
Abstract
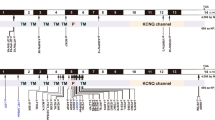
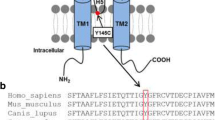
Mutations in the potassium channel gene KCNQ4 underlie DFNA2, a subtype of autosomal dominant progressive, high-frequency hearing loss. Based on a phenotype-guided mutational screening we have identified a novel mutation c.886G>A, leading to the p.G296S substitution in the pore region of KCNQ4 channel. The possible impact of this mutation on total KCNQ4 protein expression, relative surface expression and channel function was investigated. When the G296S mutant was expressed in Xenopus oocytes, electrophysiological recordings did not show voltage-activated K+ currents. The p.G296S mutation impaired KCNQ4 channel activity in two manners. It greatly reduced surface expression and, secondarily, abolished channel function. The deficient expression at the cell surface membrane was further confirmed in non-permeabilized NIH-3T3 cells transfected with the mutant KCNQ4 tagged with the hemagglutinin epitope in the extracellular S1–S2 linker. Co-expression of mutant and wild type KCNQ4 in oocytes was performed to mimic the heterozygous condition of the p.G296S mutation in the patients. The results showed that the G296S mutant exerts a strong dominant-negative effect on potassium currents by reducing the wild type KCNQ4 channel expression at the cell surface. This is the first study to identify a trafficking-dependent dominant mechanism for the loss of KCNQ4 channel function in DFNA2.





Similar content being viewed by others

References
Akita J, Abe S, Shinkawa H, Kimberling WJ, Usami S (2001) Clinical and genetic features of nonsyndromic autosomal dominant sensorineural hearing loss: KCNQ4 is a gene responsible in Japanese. J Hum Genet 46:355–361
Anderson CL, Delisle BP, Anson BD, Kilby JA, Will ML, Tester DJ, Gong Q, Zhou Z, Ackerman MJ, January CT (2006) Most LQT2 mutations reduce Kv11.1 (hERG) current by a class 2 (trafficking-deficient) mechanism. Circulation 113:365–373
Balatsouras D, Kaberos A, Karapantzos E, Homsioglou E, Economou NC, Korres S (2004) Correlation of transiently evoked otoacoustic emission measures to auditory thresholds. Med Sci Monit 10:MT24–30
Beisel KW, Nelson NC, Delimont DC, Fritzsch B (2000) Longitudinal gradients of KCNQ4 expression in spiral ganglion and cochlear hair cells correlate with progressive hearing loss in DFNA2. Mol Brain Res 82:137–149
Beisel KW, Rocha-Sánchez S, Morris KA, Nie L, Feng F, Kachar B, Yamoah EN, Fritzsch B (2005) Differential expression of KCNQ4 in inner hair cells and sensory neurons is the basis of progressive high-frequency hearing loss. J Neurosci 25:9285–9293
Coucke P, Van Camp G, Djoyodiharjo B, Smith SD, Frants RR, Padberg GW, Darby JK, Huizing EH, Cremers CW, Kimberling WJ, Oostra BA, Van de Heyning PH, Willems PJ (1994) Linkage of autosomal dominant hearing loss to the short arm of chromosome 1 in two families. N Engl J Med 331:425–431
Coucke PJ, Van Hauwe P, Kelley PM, Kunst H, Schatteman I, Van Velzen D, Meyers J, Ensink RJ, Verstreken M, Declau F, Marres H, Kastury K, Bhasin S, McGuirt WT, Smith RJH, Cremers CWRJ, Van de Heyning P, Willems PJ, Smith SD, Van Camp G (1999) Mutations in the KCNQ4 gene are responsible for autosomal dominant deafness in four DFNA2 families. Hum Mol Genet 8:1321–1328
Dib C, Faure S, Fizames C, Samson D, Drouot N, Vignal A, Millasseau P, Marc S, Hazan J, Seboun E, Lathrop M, Gyapay G, Morissette J, Weissenbach J (1996) A comprehensive genetic map of the human genome based on 5,264 microsatellites. Nature 380:152–154
Donger C, Denjoy I, Berthet M, Neyroud N, Cruaud C, Bennaceur M, Chivoret G, Schwartz K, Coumel P, Guicheney P (1997) KVLQT1 C-terminal missense mutation causes a forme fruste long-QT syndrome. Circulation 96:2778–2781
Doyle DA, Morais-Cabral J, Pfuetzner RA, KuoA, Gulbis JM, Cohen SL, Chait BT, MacKinnon R (1998) The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science 280:69–77
Ellgaard L, Helenius A (2003) Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol 4: 181–191
Etxeberria A, Santana-Castro I, Regalado MP, Aivar P, Villarroel A (2004) Three mechanisms underlie KCNQ2/3 heteromeric potassium M-channel potentiation. J Neurosci 24:9146–9152
Friedman TB, Griffith AJ (2003) Human nonsyndromic sensorineural deafness. Annu Rev Genom Hum Genet 4:341–412
Howard RJ, Clark KA, Holton JM, Minor DL Jr (2007) Structural insight into KCNQ (Kv7) channel assembly and channelopathy. Neuron 53:663–675
Jentsch TJ (2000) Neuronal KCNQ potassium channels: physiology and role in disease. Nat Rev 1:21–30
Kamada F, Kure S, Kudo T, Suzuki Y, Oshima T, Ichinohe A, Kojima K, Niihori T, Kanno J, Narumi Y, Narisawa A, Kato K, Auki Y, Ikeda K, Kobayashi T, Matsubara Y (2006) A novel KCNQ4 one-base deletion in a large pedigree with hearing loss: implication for the genotype-phenotype correlation. J Hum Genet 51:455–460
Kanki H, Kupershmidt S, Yang T, Wells S, Roden DM (2004) A structural requirement for processing the cardiac K+ channel KCNQ1. J Biol Chem 279:33976–33983
Kharkovets T, Hardelin JP, Safieddine S, Schweizer M, El-Amraoui A, Petit C., Jentsch TJ (2000) KCNQ4, a K+ channel mutated in a form of dominant deafness, is expressed in the inner ear and the central auditory pathway. Proc Natl Acad Sci USA 97:4333–4338
Kharkovets T, Dedek K, Maier H, Schweizer M, Khimich D, Nouvian R, Vardanyan V, Leuwer R, Moser T, Jentsch TJ (2006) Mice with altered KCNQ4 K+ channels implicate sensory outer hair cells in human progressive deafness. EMBO J 25:642–652
Kubisch C, Schroeder BC, Friedrich T, Lutjohann B, El-Amraoui A, Marlin S, Petit C, Jentsch TJ (1999) KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness. Cell 96:437–446
Lorenz C, Pusch M, Jentsch TJ (1996) Heteromultimeric CLC chloride channels with novel properties. Proc Natl Acad Sci USA 93:13362–13366
Ma D, Jan LY (2002) ER transport signals and trafficking of potassium channels and receptors. Curr Opin Neurobiol 12:287–292
MacKinnon R (1991) Determination of the subunits stoichiometry of a voltage-activated potassium channel. Nature 350:232–235
Manganas LN, Wang Q, Scannevin RH, Antonucci DE, Rhodes KJ, Trimmer JS (2001) Identification of a trafficking determinant localized to the Kv1 potassium channel pore. Proc Natl Acad USA 98:14055–14059
Oliver D, Knipper M, Derst C, Fakler B (2003) Resting potential and submembrane calcium concentration of inner hair cells in the isolated mouse cochlea are set by KCNQ-type potassium channels. J Neurosci 23:2141–2149
Petersen MB (2002) Non-syndromic autosomal-dominant deafness. Clin Genet 62:1–13
Petersen MB, Willems PJ (2006) Non-syndromic, autosomal-recessive deafness. Clin Genet 69:371–392
Rowe SM, Miller S, Sorscher EJ (2005) Cystic fibrosis. N Engl J Med 352:1992–2001
Schwake M, Pusch M, Kharkovets T, Jentsch TJ (2000) Surface expression and single channel properties of KCNQ2/KCNQ3, M-type K+ channels involved in epilepsy. J Biol Chem 18:13343–13348
Schwake M, Jentsch TJ, Friedrich T (2003) A carboxy-terminal domain determines the subunit specificity of KCNQ K+ channel assembly. EMBO Rep 4:76–81
Schwake M, Athanasiadu D, Beimgraben C, Blanz J, Beck C, Jentsch TJ, Saftig P, Friedrich T (2006) Structural determinants of M-type KCNQ (Kv7) K+ channel assembly. J Neurosci 26:3757–3766
Shalaby FY, Levesque PC, Yang WP, Little WA, Conder ML, Jenkins-West T, Blanar MA (1997) Dominant-negative KvLQT1 mutations underlie the LQT1 form of long QT syndrome. Circulation 96:1733–1736
Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL, Moss AJ, Schwartz PJ, Towbin JA, Vincent GM, Keating MT (2000) Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation 102:1178–1185
Strutz-Seebohm M, Seebohm G, Fedorenko O, Baltaev R, Engel J, Knirsch M, Lang F (2006) Functional co-assembly of KCNQ4 with KCNE-β-subunits in Xenopus oocytes. Cell Physiol Biochem 18:57–66
Su CC, Yang JJ, Shieh JC, Su MC, Li SY (2007) Identification of novel mutations in the KCNQ4 gene of patients with nonsyndromic deafness from Taiwan. Audiol Neurootol 12:20–26
Talebizadeh Z, Kelley PM, Askew JW, Beisel KW, Smith SD (1999) Novel mutation in the KCNQ4 gene in a large kindred with dominant progressive hearing loss. Hum Mutat 14:493–501
Tanaka T, Nagai R, Tomoike H, Takata S, Yano K, Yabuta K, Haneda N, Nakano O, Shibata A, Sawayama T, Kasai H, Yazaki Y, Nakamura Y (1997) Four novel KVLQT1 and four novel HERG mutations in familial long-QT syndrome. Circulation 95:565–567
Topsakal V, Pennings RJ, te Brinke H, Hamel B, Huygen PL, Kremer H, Cremers CW (2005) Phenotype determination guides swift genotyping of a DFNA2/KCNQ4 family with a hot spot mutation (W276S). Otol Neurotol 26:52–58
Van Camp G, Coucke PJ, Kunst H, Schatteman I, Van Velzen D, Marres H, Van Ewijk M, Declau F, Van Hauwe P, Meyers J, Kenyon J, Smith SD, Smith RJH, Djelantik B, Cremers CWRJ, Van de Heyning PH, Willems PJ (1997) Linkage analysis of progressive hearing loss in five extended families maps the DFNA2 gene to a 1.25-Mb region on chromosome 1p. Genomics 41:70–74
Van Camp G, Coucke PJ, Akita J, Fransen E, Abe S, De Leenheer EM, Huygen PL, Cremers CW, Usami S (2002) A mutational hot spot in the KCNQ4 gene responsible for autosomal dominant hearing impairment. Hum Mutat 20:15–19
Van Hauwe P, Coucke PJ, Ensink RJ, Huygen P, Cremers CW, Van Camp G (2000) Mutations in the KCNQ4 K+ channel gene, responsible for autosomal dominant hearing loss, cluster in the channel pore region. Am J Med Genet 93:184–187
Wollnik B, Schroeder BC, Kubish C, Esperer HD, Wieacker P, Jentsch TJ (1997) Pathophysiological mechanisms of dominant and recessive KvLQT1 K+ channel mutations found in inherited cardiac arrhythmias. Hum Mol Genet 6:1943–1949
Xia JH, Liu CY, Tang BS, Pan Q, Huang L, Dai HP, Zhang BR, Xie W, Hu DX, Zheng D, Shi X, Wang D, Xia K, Yu K, Liao X, Fena Y, Yang Y, Xiao J, Xie D, Huang J (1998) Mutations in the gene encoding gap junction protein beta-3 associated with autosomal dominant hearing impairment. Nat Genet 20:370–373
Acknowledgments
We thank members of the Spanish family, whose participation made this study possible. We also express our gratitude to T.J. Jentsch for kindly providing antibodies and to A. Muñoz, PhD, for technical assistance. This work was supported by grants from the Spanish Ministerio de Ciencia y Tecnología (SAF 2002-03966; BFI 2003-00693; SAF 2005-03414 to L.C.B.), Spanish Fondo de Investigaciones Sanitarias (FIS G03/203; CP03/00014), Programa Ramón y Cajal (to I.d.C); Fundación Ramón Areces and European Union (FP6 Integrated Project EUROHEAR, LSHG-CT-2004-512063; SLMM-CT-2004-50303) and from the Comunidad Autónoma de Madrid (Programa de Biociencias en Ingeniería Biomédica MADR.IB, S2006/SAL-0312 to L.C.B).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mencía, Á., González-Nieto, D., Modamio-Høybjør, S. et al. A novel KCNQ4 pore-region mutation (p.G296S) causes deafness by impairing cell-surface channel expression. Hum Genet 123, 41–53 (2008). https://doi.org/10.1007/s00439-007-0447-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-007-0447-7