
Abstract
Background
Anastomotic leakage (AL) after colorectal surgery is a severe complication, resulting in morbidity, reinterventions, prolonged hospital stay and, in some cases, death. Some technical and patient-related aetiological factors of AL are well established. In many cases, however, none of these factors seem to explain the occurrence of AL. Recent studies suggest that the intestinal microbiome plays a role in wound healing, diabetes and Crohn’s disease. The aim of this study was to compare the intestinal microbiota of patients who developed AL with matched patients with healed colorectal anastomoses.
Methods
We investigated the microbiome in the doughnuts collected from 16 patients participating in the C-seal trial. We selected eight patients who developed AL requiring reintervention and eight matched controls without AL. We analysed the bacterial 16S rDNA of both groups with MiSeq sequencing.
Results
The abundance of Lachnospiraceae is statistically higher (P = 0.001) in patient group who did develop AL, while microbial diversity levels were higher in the group who did not develop AL (P = 0.037). Body mass index (BMI) was also positively associated with the abundance of the Lachnospiraceae family (P = 0.022).
Conclusion
A correlation between the bacterial family Lachnospiraceae, low microbial diversity and anastomotic leakage, possibly in association with the BMI, was found. The relative abundance of the Lachnospiraceae family is possibly explained by the higher abundance of mucin-degrading Ruminococci within that family in AL cases (P = 0.011) as is similarly the case in IBD.
Similar content being viewed by others
Anastomotic leakage (AL) after colorectal surgery is a severe complication, resulting in morbidity, reinterventions, prolonged hospital stay and, in some cases, death. AL can be defined as a defect of the integrity of the intestinal wall at the anastomotic site leading to a communication of the intra- and extraluminal compartments. It may present as a subclinical abscess that drains spontaneously and needs no further treatment to a completely dehiscent anastomosis leading to a faecal peritonitis and sepsis. In many cases, a temporary or definitive ostomy is made. A nationwide Dutch audit revealed an AL rate (requiring reintervention) of 12 % in primary colorectal anastomoses and 9 % in anastomoses with a deviating ostomy [1]. In the literature, AL rates after colorectal surgery are reported in the range of 4–20 % [2].
Some aetiological factors of AL are well established. The anastomosis may be poorly constructed, with tension between the afferent and efferent loop, insufficient circulation or incomplete doughnuts. Healing of the anastomosis may be compromised, as could be the case in patients with diabetes, atherosclerosis or corticosteroid use. However, in many cases, none of these factors seem to explain the occurrence of AL. Therefore, it remains difficult to predict the occurrence of post-operative AL for the individual patient.
Recent studies showed that the composition of the bacterial growth in the intestine influences various processes in the body. For example, bacteria in the intestine are known to influence wound healing [3], and the intestinal microbiome has recently been linked to the origin of diabetes [4, 5]. The development of chemotherapy-induced mucositis is associated with an altered intestinal microbiome [6]. Even the recurrence of Crohn’s disease after resection is suggested to be under influence of microbes [7]. There is also a strong suggestion that the composition of the intestinal microbes affects the healing of the anastomosis and might hence be influenced by antibiotics [8]. In addition, selective decontamination of the digestive tract reduces infections and appears to have a beneficial effect on AL in colorectal surgery [9]. However, there are no publications relating the intestinal bacterial growth with surgical outcome of colorectal resections.
We hypothesized that the composition of the intestinal microbiome could play a significant role in anastomotic healing and the occurrence of leakage. The aim of this study was to compare the intestinal microbiota of patients who developed AL with matched patients with healed colorectal anastomoses, without clinical signs of AL.
Materials and methods
Patients
For this study, eight patients who developed AL requiring reintervention were selected and matched with eight patients without AL. Matching was done on gender, age and pre-operative chemotherapy and radiotherapy. All patients were included in the C-seal trial [2]. This multicentre trial was designed to evaluate the efficacy of the C-seal; the primary endpoint was AL requiring reintervention. This trial was open for inclusion from December 2011 until January 2014. Inclusion criteria were elective colorectal surgery with a circular stapled colorectal anastomosis, age ≥18 years, ASA score <4, mechanical pre-operative bowel preparation and no clinical signs of peritonitis. Exclusion criteria were major surgical or interventional procedures in the 30 days prior to this surgery or other interventional procedures planned within 30 days of entry in this study, and psychological, familial, sociological or geographical conditions which could potentially hamper compliance with the study protocol or the follow-up schedule.
The study was approved by the Medical Ethics Committee of the University Medical Center Groningen—University of Groningen and all participating centres and registered in the Dutch Trial Registry under the number NTR3080. All the patients provided written informed consent. All data were collected anonymously, encoded and saved in a database.
Sample collection
For all patients who consented to be enrolled in this study, we retrieved and stored the ‘doughnut’. This ‘doughnut’ is the small ring of colon and rectum that is cut by the circular stapler to make the anastomosis. Bacterial DNA was isolated from the doughnut and was subsequently analysed using MiSeq sequencing to see whether the microbial composition could be linked with clinical outcome.
DNA extraction
Total DNA was extracted from 0.25 g of a ‘doughnut’ using the repeated bead-beating method described in detail by Yu and Morrison [10], with a number of modifications. In brief, four 3-mm instead of 0.5-mm glass beads were added during the homogenization step. Bead beating was performed using a Precellys 24 (Bertin Technologies, Montigny-le-Bretonneux, France) at 5.5 beats per millisecond in three rounds of 1 min each with 30-s pauses at room temperature in between. The incubation temperature after the bead beating was raised from 70 to 95 °C. Importantly, protein precipitation with 260 μl of ammonium acetate was carried out twice instead of only once. Additional purification steps using columns were not needed after DNA precipitation.
MiSeq preparation sequencing pipeline
The V3–V4 region of the 16S rRNA gene was amplified from the bacterial DNA by polymerase chain reaction (PCR) using modified 341F and 806R primers (Supplementary Table 1) with a six-nucleotide barcode on the 806R primer as described elsewhere [11, 12]. Reaction conditions consisted of an initial 94 °C for 3 min followed by 32 cycles of 94 °C for 45 s, 50 °C for 60 s and 72 °C for 90 s, and a final extension of 72 °C for 10 min. An agarose gel confirmed the presence of product (band at ~465 base pairs) in successfully amplified samples. The remainder of the PCR product (~45 μl) of each sample was mixed thoroughly with 25 μl Agencourt AMPure XP magnetic beads and was incubated at room temperature for 5 min. Beads were subsequently separated from the solution by placing the tubes in a magnetic bead separator for 2 min. After discarding the cleared solution, the beads were washed twice by resuspending the beads in 200 μl freshly prepared 80 % ethanol, incubating the tubes for 30 s in the magnetic bead separator and subsequently discarding the cleared solution. The pellet was subsequently dried for 15 min and resuspended in 52.5 μl 10 mM Tris HCl pH 8.5 buffer. Fifty microlitres of the cleared-up solution is subsequently transferred to a new tube. The DNA concentration of each sample was determined using a Qubit® 2.0 fluorometer (www.invitrogen.com/qubit), and the remainder of the sample was stored at −20 °C until library normalization. Library normalization was done the day before running samples on the MiSeq by making 2 nM dilutions of each sample. Samples were pooled together by combining 5 μl of each diluted sample. Ten microlitres of the sample pool and 10 μl 0.2 M NaOH were subsequently combined and incubated for 5 min to denature the sample DNA. To this, 980 μl of the HT1 buffer from the MiSeq 2 × 300 kit was subsequently added. A denatured diluted PhiX solution was made by combining 2 μl of a 10 nM PhiX library with 3 μl 10 mM Tris HCl pH 8.5 buffer with 0.1 % Tween 20. This 5 μl mixture was mixed with 5 μl 0.2 M NaOH and incubated for 5 min at room temperature. This 10 μl mixture was subsequently mixed with 990 μl HT1 buffer. One hundred and fifty microlitres of the diluted sample pool was combined with 50 μl of the diluted PhiX solution and was further diluted by adding 800 μl HT1 buffer. Six hundred microlitres of the prepared library was loaded into the sample loading reservoir of the MiSeq 2 × 300 cartridge.
MiSeq sequencing pipeline and statistical analysis
Software that was used to analyse the data received from Illumina paired-end sequencing, included PANDAseq [13], QIIME and ARB [14]. Reads with a quality score lower than 0.9 were discarded by PANDAseq. Statistical analyses were performed on the family, genus and species level. QIIME identified sequences down to the family and genus level and was used to perform weighted alpha-diversity analyses, while ARB was used to identify sequences down to the species level. Principal component analysis (PCA) was performed to describe the variation in all of the bacterial groups into a very limited amount of new relevant dimensions of variability in order to address the issue of multiple testing. In this study only, principal component 1, which describes over 67 % of the variation in the data, was correlated with the occurrence of AL. The hierarchical clustering analysis was performed with the Hierarchical Clustering Explorer rmed (http://www.cs.umd.edu/hcil/multi-cluster/). The Simpson index was used as a measure of microbial diversity. Nonparametric tests were used, as microbial abundances are rarely normally distributed. Mann–Whitney U or Spearman’s ρ tests were used as indicated. The use ± indicates that a standard deviation is given. All tests were two-tailed, and a P < 0.05 was considered to indicate statistical significance. All statistical analyses were performed using IBM ® SPSS ® Statistics 20.0.
Results
The doughnuts of eight patients with AL and eight patients without AL were analysed. Patient characteristics are listed in Table 1. Body mass index (BMI) was slightly higher in the group of patients with AL, but was not a significant or independent factor for AL in this study group (P = 0.074, Mann–Whitney U test).
The microbial composition was successfully determined of 15 of the doughnuts. In one doughnut of a patient in the control group, the microbial identification was not successful, probably due to insufficient extraction of bacterial DNA.
Bacterial composition in relation to AL
The strongest and most straightforward correlation that could be found between the bacterial composition and AL was that the abundance of the Lachnospiraceae family (27.3 ± 15.9 %) was significantly higher in patients who developed AL as compared to patients who did not develop AL (P = 0.001, Mann–Whitney U test). The most predominant genera of the Lachnospiraceae family (Ruminococcus (6.1 ± 11.9 %), Blautia (5.2 ± 4.5 %), Roseburia (4.4 ± 4.3 %) and Coprococcus (4.4 ± 4.4 %)) all contributed to this particular association. A receiver operator characteristic (ROC) curve showed that the sensitivity and specificity for the clinical outcome of this finding are high (as can be seen in the Supplementary Fig. 1). BMI was also positively associated with the abundance of the Lachnospiraceae family (P = 0.022, Spearman’s ρ test).
Complete linkage clustering analysis of the sample on the family level furthermore identified one particular cluster of five samples from patients who developed AL who could be distinguished from the other samples mainly by having the highest Lachnospiraceae abundances of all samples. Samples not within this particular cluster (left cluster, Fig. 1) had lower Lachnospiraceae abundances (P = 0.002, Mann–Whitney U test).

Hierarchical clustering analysis (top) in combination with the relative abundances of the different microbial families in samples from patients in whom AL occurred (red circles) and of those with no AL (green circles) developed (Color figure online)
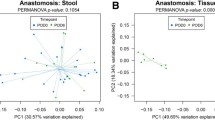
Principal component and alpha-diversity analyses on the family level, however, put the Lachnospiraceae association in a slightly different perspective. A principal component analysis shows that principal component 1 (PC1), which accounts for 67 % of the variation within the data, is positively correlated with patients developing AL (P = 0.021, Mann–Whitney U test), while the Simpson index, a measure of within (α) sample diversity, is negatively correlated with developing AL (P = 0.037, Mann–Whitney U test). Together, these two variables separate most AL and non-AL patients from one another (Fig. 2). Both the Simpson index and PC1 are strongly associated with the abundance of the two most abundant families, Lachnospiraceae and Bacteroidaceae (23.8 ± 15.5 %). The Lachnospiraceae abundance is positively correlated with PC1 (P = 0.003, Spearman’s ρ test) and negatively with the Simpson index (P = 0.007, Spearman’s ρ test), and the same is true for Bacteroidaceae. (P = 0.017 and P = 0.015, respectively). As earlier, a ROC curve showed here as well that the sensitivity and specificity of these tests are high, despite the group of only 16 patients (see Supplementary Fig. 2).

Principal component analysis (PC1, x-axis) in combination with a diversity analysis (y-axis) with respect to the occurrence (red circles) or absence (green circles) of AL in patients. AL is in general associated with a high score on PC1 and/or a low microbial diversity (Color figure online)
When dissecting the Lachnospiraceae finding (37 vs. 17 % average abundance, P = 0.001) down to the species level, it is found that much of the Lachnospiraceae pattern can be attributed to the variation in the amount of Ruminococcus obeum, a mucin-degrading bacterium (6.5 vs. 1.7 % average abundance, P = 0.021). Mucin-degrading Ruminococci from the Lachnospiraceae family as a whole, which include R. gnavus and R. torques, represent the most compelling suggestion for a clinically relevant finding, as their abundance is higher in AL cases (15.5 vs. 3.8 % average abundance, P = 0.011). The BMI, however, was only associated (yet not significantly) with R. obeum (P = 0.068), but not with the other Ruminococci or with any of the other individual bacterial species.
Discussion
This pilot study on the possible role of the intestinal microbiota in the development of AL after colorectal resection with stapled anastomosis revealed interesting patterns. The correlation that was found between AL and the abundance of Lachnospiraceae (Fig. 1) was of particular interest as the association between the Lachnospiraceae family and AL was unexpected: most of the bacteria from this family are not particularly known to have a negative influence on the bowel. In fact, many butyrate-producing genera are found within the Lachnospiraceae family.
Butyrate is thought to be beneficial as it is the main energy source for colonic epithelial cells [15]. Furthermore, butyrate has been shown to regulate the assembly of tight junctions and to correlate with reduced gut permeability [16]. It also decreases intestinal inflammation by reducing oxidative stress in the colonic mucosa [17]. The Roseburia genus in particular is a well-known butyrate producer, which, similar to Faecalibacteria, is associated with protection against inflammatory bowel diseases [18]. However, a large fraction of the Lachnospiraceae reads was identified on the species level to be of mucin-degrading Lachnospiraceae (R. obeum, R. gnavus and R. torques) [19, 20]. The abundance of these mucin-degrading bacteria is commonly observed to be elevated in various inflammatory bowel diseases, such as Crohn’s disease, ulcerative colitis or irritable bowel syndrome [21–24].
On closer inspection, the association between Lachnospiraceae and AL could also be the result of the association between obesity and a lower microbial diversity. Obesity is known to be associated with a lower microbial diversity and with a low-grade systemic inflammation [25–27]. In addition, patients with an inflammatory bowel disease are known to have a low microbial diversity in the gut [27]. Besides that, a high BMI is associated with the development of AL [28–30]. Though the number of obese individuals in this study was limited, an association was found between BMI and Lachnospiraceae levels. Lachnospiraceae levels were strongly negatively correlated with microbial diversity levels that are in turn associated with AL (Fig. 2). So, the overabundance of Lachnospiraceae (or Bacteroidaceae is some cases) might not necessarily be directly linked with the development of AL, but with the absence of other (beneficial) microbial groups.
Alternatively, Lachnospiraceae could also be directly linked with AL as an increase in Firmicutes, of which Lachnospiraceae are an important member, is commonly found in obese people [25]. Butyric acid is also associated with obesity [31]. While butyrate is commonly associated with many beneficial effects stated earlier, an excess of butyrate might present the body with an excess of energy.
It could also be hypothesized that a poorly diversified microbiome is less stable than a well-diversified microbiome. The administration of prophylactic intravenous antibiotics, for instance, as is routinely done in colorectal surgery may cause larger shifts in the bacterial population in a poorly diversified microbiome, offering the opportunity for pathogenic bacteria to repopulate the lumen. The findings of Ohigashi et al. [32] contribute to this theory, as they found that after colorectal surgery the amount of possible pathogenic bacteria, as Enterobacteriaceae, Enterococcus, Staphylococcus and Pseudomonas, was significantly increased. We do not know, however, whether this also happened during the development of AL in our patients.
The main limitation of this study is the small number of included patients. Beside that, we only investigated AL and obesity in relation to the intestinal microbiota. In a follow-up study, we plan to include a much larger group of C-seal trial patients and perform a more detailed analysis of patient and treatment factors in relation to the intestinal microbiome.
References
Bakker IS, Snijders HS, Wouters MW, Havenga K, Tollenaar RA, Wiggers T, Dekker JW (2014) High complication rate after low anterior resection for mid and high rectal cancer; results of a population-based study. Eur J Surg Oncol 40:692–698. doi:10.1016/j.ejso.2014.02.234
Bakker IS, Morks AN, Hoedemaker HO, Burgerhof JG, Leuvenink HG, Ploeg RJ, Havenga K (2012) The C-seal trial: colorectal anastomosis protected by a biodegradable drain fixed to the anastomosis by a circular stapler, a multi-center randomized controlled trial. BMC Surg 12:23-2482-12-23, doi:10.1186/1471-2482-12-23
Scales BS, Huffnagle GB (2013) The microbiome in wound repair and tissue fibrosis. J Pathol 229:323–331. doi:10.1002/path.4118
de Goffau MC, Luopajarvi K, Knip M, Ilonen J, Ruohtula T, Harkonen T, Orivuori L, Hakala S, Welling GW, Harmsen HJ, Vaarala O (2013) Fecal microbiota composition differs between children with beta-cell autoimmunity and those without. Diabetes 62:1238–1244. doi:10.2337/db12-0526
de Goffau MC, Fuentes S, van den Bogert B, Honkanen H, de Vos WM, Welling GW, Hyoty H, Harmsen HJ (2014) Aberrant gut microbiota composition at the onset of type 1 diabetes in young children. Diabetologia 57:1569–1577. doi:10.1007/s00125-014-3274-0
van Vliet MJ, Harmsen HJ, de Bont ES, Tissing WJ (2010) The role of intestinal microbiota in the development and severity of chemotherapy-induced mucositis. PLoS Pathog 6:e1000879. doi:10.1371/journal.ppat.1000879
De Cruz P, Kang S, Wagner J, Buckley M, Sim WH, Prideaux L, Lockett T, McSweeney C, Morrison M, Kirkwood CD, Kamm MA (2014) Specific mucosa-associated microbiota in Crohn’s disease at the time of resection are associated with early disease recurrence: a pilot study. J Gastroenterol Hepatol. doi:10.1111/jgh.12694
Shogan BD, Carlisle EM, Alverdy JC, Umanskiy K (2013) Do we really know why colorectal anastomoses leak? J Gastrointest Surg 17:1698–1707. doi:10.1007/s11605-013-2227-0
Abis GS, Stockmann HB, van Egmond M, Bonjer HJ, Vandenbroucke-Grauls CM, Oosterling SJ (2013) Selective decontamination of the digestive tract in gastrointestinal surgery: Useful in infection prevention? A systematic review. J Gastrointest Surg 17:2172–2178. doi:10.1007/s11605-013-2379-y
Yu Z, Morrison M (2004) Improved extraction of PCR-quality community DNA from digesta and fecal samples. Biotechniques 36:808–812
Bokulich NA, Joseph CM, Allen G, Benson AK, Mills DA (2012) Next-generation sequencing reveals significant bacterial diversity of botrytized wine. PLoS ONE 7:e36357. doi:10.1371/journal.pone.0036357
Bartram AK, Lynch MD, Stearns JC, Moreno-Hagelsieb G, Neufeld JD (2011) Generation of multimillion-sequence 16S rRNA gene libraries from complex microbial communities by assembling paired-end illumina reads. Appl Environ Microbiol 77:3846–3852. doi:10.1128/AEM.02772-10
Masella AP, Bartram AK, Truszkowski JM, Brown DG, Neufeld JD (2012) PANDAseq: paired-end assembler for illumina sequences. BMC Bioinform 13:31-2105-13-31. doi:10.1186/1471-2105-13-31
Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar Buchner A, Lai T, Steppi S, Jobb G, Forster W, Brettske I, Gerber S, Ginhart AW, Gross O, Grumann S, Hermann S, Jost R, Konig A, Liss T, Lussmann R, May M, Nonhoff B, Reichel B, Strehlow R, Stamatakis A, Stuckmann N, Vilbig A, Lenke M, Ludwig T, Bode A, Schleifer KH (2004) ARB: a software environment for sequence data. Nucleic Acids Res 32:1363–1371. doi:10.1093/nar/gkh293
Hague A, Butt AJ, Paraskeva C (1996) The role of butyrate in human colonic epithelial cells: an energy source or inducer of differentiation and apoptosis? Proc Nutr Soc 55:937–943
Peng L, Li ZR, Green RS, Holzman IR, Lin J (2009) Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J Nutr 139:1619–1625. doi:10.3945/jn.109.104638
Hamer HM, Jonkers DM, Bast A, Vanhoutvin SA, Fischer MA, Kodde A, Troost FJ, Venema K, Brummer RJ (2009) Butyrate modulates oxidative stress in the colonic mucosa of healthy humans. Clin Nutr 28:88–93. doi:10.1016/j.clnu.2008.11.002
Tilg H, Danese S (2014) Roseburia hominis: A novel guilty player in ulcerative colitis pathogenesis? Gut 63:1204–1205. doi:10.1136/gutjnl-2013-305799
Salyers AA, West SE, Vercellotti JR, Wilkins TD (1977) Fermentation of mucins and plant polysaccharides by anaerobic bacteria from the human colon. Appl Environ Microbiol 34:529–533
Derrien M, Adawi D, Ahrné S, Jeppsson B, Molin G, Osman N, Štšepetova J, Vaughan EE, Vos WMD, Ouwehand AC (2004) The intestinal mucosa as a habitat of the gut microbiota and a rational target for probiotic functionality and safety. Microb Ecol Health Dis. doi:10.3402/mehd.v16i2-3.7934
Willing BP, Dicksved J, Halfvarson J, Andersson AF, Lucio M, Zheng Z, Jarnerot G, Tysk C, Jansson JK, Engstrand L (2010) A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes. Gastroenterology 139(1844–1854):e1. doi:10.1053/j.gastro.2010.08.049
Joossens M, Huys G, Cnockaert M, De Preter V, Verbeke K, Rutgeerts P, Vandamme P, Vermeire S (2011) Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut 60:631–637. doi:10.1136/gut.2010.223263
Malinen E (2010) Association of symptoms with gastrointestinal microbiota in irritable bowel syndrome. World J Gastroenterol 16:4532. doi:10.3748/wjg.v16.i36.4532
Png CW, Linden SK, Gilshenan KS, Zoetendal EG, McSweeney CS, Sly LI, McGuckin MA, Florin TH (2010) Mucolytic bacteria with increased prevalence in IBD mucosa augment in vitro utilization of mucin by other bacteria. Am J Gastroenterol 105:2420–2428. doi:10.1038/ajg.2010.281
de La Serre CB, Ellis CL, Lee J, Hartman AL, Rutledge JC, Raybould HE (2010) Propensity to high-fat diet-induced obesity in rats is associated with changes in the gut microbiota and gut inflammation. Am J Physiol Gastrointest Liver Physiol 299:G440–G448. doi:10.1152/ajpgi.00098.2010
Verdam FJ, Fuentes S, de Jonge C, Zoetendal EG, Erbil R, Greve JW, Buurman WA, de Vos WM, Rensen SS (2013) Human intestinal microbiota composition is associated with local and systemic inflammation in obesity. Obesity (Silver Spring) 21:E607–E615. doi:10.1002/oby.20466
Ott SJ, Schreiber S (2006) Reduced microbial diversity in inflammatory bowel diseases. Gut 55:1207
Buchs NC, Gervaz P, Secic M, Bucher P, Mugnier-Konrad B, Morel P (2008) Incidence, consequences, and risk factors for anastomotic dehiscence after colorectal surgery: a prospective monocentric study. Int J Colorectal Dis 23:265–270. doi:10.1007/s00384-007-0399-3
Biondo S, Pares D, Kreisler E, Rague JM, Fraccalvieri D, Ruiz AG, Jaurrieta E (2005) Anastomotic dehiscence after resection and primary anastomosis in left-sided colonic emergencies. Dis Colon Rectum 48:2272–2280. doi:10.1007/s10350-005-0159-9
Gendall KA, Raniga S, Kennedy R, Frizelle FA (2007) The impact of obesity on outcome after major colorectal surgery. Dis Colon Rectum 50:2223–2237. doi:10.1007/s10350-007-9051-0
Duncan SH, Barcenilla A, Stewart CS, Pryde SE, Flint HJ (2002) Acetate utilization and butyryl coenzyme A (CoA): acetate-CoA transferase in butyrate-producing bacteria from the human large intestine. Appl Environ Microbiol 68:5186–5190
Ohigashi S, Sudo K, Kobayashi D, Takahashi T, Nomoto K, Onodera H (2013) Significant changes in the intestinal environment after surgery in patients with colorectal cancer. J Gastrointest Surg 17:1657–1664. doi:10.1007/s11605-013-2270-x
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosures
Jasper B. van Praagh, Marcus C. de Goffau, Ilsalien S. Bakker, Hermie J.M. Harmsen, Peter Olinga and Klaas Havenga have no conflicts of interest or financial ties to disclose.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
van Praagh, J.B., de Goffau, M.C., Bakker, I.S. et al. Intestinal microbiota and anastomotic leakage of stapled colorectal anastomoses: a pilot study. Surg Endosc 30, 2259–2265 (2016). https://doi.org/10.1007/s00464-015-4508-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00464-015-4508-z