet FE (2000) Mutations in ATP6N1B, encoding a new kidney vacuolar proton pump 116-kD subunit, cause recessive distal renal tubular acidosis with preserved hearing. Nat Genet 26:71–75
Song HK, Kim WY, Lee HW, Park EY, Han KH, Nielsen S, Madsen KM, Kim J (2007) Origin and fate of pendrin-positive intercalated cells in developing mouse kidney. J Am Soc Nephrol 18:2672–2682
Soupene E, Inwood W, Kustu S (2004) Lack of the Rhesus protein Rh1 impairs growth of the green alga Chlamydomonas reinhardtii at high CO2. Proc Natl Acad Sci U S A 101:7787–7792
Stahl RA, Kanz L, Maier B, Schollmeyer P (1986) Hyperchloremic metabolic acidosis with high serum potassium in renal transplant recipients: a cyclosporine A associated side effect. Clinical nephrology 25:245–248
Stehberger P, Schulz N, Finberg KE, Karet FE, Giebisch G, Lifton RP, Geibel JP, Wagner CA (2003) Localization and regulation of the ATP6V0A4 (a4) vacuolar H+-ATPase subunit defective in an inherited form of distal renal tubular acidosis. J Am Soc Nephrol 14:3027–3038
Stehberger PA, Shmukler BE, Stuart-Tilley AK, Peters LL, Alper SL, Wagner CA (2007) Distal renal tubular acidosis in mice lacking the AE1 (band3) Cl-/HCO -3 exchanger (slc4a1). J Am Soc Nephrol 18:1408–1418
Sterling D, Alvarez BV, Casey JR (2002) The extracellular component of a transport metabolon. Extracellular loop 4 of the human AE1 Cl-/HCO -3 exchanger binds carbonic anhydrase IV. J Biol Chem 277:25239–25246
Sterling D, Reithmeier RA, Casey JR (2001) A transport metabolon. Functional interaction of carbonic anhydrase II and chloride/bicarbonate exchangers. J Biol Chem 276:47886–47894
Stover EH, Borthwick KJ, Bavalia C, Eady N, Fritz DM, Rungroj N, Giersch AB, Morton CC, Axon PR, Akil I, Al-Sabban EA, Baguley DM, Bianca S, Bakkaloglu A, Bircan Z, Chauveau D, Clermont MJ, Guala A, Hulton SA, Kroes H, Li VG, Mir S, Mocan H, Nayir A, Ozen S, Rodriguez SJ, Sanjad SA, Tasic V, Taylor CM, Topaloglu R, Smith AN, Karet FE (2002) Novel ATP6V1B1 and ATP6V0A4 mutations in autosomal recessive distal renal tubular acidosis with new evidence for hearing loss. J Med Genet 39:796–803
Su Y, Blake-Palmer KG, Sorrell S, Javid B, Bowers K, Zhou A, Chang SH, Qamar S, Karet FE (2008) Human H+ATPase a4 subunit mutations causing renal tubular acidosis reveal a role for interaction with phosphofructokinase-1. Am J Physiol Renal Physiol 295:F950–F958
Su Y, Zhou A, Al-Lamki RS, Karet FE (2003) The 'a' subunit of the V-type H+-ATPase interacts with phosphofructokinase-1 in humans. J Biol Chem 278:20013–20018
Swarts HG, Koenderink JB, Willems PH, De Pont JJ (2005) The non-gastric H, K-ATPase is oligomycin-sensitive and can function as an H+, NH4(+)-ATPase. J Biol Chem 280:33115–33122
Taher SM, Anderson RJ, McCartney R, Popovtzer MM, Schrier RW (1974) Renal tubular acidosis associated with toluene "sniffing". N Eng J Med 290:765–768
Takito J, Hikita C, Al-Awqati Q (1996) Hensin, a new collecting duct protein involved in the in vitro plasticity of intercalated cell polarity. J Clin Invest 98:2324–2331
Tanphaichitr VS, Sumboonnanonda A, Ideguchi H, Shayakul C, Brugnara C, Takao M, Veerakul G, Alper SL (1998) Novel AE1 mutations in recessive distal renal tubular acidosis. Loss-of-function is rescued by glycophorin A. J Clin Invest 102:2173–2179
Teng-umnuay P, Verlander JW, Yuan W, Tisher CC, Madsen KM (1996) Identification of distinct subpopulations of intercalated cells in the mouse collecting duct. J Am Soc Nephrol 7:260–274
Tessitore N, Ortalda V, Fabris A, D'Angelo A, Rugiu C, Oldrizzi L, Lupo A, Valvo E, Gammaro L, Loschiavo C et al (1985) Renal acidification defects in patients with recurrent calcium nephrolithiasis. Nephron 41:325–332
Teta D, Bevington A, Brown J, Pawluczyk I, Harris K, Walls J (2003) Acidosis downregulates leptin production from cultured adipocytes through a glucose transport-dependent post-transcriptional mechanism. J Am Soc Nephrol 14:2248–2254
Toye AM (2005) Defective kidney anion-exchanger 1 (AE1, Band 3) trafficking in dominant distal renal tubular acidosis (dRTA). Biochem Soc Symp 72:47–63
Toye AM, Banting G, Tanner MJ (2004) Regions of human kidney anion exchanger 1 (kAE1) required for basolateral targeting of kAE1 in polarised kidney cells: mis-targeting explains dominant renal tubular acidosis (dRTA). J Cell Sci 117:1399–1410
Toye AM, Bruce LJ, Unwin RJ, Wrong O, Tanner MJ (2002) Band 3 Walton, a C-terminal deletion associated with distal renal tubular acidosis, is expressed in the red cell membrane but retained internally in kidney cells. Blood 99:342–347
Tsuruoka S, Schwartz GJ (1998) Adaptation of the outer medullary collecting duct to metabolic acidosis in vitro. Am J Physiol 275:F982–F990
Ullrich KJ, Eigler FW (1958) Renal tubular hydrogen-ion secretion in mammals. Pflugers Arch 267:491–496
Vallet M, Picard N, Loffing-Cueni D, Fysekidis M, Bloch-Faure M, Deschenes G, Breton S, Meneton P, Loffing J, Aronson PS, Chambrey R, Eladari D (2006) Pendrin regulation in mouse kidney primarily is chloride-dependent. J Am Soc Nephrol 17:2153–2163
Van Huyen JP, Cheval L, Bloch-Faure M, Belair MF, Heudes D, Bruneval P, Doucet A (2008) GDF15 triggers homeostatic proliferation of acid-secreting collecting duct cells. J Am Soc Nephrol 19:1965–1974
Vargas-Poussou R, Houillier P, Le Pottier N, Strompf L, Loirat C, Baudouin V, Macher MA, Dechaux M, Ulinski T, Nobili F, Eckart P, Novo R, Cailliez M, Salomon R, Nivet H, Cochat P, Tack I, Fargeot A, Bouissou F, Kesler GR, Lorotte S, Godefroid N, Layet V, Morin G, Jeunemaitre X, Blanchard A (2006) Genetic investigation of autosomal recessive distal renal tubular acidosis: evidence for early sensorineural hearing loss associated with mutations in the ATP6V0A4 gene. J Am Soc Nephrol 17:1437–1443
Vasuvattakul S, Yenchitsomanus PT, Vachuanichsanong P, Thuwajit P, Kaitwatcharachai C, Laosombat V, Malasit P, Wilairat P, Nimmannit S (1999) Autosomal recessive distal renal tubular acidosis associated with Southeast Asian ovalocytosis. Kidney Int 56:1674–1682
Verlander JW, Hassell KA, Royaux IE, Glapion DM, Wang ME, Everett LA, Green ED, Wall SM (2003) Deoxycorticosterone upregulates PDS (Slc26a4) in mouse kidney: role of pendrin in mineralocorticoid-induced hypertension. Hypertension 42:356–362
Verlander JW, Kim YH, Shin W, Pham TD, Hassell KA, Beierwaltes WH, Green ED, Everett L, Matthews SW, Wall SM (2006) Dietary Cl(-) restriction upregulates pendrin expression within the apical plasma membrane of type B intercalated cells. Am J Physiol Renal Physiol 291:F833–F839
Verlander JW, Miller RT, Frank AE, Royaux IE, Kim YH, Weiner ID (2003) Localization of the ammonium transporter proteins RhBG and RhCG in mouse kidney. Am J Physiol Renal Physiol 284:F323–F337
Vieira FL, Malnic G (1968) Hydrogen ion secretion by rat renal cortical tubules as studied by an antimony microelectrode. Am J Physiol 214:710–718
Vijayakumar S, Erdjument-Bromage H, Tempst P, Al-Awqati Q (2008) Role of integrins in the assembly and function of hensin in intercalated cells. J Am Soc Nephrol 19:1079–1091
Vijayakumar S, Takito J, Hikita C, Al-Awqati Q (1999) Hensin remodels the apical cytoskeleton and induces columnarization of intercalated epithelial cells: processes that resemble terminal differentiation. J Cell Biol 144:1057–1067
Wagner CA, Finberg KE, Breton S, Marshansky V, Brown D, Geibel JP (2004) Renal vacuolar H+-ATPase. Physiol Rev 84:1263–1314
Wagner CA, Finberg KE, Stehberger PA, Lifton RP, Giebisch GH, Aronson PS, Geibel JP (2002) Regulation of the expression of the Cl-/anion exchanger pendrin in mouse kidney by acid–base status. Kidney Int 62:2109–2117
Wall SM, Davis BS, Hassell KA, Mehta P, Park SJ (1999) In rat tIMCD, NH +4 uptake by Na+-K+-ATPase is critical to net acid secretion during chronic hypokalemia. Am J Physiol 277:F866–F874
Wall SM, Fischer MP (2002) Contribution of the Na(+)-K(+)-2Cl(-) cotransporter (NKCC1) to transepithelial transport of H(+), NH(4)(+), K(+), and Na(+) in rat outer medullary collecting duct. J Am Soc Nephrol 13:827–835
Wall SM, Fischer MP, Mehta P, Hassell KA, Park SJ (2001) Contribution of the Na+-K+-2Cl- cotransporter NKCC1 to Cl- secretion in rat OMCD. Am J Physiol Renal Physiol 280:F913–F921
Wall SM, Hassell KA, Royaux IE, Green ED, Chang JY, Shipley GL, Verlander JW (2002) Localization of Pendrin in Mouse Kidney. Am J Physiol Renal Physiol 284:F229–F241
Wall SM, Kim YH, Stanley L, Glapion DM, Everett LA, Green ED, Verlander JW (2004) NaCl restriction upregulates renal Slc26a4 through subcellular redistribution: role in Cl- conservation. Hypertension 44:982–987
Wall SM, Knepper MA, Hassell KA, Fischer MP, Shodeinde A, Shin W, Pham TD, Meyer JW, Lorenz JN, Beierwaltes WH, Dietz JR, Shull GE, Kim YH (2006) Hypotension in NKCC1 null mice: role of the kidneys. Am J Physiol Renal Physiol 290:F409–F416
Walsh S, Turner CM, Toye A, Wagner C, Jaeger P, Laing C, Unwin R (2007) Immunohistochemical comparison of a case of inherited distal renal tubular acidosis (with a unique AE1 mutation) with an acquired case secondary to autoimmune disease. Nephrol Dial Transplant 22:807–812
Watanabe S, Tsuruoka S, Vijayakumar S, Fischer G, Zhang Y, Fujimura A, Al-Awqati Q, Schwartz GJ (2005) Cyclosporin A produces distal renal tubular acidosis by blocking peptidyl prolyl cis-trans isomerase activity of cyclophilin. Am J Physiol 288:F40–F47
Wehrli P, Loffing-Cueni D, Kaissling B, Loffing J (2007) Replication of segment-specific and intercalated cells in the mouse renal collecting system. Histochemistry and Cell Biology 127:389–398
Weiner ID (2004) The Rh gene family and renal ammonium transport. Curr Opin Nephrol Hypertens 13:533–540
Weiner ID, Hamm LL (2007) Molecular mechanisms of renal ammonia transport. Annu Rev Physiol 69:317–340
Williamson RC, Brown AC, Mawby WJ, Toye AM (2008) Human kidney anion exchanger 1 localisation in MDCK cells is controlled by the phosphorylation status of two critical tyrosines. J Cell Sci 121:3422–3432
Winter C, Schulz N, Giebisch G, Geibel JP, Wagner CA (2004) Nongenomic stimulation of vacuolar H+-ATPases in intercalated renal tubule cells by aldosterone. Proc Nat Acad Sci USA 101:2636–2641
Woo AL, Noonan WT, Schultheis PJ, Neumann JC, Manning PA, Lorenz JN, Shull GE (2003) Renal function in NHE3-deficient mice with transgenic rescue of small intestinal absorptive defect. Am J Physiol Renal Physiol 284:F1190–F1198
Wrong O, Bruce LJ, Unwin RJ, Toye AM, Tanner MJ (2002) Band 3 mutations, distal renal tubular acidosis, and Southeast Asian ovalocytosis. Kidney Int 62:10–19
Wrong OM, Feest TG, MacIver AG (1993) Immune-related potassium-losing interstitial nephritis: a comparison with distal renal tubular acidosis. Q J Med 86:513–534
Yakupoglu HY, Corsenca A, Wahl P, Wuthrich RP, Ambuhl PM (2007) Posttransplant acidosis and associated disorders of mineral metabolism in patients with a renal graft. Transplantation 84:1151–1157
Yang Q, Li G, Singh SK, Alexander EA, Schwartz JH (2006) Vacuolar H+-ATPase B1 subunit mutations that cause inherited distal renal tubular acidosis affect proton pump assembly and trafficking in inner medullary collecting duct cells. J Am Soc Nephrol 17:1858–1866
Yip KP, Kurtz I (1995) NH3 permeability of principal cells and intercalated cells measured by confocal fluorescence imaging. Am J Physiol 269:F545–F550
Zheng L, Kostrewa D, Berneche S, Winkler FK, Li XD (2004) The mechanism of ammonia transport based on the crystal structure of AmtB of Escherichia coli. Proc Natl Acad Sci U S A 101:17090–17095
Zidi-Yahiaoui N, Mouro-Chanteloup I, D'Ambrosio AM, Lopez C, Gane P, Le van Kim C, Cartron JP, Colin Y, Ripoche P (2005) Human Rhesus B and Rhesus C glycoproteins: properties of facilitated ammonium transport in recombinant kidney cells. Biochem J 391:33–40
Acknowledgements
Work in the laboratory of the authors has been supported by grants from the Swiss National Science Foundation and FP6 and FP7 work program projects of the European Community (EuReGene, EUNEFRON). N. Mohebbi is the recipient of an ERA-EDTA long-term fellowship.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wagner, C.A., Devuyst, O., Bourgeois, S. et al. Regulated acid–base transport in the collecting duct. Pflugers Arch - Eur J Physiol 458, 137–156 (2009). https://doi.org/10.1007/s00424-009-0657-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-009-0657-z