
Abstract
Recombinant baculovirus (BV) expression systems are widely applied in the production of viral capsid proteins and virus-like particles (VLPs) for use as immunogens and vaccine candidates. Traditional density gradient purification of VLPs does not enable complete elimination of BV-derived impurities, including live viruses, envelope glycoprotein gp64 and baculoviral DNA. We used an additional purification system based on ionic strength to purify norovirus (NoV) GII-4 capsid-derived VLPs. The anion exchange chromatography purification led to highly purified VLPs free from BV impurities with intact morphology. In addition, highly purified VLPs induced strong NoV-specific antibody responses in BALB/c mice. Here, we describe a method for NoV VLP purification and several methods for determining their purity, including quantitative PCR for BV DNA detection.
Similar content being viewed by others


Introduction
Baculoviruses (BVs) are dsDNA viruses that naturally infect insects and arthropods and are unable to replicate in mammalian or other vertebrate cells [3, 8, 32]. The recombinant BV expression system in insect cells was developed for protein expression in the 1980s and has since been used in a wide range of applications because many proteins require folding, subunit assembly, and extensive posttranslational modification to be biologically active [18, 54, 57]. This expression system is highly effective in producing large quantities of recombinant viral capsid and envelope proteins that spontaneously assemble into virus-like particles (VLPs) inside the cytoplasm of infected insect cells [18, 33, 42, 54]. These VLPs are morphologically and antigenically similar to native viruses [33] but lack the replicative capacity of the virus, are non-infectious, and do not contain any genetic material. VLPs, moreover, are commonly stable in storage and relatively acid and heat stable [5, 19, 33]. VLPs provide a valuable alternative in the development of diagnostic assays for immunological and epidemiological studies.
Numerous types of VLPs have been produced for various viruses, such as canine parvovirus, Ebola virus, coronavirus, rotavirus, hepatitis B virus and human immunodeficiency virus (HIV) [40, 41, 48, 53, 54, 62]. VLPs have been demonstrated to be highly effective in stimulating adaptive B- and T cell immune responses in mice [19, 37, 62]. Thus, there is considerable potential for using VLPs as vaccine candidates. Indeed, two human papilloma virus (HPV)-based VLP vaccines have been licensed and are currently widely used [49, 51].
Human noroviruses (NoVs) belong to the family Caliciviridae. NoVs cause the second most common nonbacterial gastroenteritis after rotavirus in humans of all age groups [21, 38, 44] and are associated with outbreaks of gastroenteritis worldwide [12, 39]. NoVs cause approximately 1 million hospitalizations annually and more than 200,000 deaths worldwide in children under 5 years of age [22, 46]. Most NoVs affecting humans belong to two genogroups (GI and GII), and these two genogroups are divided into at least 8 GI and 17 GII genotypes [66]. Despite this diversity, in recent years, genotype GII-4 has primarily been responsible for the majority of sporadic gastroenteritis cases and outbreaks [39, 56].
NoVs are difficult to study because they do not grow in a cell culture system, and no animal model is available. The major capsid protein VP1 is the main target for neutralizing antibodies to NoV [14, 25, 58]. The cloning and expression of the NoV capsid gene VP1 in a BV expression system has been shown to result in the assembly of VLPs that are similar to native virus in size and appearance [33]. NoV VLPs have been used to study protein interactions [25], and virus assembly [50] and is also a source of antigen for diagnostic serological assays and for the development of candidate vaccines against NoVs gastroenteritis [6, 10, 20, 24, 29].
A major challenge for vaccine development is purification of VLPs from recombinant BVs [27, 52]. VLPs used for research purposes are commonly purified by density gradient centrifugation based on separation by size [5, 9, 16, 24, 28, 33, 50]. These methods result in particles with a purity greater than 80 % [19] but do not readily discriminate between VLPs and BVs [17, 47]. Therefore, BV is likely to be present in many of these preparations, and moreover, BVs have been shown to have an adjuvant effect—especially live BVs [27, 51]. Purification procedures based on chromatography have been shown to remove the contaminating BV in VLP preparations [35, 36, 43, 60, 61]. The purification technique affects the structural characteristics, immunogenicity, morphological integrity, antigenicity and functionality of purified VLPs [13, 28, 35, 45].
This study explores the effect of anion exchange chromatography purification on the homogeneity, morphology, antigenicity and immunological properties of NoV VLPs. In addition, several methods are applied for determining their purity.
Materials and methods
Production of NoV VLPs and mock baculoviruses
The insect cell line Sf9 (Invitrogen, Carlsbad, CA) was used to produce NoV GII-4 VLPs as described earlier [28]. A crude purification of VLPs was done using two discontinuous sucrose gradients, and sucrose was removed by overnight dialysis against phosphate-buffered saline (PBS) (pH 7.2). VLPs were concentrated using an Amicon Ultra 30 kDa centrifuge filter device (Millipore Corporation, Billerica, Germany).
Baculoviruses lacking the recombinant NoV gene (mock BV) were produced in Sf9 cells using a baculovirus expression kit (Invitrogen). The cells were seeded in Multidish 6-well plates (Nunc, Thermo Fisher, Scientific, Roskilde, Denmark) at a density of 1 × 106 cells/ml in serum-free medium (Sf 900 SFM III; Invitrogen) and transfected with bacmid DNA (1 μg) using Cellfectin (Invitrogen). The cells were grown at 27 °C and harvested 72 h post-transfection. The cell suspension was centrifuged at 500 × g for 5 min, and the supernatant (P1 BV stock) was collected and used to infect fresh Sf9 cells. Six days postinfection (dpi) the cell suspension was centrifuged as above, and the supernatant (P2 BV stock) was collected and stored at 4 °C. P2 BV stock was used to infect 200-ml cell cultures at a multiplicity of infection (MOI) of 1 and at 6 dpi, the culture was clarified by centrifugation at 3000 × g for 20 min at 4 °C. Mock BVs were concentrated by ultracentrifugation (L8-60M ultracentrifuge, Beckman SW-32.1 Ti rotor) at 100,000 × g for 1.5 h at 4 °C and resuspended in 0.2 M Tris–HCl, pH 7.3. BVs were loaded onto a 10 %–60 % discontinuous sucrose gradient and ultracentrifuged at 100,000 × g for 3 h at 4 °C. Fractions containing BVs determined by SDS-PAGE as described below were pooled, and sucrose was removed by overnight (o/n) dialysis against 1 liter of PBS. BVs were concentrated using an Amicon Ultra 50 kDa centrifuge filter device (Millipore Corporation) and stored at 4 °C in PBS.
Anion exchange chromatography purification of NoV VLPs
Chromatographic purification of NoV GII-4 VLPs was done using a column with anion exchangers (5 ml HiTrap Q, GE Healthcare, Uppsala, Sweden). The column was washed with start buffer consisting of 50 mM sodium phosphate (pH 6.6) to remove preservatives. Next, the column was washed with elution buffer consisting of 0.5 M NaCl in 50 mM sodium phosphate (pH 6.6) and equilibrated with start buffer. Crude purified NoV GII-4 VLPs in PBS were dialyzed against the start buffer before loading into the column (500 μg/column). Unbound VLPs were washed out with start buffer, and column-bound proteins were eluted using elution buffer. A total of 26 fractions (500 μl) were collected and analyzed by SDS-PAGE. The fractions containing NoV capsid were pooled and dialyzed against start buffer. The chromatographic purification was repeated using a fresh column. The fractions were collected, and the identity of the protein was confirmed by SDS-PAGE and immunoblotting. The fractions that were free of BV gp64 protein were pooled, dialyzed against PBS, and sterile filtered using a 0.2-μm syringe filter (VWR, Darmstadt, Germany).
The mock BV preparation was applied to the anion exchange chromatography column using the same method as described above. The fractions (26 fractions, 500 μl each) were collected and analyzed by SDS-PAGE.
SDS-PAGE and immunoblotting
Analysis of NoV capsid and BV proteins by SDS-PAGE was done using polyacrylamide gels with 12 % acrylamide in the separating gel and 5 % in the stacking gel (Bio-Rad Laboratories, Hercules, CA). Samples were boiled for 5 min in Laemmli sample buffer containing 2 % SDS, 5 % β-mercaptoethanol, 62.5 mM Tris-HCl (pH 6.8), 25 % glycerol and 0.01 % bromophenol blue (Bio-Rad). Gels were stained with PageBlue™ Protein Staining Solution (Fermentas, Vilnius, Lithuania). For higher sensitivity, protein detection gels were stained with PageSilver™ Silver Staining Solution (Fermentas) according to the manufacturer’s instructions. To determine the protein purity of NoV VLPs, densitometric analysis of Page Blue-stained SDS-PAGE gels was performed using AlphaEase® FC software (Alpha Innotech, San Leandro, CA). Mock BV and Sf9 cell lysate were used as controls.
For immunoblotting analysis, the proteins were transferred onto a nitrocellulose membrane (Trans-Blot transfer membrane, Bio-Rad). The NoV capsid proteins were detected using NoV GII-4 monoclonal antibody (Kim Laboratories Inc., Illinois, USA) at a 1:4000 dilution in 1 % milk and 0.05 % Tween 20 in Tris-buffered saline (TBS). The BV protein gp64 was detected using 1:400 dilution of anti gp64 monoclonal antibody (Santa Cruz Biotechnology, Santa Cruz, USA) in 1 % milk, 0.05 % Tween 20 in TBS. Horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (Sigma-Aldrich) was used as a secondary antibody at a dilution of 1:10,000 to detect NoV capsid proteins, and at a 1:1000 dilution to detect the BV gp64 protein. The membranes were developed colorimetrically using an Opti-4CN detection kit (Bio-Rad) according to the manufacturer’s instructions.
Determination of protein and total DNA content
The total protein content of the NoV VLPs was determined by Pierce BCA Protein Assay (Thermo Science, Rockford, USA) according to the manufacturer’s instructions, with bovine serum albumin (BSA) used as a standard. The optical density (OD) at 544 nm was determined using a Victor2 1420 Multilabel Counter (Perkin Elmer, Waltman, MA, USA).
The total dsDNA concentration was determined using a Quant-it dsDNA Broad-Range Assay Kit (Invitrogen) according to the manufacturer’s instructions, with λ DNA used as a standard. Fluorescence at excitation/emission 485/535 nm was determined using a Victor2 1420 Multilabel Counter (Perkin Elmer).
Determination of endotoxin content and sterility test
Endotoxin levels in the purified VLP preparations were quantified by Limulus amebocyte lysate (LAL) assay (Lonza, Walkersville, MD, USA) according to manufacturer’s instructions.
In general, more than one culture medium is recommended for sterility testing [64]. Fluid thioglycolate medium and soybean-casein digest medium (both from Sigma-Aldrich) were used for VLPs sterility testing according to manufacturer’s instructions.
Baculovirus DNA genome analysis
The amount of baculoviral DNA quantity was determined using a BacPAK™ qPCR Titration Kit (Clontech, CA, USA) according to manufacturer’s instructions. Briefly, DNA was extracted using the NucleoSpin® Virus Kit included in the BacPAK™ qPCR Titration Kit. SYBR Green detection technology was used to determine baculoviral DNA genome content using the BV AcMNPV vector as a standard (detection range from 1.4 × 100 to 1.4 × 108 copies/μl). Pure NoV VLPs, crudely purified NoV VLPs (positive control) and Sf9 insect cell lysate (negative control) were serially diluted 100–10−2. Forty-cycle PCR was run in a 7900HT Fast Real-Time PCR machine (Applied Biosystems, California, USA) using the 96-plate format with the following conditions: primary denaturation at 95 °C for 30 s, denaturation at 95 °C for 3 s and annealing/extension at 60 °C for 30 s. Because of DNA extraction and dilution of samples, a starting copy number value for the samples was back-calculated by the corresponding dilution factors according to manufacturer′s instructions.
Baculovirus titer determination
Live BV titers were determined using a BacPAK™ Rapid Titer Kit (Clontech). Early log-phase Sf9 cells were seeded on a 96-well microtiter plate (Nunc™, Roskilde, Denmark) in Sf900 cell medium (6.5 × 104 cells/well) and infected with serially diluted test samples. After a 1-hour incubation, the samples were aspirated from the wells, and the cells were covered with methyl cellulose. After incubation for 43–47 h at 27 °C, the cells were fixed with 4 % paraformaldehyde (Sigma Aldrich), stained with monoclonal gp64 antibody and detected with HRP-conjugate provided in the kit. Using an inversion microscope (Nikon, Badhoevedorp, The Netherlands), the foci of infection (clusters of infected cells) were counted in duplicate wells. Plaque-forming units per ml (ifu/ml) were calculated by multiplying the average number of foci per well by the corresponding dilution factors.
Electron microscopy (EM)
The VLP preparations were negatively-stained with 3 % uranyl acetate (UA) (pH 4.6). Three μl of the VLP sample was applied to a carbon-coated grid for 30 s. The grid was dried with filter paper and 3 μl of UA was applied to the grid for 30 s. Excess liquid was removed, and the grid was examined using an FEI Tecnai F12 electron microscope (Philips Electron Optics, The Netherlands) operating at 120 kV.
Immunization of mice
Female BALB/c mice, aged 7 weeks (4 mice/group), were immunized twice intradermally (ID) with pure NoV VLPs at weeks 0 and 3. The doses were 1 μg and 10 μg per immunization point. Blood samples were drawn at study weeks 0 (pre-bleed), 2, 3 and 4, mice were euthanized 2 weeks after the second immunization, and whole blood was collected. Negative control mice were left unvaccinated.
Enzyme-linked immunosorbent assay (ELISA)
Sera from immunized and control mice were tested for NoV GII-4 VLP-specific IgG, IgG1, and IgG2a antibodies by enzyme-linked immunosorbent assay (ELISA) as described in detail elsewhere [10, 58]. Briefly, GII-4 VLPs were coated at 0.2 μg/ml (100 μl/well). Serum samples were used at 1:200 dilution or twofold dilution series were utilized. Analyses of IgG1 and IgG2a antibodies were done using pooled sera from mice. Horseradish peroxidase (HRP)-conjugated anti-mouse IgG (Sigma-Aldrich) was used at a dilution of 1:4000. IgG subtypes were determined using goat anti-mouse IgG1 or IgG2a HRP conjugates (Invitrogen) at a dilution of 1:6000.
Results
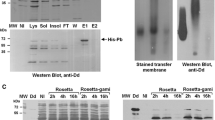
To identify the impurities in the NoV VLP preparation after crude purification with two successive sucrose gradients [28], various analyses were performed. First, NoV capsid proteins were analyzed by 12 % SDS-PAGE and Page Blue staining (Fig. 1A). As seen from the gel, NoV capsid protein appeared as a doublet protein band (58 and 64 kDa), which is typical for the capsid GII-4 protein [28]. In addition, a weaker band in the preparation of NoV capsid VLPs and mock BV, indicated by an arrowhead, was detected, corresponding to the size of the BV envelope gp64 protein (Fig. 1A, lanes a and b). No other proteins derived from BV or Sf9 cells were observed in the VLP preparation. Confirmation of protein identity in the VLP preparation was performed by immunoblotting using NoV GII-4-specific and BV gp64-specific monoclonal antibodies (Fig. 1B). The specificity of the staining was confirmed using a mixture of these monoclonal antibodies to stain a Sf9 cell lysate, with a negative result. In addition, EM analysis of the protein samples (Fig. 1C), in addition to NoV VLPs of approximately 40 nm in size, revealed the presence of residual rod-shaped BVs in the preparation.

Analysis of the identity and purity of sucrose-gradient-purified NoV capsid VLPs. (A) Protein analysis by 12 % SDS-PAGE. Lane m, protein molecular weight marker; lane a, NoV capsid protein VLPs; lane b, mock BV; lane c, Sf9 insect cell lysate. (B) Immunoblotting of NoV capsid protein shown in panel A with NoV GII-4- (lane a) and BV gp64-specific (lane b) monoclonal antibodies. An Sf9 insect cell lysate (lane c) served as a negative control and was probed with both NoV- and BV-specific monoclonal antibodies. An arrowhead in panels A and B indicates the major band of the BV gp64 protein. (C) Electron microscopic analysis of a gradient-purified VLP preparation at a magnification of ×18,500. The arrow indicates an NoV VLP, and the arrowhead, residual BV
To determine the elution times for NoV VLPs and mock BV, both samples were applied to the anion exchange chromatography columns under identical conditions. Examination of the fractions collected (500 μl each) showed that an apparent peak (fractions 15–17, respectively) of the NoV capsid protein (Fig. 2A) was eluted just prior to the BV protein (Fig. 2B). Fractions 15–17 were completely negative when analyzed by immunoblot using BV gp64-specific monoclonal antibody, indicating the absence of BV contamination (data not shown). Further optimization of the steps of the purification process were undertaken to improve the yield of the pure NoV capsid protein. First, different concentrations of NaCl (0.5 M, 1 M and 1.5 M) were used to test the effect of the ionic strength of the elution buffer (50 mM Na2HPO4). Second, the concentration of the protein sample loaded onto the column was also optimized. The best purity and yield were obtained with 50 mM Na2HPO4-0.5 M NaCl buffer, and the optimum amount of crude purified VLPs was 500 μg/column (data not shown). After the first anion exchange chromatography step, fractions 15–17 were pooled and applied to the second, fresh column. The analysis of collected fractions of the second anion exchange chromatography is shown in Fig. 3A. The three fractions (15–17) containing NoV VLPs were collected, pooled, dialyzed against PBS and sterile-filtered. Immunoblotting of each fraction confirmed the lack of BV gp64 impurities (Fig. 3B).

Anion exchange chromatography fractions 15-21 analyzed by 12 % SDS-PAGE and visualized by silver staining. (A) Fractions from NoV capsid VLPs and (B) fractions from control mock BV run through the chromatography columns. m, protein molecular weight marker

Anion exchange chromatography fractions 15–21 of NoV capsid VLPs after the second column purification analyzed by SDS-PAGE and visualized by Page Blue (A) Immunoblotting of fractions 15-17 using BV-gp64-specific monoclonal antibodies (B) Mock BV was used as positive control (lane c). An arrowhead indicates BV. m, protein molecular weight marker
From the starting material (2 mg crude purified VLPs), 0.226 mg anion-exchange-chromatography-purified NoV capsid VLPs was obtained and subjected to several analyses (Figs. 4, 5; Table 1). The identity of the NoV VLPs was demonstrated by immunoblotting using an NoV GII-4-specific monoclonal antibody (Fig. 4A). The results showed doublets of NoV capsid proteins [28] without detectable degradation products. Probing with a BV-gp64-specific monoclonal antibody did not detect any BV gp64 protein (Fig. 4B). Densitometric analysis of the SDS-PAGE confirmed that the NoV VLPs were >90 % pure. The presence of live BV was analyzed as well. As seen in Fig. 4C, duplicate wells of the purified VLP sample completely lacked live BVs in contrast to the mock BV sample wells, which contained ~109 pfu/ml live BV, as determined using a BacPak Rapid Titer Kit. The total DNA content in the pure VLP sample was also quantified, and some residual DNA was detected, but the DNA content was very low (0.41 ng/μl). In addition, the genomic BV DNA was quantified using BV Q-PCR. BacPAK control BV DNA template was used to create a standard curve (Fig. 5). Purified NoV VLPs contained only a few copies of the BV DNA (Table 1; Fig. 5). From the copies of BV DNA in a pure sample, the amount of BV DNA was calculated to be <0.001 pg/μl.

Identity, purity and morphology of anion-exchange-chromatography-purified NoV capsid VLPs. Immunoblot analysis of (A) pure NoV VLPs detected by GII-4-capsid-specific and (B) BV-gp64-specific monoclonal antibodies. Sucrose-gradient-purified NoV VLPs were used as a control (lane c). An arrowhead indicates BV. m, protein molecular weight marker. (C) Live BV detection using a BacPAK™ Rapid Titer Kit. Replicate wells a1 and a2 contain an anion-exchange-chromatography-purified NoV VLPs sample. Dark spots (foci) in the b1 and b2 wells indicate a sample positive for live BV (mock BV). EM of chromatographically purified NoV VLPs (D) and sucrose gradient purified NoV VLPs (E) observed at a magnification of ×30000. Bar, 100 nm

Baculovirus Q-PCR. Standard curve created using serial dilutions of the BacPAK control template (1.4 × 108–1.4 × 100), demonstrating a strong correlation between Ct values and the DNA copy number (log scale) with R2 = 0.975, slope = −3.20, y-Intercept = 34.15. Pure NoV VLP samples, positive control samples (crude purified NoV VLPs) and negative control sample (Sf9 insect cell lysate) were serially diluted 100–10−2. The number of copies BV DNA in the negative control sample were under the detection limit of the assay
EM examination of the purified samples revealed that the structural characteristics (protein assembly into VLPs) and morphological integrity were not affected by the purification process (Fig. 4D) and were similar to those of the gradient-purified VLPs (Fig. 4E). Chromatographically purified NoV VLPs of approx. 40 nm in size were seen in every grid square. NoV VLPs were not contaminated with bacterial endotoxin, as we detected <0.1 EU/10 μg of protein; an amount that is below the international standard of ≤30 EU/20 μg of protein [41]. The purified VLPs were sterile and stable for at least 5 months at 4 °C in PBS, pH 7.4 (data not shown).
To determine the effect of anion exchange chromatographic purification on the immunogenicity of the VLPs, BALB/c mice were immunized with pure NoV GII-4 VLPs at 1 and 10 μg doses without external adjuvants. NoV-GII-4-specific serum antibody responses were induced by each dose of pure NoV VLP (Fig. 6). The IgG response was already quite high after the first immunization and was increased after the second dose at three weeks (Fig. 6A). NoV-specific IgG subtypes were measured to determine the Th1 and Th2 responses [10]. All test groups had similar levels of NoV-specific IgG1 (Fig. 6B) and IgG2a (Fig. 6C) antibodies. Negative control mice lacked detectable NoV-specific antibodies.

NoV capsid-specific antibody responses of BALB/c mice immunized twice intradermally (ID) with 1 μg or 10 μg of pure NoV GII-4 VLPs. (A) Serum samples collected at study weeks 0 (pre-bleed), 2, 3, 4 and 5 were tested by ELISA for NoV-specific IgG. The arrows indicate immunization points at study weeks 0 and 3. NoV-specific serum IgG1 (B) and IgG2a (C) subtype antibody responses. Mean ODs of the groups with standard errors are shown. Control mice were left unvaccinated
Discussion
NoV VLPs are commonly used in different serological assays and animal immunogenicity studies [7, 24, 25, 30]. Density gradient purification of VLPs based on size discrimination is most commonly used in laboratory research [28, 52]. These gradient-purified VLPs contain recombinant BVs and other impurities originating from the expression system [19]. BVs are multiplied to very high titers in suspension culture, complicating the purification procedure. We used two successive anion exchange chromatography steps to purify the VLPs. Our data demonstrate that chromatographically purified VLPs derived from NoV genotype GII-4 possess excellent purity, morphology, antigenicity and immunogenicity.
An adjuvant effect on the immune system of live BVs present in the VLPs has been suggested, specifically in parvovirus and influenza virus VLP preparations [26, 27, 52]. We investigated BV-derived impurities in the sucrose-gradient-purified NoV GII-4 VLPs. The major protein impurity (close to 10 %, determined by densitometric analysis) came from envelope glycoprotein gp64 of BV. The BV gp64 is also linked to adjuvant behavior [2, 23]. gp64 contains a mannose-binding residue, which is expressed on macrophages and dendritic cells. Abe et al. [2] have suggested that gp64 recognizes a Toll-like receptor (TLR) and activates the immune response by inducing inflammatory cytokines such as tumor necrosis factor-γ (TNF-γ) and interleukin-6 (IL-6). The anion-exchange-chromatography-purified NoV VLP preparations were examined for expression-vector-related protein impurities. Densitometrical SDS-PAGE analyses as well as immunoblotting identified NoV capsid protein with >90 % purity and without the traces of BV gp64 protein.
To obtain extremely pure VLPs, several chromatography steps are required [35, 45, 63], but repeated chromatography steps can affect the conformation of VLPs [11]. Purification procedures that preserve the intact conformation of VLPs are important because recombinant VLPs are inherently unstable and tend to denature and aggregate in solution [11, 55]. We used a purification procedure with a combination of two anion exchange chromatography steps in order to minimize impurities and to obtain structurally intact NoV VLPs. Our results show that NoV VLPs purified by two anion exchange chromatography steps retain the structure of the icosahedral capsids and show excellent stability, comparable to density-gradient-purified VLPs (Fig. 4D, E).
It has been reported that ion exchange chromatography is the process of choice for removing to the flowthrough pool host-cell protein and DNA [34, 60, 65]. The anion exchange chromatography method is based on adsorption and reversible binding of charged samples. The major DNA impurities in the VLP preparations come from the baculoviral DNA and not the host cells [27]. Abe and colleagues [1] have shown that BV DNA contains unmethylated CpG motifs that induce proinflammatory cytokines through the TLR-9/MYD88-dependent signaling pathway. Another group has also reported that BV DNA may be responsible for the adjuvant properties of BVs [27]. Our results show that the number of copies of BV DNA was extremely low (Fig. 5; Table 1) in the pure NoV VLP preparation. To the best of our knowledge, this is the first study to determine the number of copies of BV DNA in an NoV VLP preparation by the quantitative BV Q-PCR method. The total dsDNA content in the pure NoV VLP preparation was low (0.9 ng/1-μg dose). In the case of VLP-based vaccine production, the residual DNA needs to be removed to reach acceptable threshold values, typically 10 ng/dose [15]. One group has shown that only live BV was able to stimulate innate immunity, which was not due to the presence of viral DNA [23]. Therefore the presence and quantity of live BVs in the pure VLP preparation were determined. The infectious BV titer was 0 pfu/ml, confirming the complete absence of recombinant live BV in the preparations of pure NoV VLP.
BALB/c mice were immunized with pure NoV VLPs with two relatively low doses. A strong NoV-GII-4-specific immune response was induced. Balanced Th1-type and Th2-type immune response (reflected as IgG2a and IgG1 antibody production) [10] was generated with the VLPs in the absence of external adjuvants. Although we performed no head-to-head comparison of immunogenicity of classical density-gradient-purified and chromatographically purified NoV VLPs, the results obtained in the present study are comparable to those obtained previously with a crude preparation of purified VLPs [10, 58]. These results indicate that pure NoV VLPs are extremely potent immunogens. Others have also shown that compared to soluble individual proteins, multivalent structures of VLPs induce strong B cell and T cell responses in the absence of adjuvants [19, 31].
Finally, we have described a relatively straightforward chromatography method for purification of NoV VLP and methods to confirm their purity. The purified VLPs were tested for impurities including BV protein, DNA, live BV and other impurities related to the expression system. Most significantly, the purified NoV capsid VLPs retained their morphological and antigenic features as well as their immunogenicity. Recombinant NoV VLP vaccine candidates are in the early phases of development [4, 10, 59]. Some research groups have used intranasally delivered NoV VLPs in small animals as well as in human challenge studies [4, 59]. In addition, a vaccine against acute gastroenteritis in young children containing a combination of NoV VLPs and rotavirus VP6 protein is under development [10]. We suggest that this small laboratory-scale purification procedure may potentially be applied for large-scale production of NoV VLPs.
References
Abe T, Hemmi H, Miyamoto H et al (2005) Involvement of the Toll-like receptor 9 signaling pathway in the induction of innate immunity by baculovirus. J Virol 79:2847–2858. doi:10.1128/JVI.79.5.2847-2858.2005
Abe T, Takahashi H, Hamazaki H et al (2003) Baculovirus induces an innate immune response and confers protection from lethal influenza virus infection in mice. J Immunol 171:1133–1139
Andreadis TG, Becnel JJ, White SE (2003) Infectivity and pathogenicity of a novel baculovirus, CuniNPV from Culex nigripalpus (Diptera: Culicidae) for thirteen species and four genera of mosquitoes. J Med Entomol 40:512–517
Atmar RL, Bernstein DI, Harro CD et al (2011) Norovirus vaccine against experimental human Norwalk Virus illness. N Engl J Med 365:2178–2187. doi:10.1056/NEJMoa1101245
Ausar SF, Foubert TR, Hudson MH et al (2006) Conformational stability and disassembly of Norwalk virus-like particles. Effect of pH and temperature. J Biol Chem 281:19478–19488. doi:10.1074/jbc.M603313200
Ball JM, Graham DY, Opekun AR et al (1999) Recombinant Norwalk virus-like particles given orally to volunteers: phase I study. Gastroenterology 117:40–48
Ball JM, Hardy ME, Atmar RL et al (1998) Oral immunization with recombinant Norwalk virus-like particles induces a systemic and mucosal immune response in mice. J Virol 72:1345–1353
Becnel J, White S, Moser B et al (2001) Epizootiology and transmission of a newly discovered baculovirus from the mosquitoes Culex nigripalpus and C. quinquefasciatus. J Gen Virol 82:275–282
Bellier B, Dalba C, Clerc B et al (2006) DNA vaccines encoding retrovirus-based virus-like particles induce efficient immune responses without adjuvant. Vaccine 24:2643–2655. doi:10.1016/j.vaccine.2005.11.034
Blazevic V, Lappalainen S, Nurminen K et al (2011) Norovirus VLPs and rotavirus VP6 protein as combined vaccine for childhood gastroenteritis. Vaccine 29:8126–8133. doi:10.1016/j.vaccine.2011.08.026
Buck CB, Thompson CD, Pang YY et al (2005) Maturation of papillomavirus capsids. J Virol 79:2839–2846. doi:10.1128/JVI.79.5.2839-2846.2005
Buesa J, Collado B, Lopez-Andujar P et al (2002) Molecular epidemiology of caliciviruses causing outbreaks and sporadic cases of acute gastroenteritis in Spain. J Clin Microbiol 40:2854–2859
Burova E, Ioffe E (2005) Chromatographic purification of recombinant adenoviral and adeno-associated viral vectors: methods and implications. Gene Ther 12(Suppl 1):S5–17. doi:10.1038/sj.gt.3302611
Cannon JL, Lindesmith LC, Donaldson EF et al (2009) Herd immunity to GII.4 noroviruses is supported by outbreak patient sera. J Virol 83:5363–5374. doi:10.1128/JVI.02518-08
CBER g (2007) US Food and Drug Administration, Center for Biologics Evaluation and Research
Crawford SE, Labbe M, Cohen J et al (1994) Characterization of virus-like particles produced by the expression of rotavirus capsid proteins in insect cells. J Virol 68:5945–5952
Cruz PE, Maranga L, Carrondo MJ (2002) Integrated process optimization: lessons from retrovirus and virus-like particle production. J Biotechnol 99:199–214
Delchambre M, Gheysen D, Thines D et al (1989) The GAG precursor of simian immunodeficiency virus assembles into virus-like particles. EMBO J 8:2653–2660
Deml L, Speth C, Dierich MP et al (2005) Recombinant HIV-1 Pr55gag virus-like particles: potent stimulators of innate and acquired immune responses. Mol Immunol 42:259–277. doi:10.1016/j.molimm.2004.06.028
El-Kamary SS, Pasetti MF, Mendelman PM et al (2010) Adjuvanted intranasal Norwalk virus-like particle vaccine elicits antibodies and antibody-secreting cells that express homing receptors for mucosal and peripheral lymphoid tissues. J Infect Dis 202:1649–1658. doi:10.1086/657087
Glass RI, Noel J, Ando T et al (2000) The epidemiology of enteric caliciviruses from humans: a reassessment using new diagnostics. J Infect Dis 181(Suppl 2):S254–261. doi:10.1086/315588
Glass RI, Parashar UD, Estes MK (2009) Norovirus gastroenteritis. N Engl J Med 361:1776–1785. doi:10.1056/NEJMra0804575
Gronowski AM, Hilbert DM, Sheehan KC et al (1999) Baculovirus stimulates antiviral effects in mammalian cells. J Virol 73:9944–9951
Hansman GS, Natori K, Shirato-Horikoshi H et al (2006) Genetic and antigenic diversity among noroviruses. J Gen Virol 87:909–919. doi:10.1099/vir.0.81532-0
Harrington PR, Lindesmith L, Yount B et al (2002) Binding of Norwalk virus-like particles to ABH histo-blood group antigens is blocked by antisera from infected human volunteers or experimentally vaccinated mice. J Virol 76:12335–12343
Haynes JR, Dokken L, Wiley JA et al (2009) Influenza-pseudotyped Gag virus-like particle vaccines provide broad protection against highly pathogenic avian influenza challenge. Vaccine 27:530–541. doi:10.1016/j.vaccine.2008.11.011
Hervas-Stubbs S, Rueda P, Lopez L et al (2007) Insect baculoviruses strongly potentiate adaptive immune responses by inducing type I IFN. J Immunol 178:2361–2369
Huhti L, Blazevic V, Nurminen K et al (2010) A comparison of methods for purification and concentration of norovirus GII-4 capsid virus-like particles. Arch Virol 155:1855–1858. doi:10.1007/s00705-010-0768-z
Iritani N, Seto T, Hattori H et al (2007) Humoral immune responses against norovirus infections of children. J Med Virol 79:1187–1193. doi:10.1002/jmv.20897
Iritani N, Seto Y, Kubo H et al (2003) Prevalence of Norwalk-like virus infections in cases of viral gastroenteritis among children in Osaka City, Japan. J Clin Microbiol 41:1756–1759
Jegerlehner A, Storni T, Lipowsky G et al (2002) Regulation of IgG antibody responses by epitope density and CD21-mediated costimulation. Eur J Immunol 32:3305–3314
Jehle JA, Blissard GW, Bonning BC et al (2006) On the classification and nomenclature of baculoviruses: a proposal for revision. Arch Virol 151:1257–1266. doi:10.1007/s00705-006-0763-6
Jiang X, Wang M, Graham DY et al (1992) Expression, self-assembly, and antigenicity of the Norwalk virus capsid protein. J Virol 66:6527–6532
Kalbfuss B, Wolff M, Geisler L et al (2007) Direct capture of influenza A virus from cell culture supernatant with Sartobind anion-exchange membrane adsorbers. J Membr Sci 299:251–260. doi:10.1016/j.memsci.2007.04.048
Kim HJ, Lim SJ, Kwag HL et al (2012) The choice of resin-bound ligand affects the structure and immunogenicity of column-purified human papillomavirus type 16 virus-like particles. PLoS ONE 7:e35893. doi:10.1371/journal.pone.0035893
Koho T, Mantyla T, Laurinmaki P et al (2012) Purification of norovirus-like particles (VLPs) by ion exchange chromatography. J Virol Methods 181:6–11. doi:10.1016/j.jviromet.2012.01.003
Lenz P, Day PM, Pang YY et al (2001) Papillomavirus-like particles induce acute activation of dendritic cells. J Immunol 166:5346–5355
Lew JF, Valdesuso J, Vesikari T et al (1994) Detection of Norwalk virus or Norwalk-like virus infections in Finnish infants and young children. J Infect Dis 169:1364–1367
Lopman B, Vennema H, Kohli E et al (2004) Increase in viral gastroenteritis outbreaks in Europe and epidemic spread of new norovirus variant. Lancet 363:682–688. doi:10.1016/S0140-6736(04)15641-9
Lu X, Chen Y, Bai B et al (2007) Immune responses against severe acute respiratory syndrome coronavirus induced by virus-like particles in mice. Immunology 122:496–502. doi:10.1111/j.1365-2567.2007.02676.x
Makidon PE, Bielinska AU, Nigavekar SS et al (2008) Pre-clinical evaluation of a novel nanoemulsion-based hepatitis B mucosal vaccine. PLoS ONE 3:e2954. doi:10.1371/journal.pone.0002954
Maranga L, Rueda P, Antonis AF et al (2002) Large scale production and downstream processing of a recombinant porcine parvovirus vaccine. Appl Microbiol Biotechnol 59:45–50. doi:10.1007/s00253-002-0976-x
Morenweiser R (2005) Downstream processing of viral vectors and vaccines. Gene Ther 12(Suppl 1):S103–110. doi:10.1038/sj.gt.3302624
Pang XL, Joensuu J, Vesikari T (1999) Human calicivirus-associated sporadic gastroenteritis in Finnish children less than two years of age followed prospectively during a rotavirus vaccine trial. Pediatr Infect Dis J 18:420–426
Park MA, Kim HJ, Kim HJ (2008) Optimum conditions for production and purification of human papillomavirus type 16 L1 protein from Saccharomyces cerevisiae. Protein Expr Purif 59:175–181. doi:10.1016/j.pep.2008.01.021
Patel MM, Widdowson MA, Glass RI et al (2008) Systematic literature review of role of noroviruses in sporadic gastroenteritis. Emerg Infect Dis 14:1224–1231
Pattenden LK, Middelberg AP, Niebert M et al (2005) Towards the preparative and large-scale precision manufacture of virus-like particles. Trends Biotechnol 23:523–529. doi:10.1016/j.tibtech.2005.07.011
Peixoto C, Sousa MF, Silva AC et al (2007) Downstream processing of triple layered rotavirus like particles. J Biotechnol 127:452–461. doi:10.1016/j.jbiotec.2006.08.002
Pomfret TC, Gagnon JM Jr, Gilchrist AT (2011) Quadrivalent human papillomavirus (HPV) vaccine: a review of safety, efficacy, and pharmacoeconomics. J Clin Pharm Ther 36:1–9. doi:10.1111/j.1365-2710.2009.01150.x
Prasad BV, Rothnagel R, Jiang X et al (1994) Three-dimensional structure of baculovirus-expressed Norwalk virus capsids. J Virol 68:5117–5125
Roldao A, Mellado MC, Castilho LR et al (2010) Virus-like particles in vaccine development. Expert Rev Vaccines 9:1149–1176. doi:10.1586/erv.10.115
Rueda P, Fominaya J, Langeveld JP et al (2000) Effect of different baculovirus inactivation procedures on the integrity and immunogenicity of porcine parvovirus-like particles. Vaccine 19:726–734
Sailaja G, Skountzou I, Quan FS et al (2007) Human immunodeficiency virus-like particles activate multiple types of immune cells. Virology 362:331–341. doi:10.1016/j.virol.2006.12.014
Saliki JT, Mizak B, Flore HP et al (1992) Canine parvovirus empty capsids produced by expression in a baculovirus vector: use in analysis of viral properties and immunization of dogs. J Gen Virol 73(Pt 2):369–374
Shi L, Sanyal G, Ni A et al (2005) Stabilization of human papillomavirus virus-like particles by non-ionic surfactants. J Pharm Sci 94:1538–1551. doi:10.1002/jps.20377
Siebenga J, Kroneman A, Vennema H et al (2008) Food-borne viruses in Europe network report: the norovirus GII.4 2006b (for US named Minerva-like, for Japan Kobe034-like, for UK V6) variant now dominant in early seasonal surveillance. Euro Surveill 13:8009
Smith GE, Summers MD, Fraser MJ (1983) Production of human beta interferon in insect cells infected with a baculovirus expression vector. Mol Cell Biol 3:2156–2165
Tamminen K, Huhti L, Koho T et al (2012) A comparison of immunogenicity of norovirus GII-4 virus-like particles and P-particles. Immunology 135:89–99. doi:10.1111/j.1365-2567.2011.03516.x
Velasquez LS, Shira S, Berta AN et al (2011) Intranasal delivery of Norwalk virus-like particles formulated in an in situ gelling, dry powder vaccine. Vaccine 29:5221–5231. doi:10.1016/j.vaccine.2011.05.027
Vicente T, Roldao A, Peixoto C et al (2011) Large-scale production and purification of VLP-based vaccines. J Invertebr Pathol 107(Suppl):S42–48. doi:10.1016/j.jip.2011.05.004
Vicente T, Sousa MFQ, Peixoto C et al (2008) Anion-exchange membrane chromatography for purification of rotavirus-like particles. J Membr Sci 311:270–283. doi:10.1016/j.memsci.2007.12.021
Warfield KL, Bosio CM, Welcher BC et al (2003) Ebola virus-like particles protect from lethal Ebola virus infection. Proc Natl Acad Sci USA 100:15889–15894. doi:10.1073/pnas.2237038100
Woo MK, An JM, Kim JD et al (2008) Expression and purification of human papillomavirus 18 L1 virus-like particle from saccharomyces cerevisiae. Arch Pharm Res 31:205–209
World Health Organization (2012) Document QAS/11.413 Final. March 2012
Wu C, Soh KY, Wang S (2007) Ion-exchange membrane chromatography method for rapid and efficient purification of recombinant baculovirus and baculovirus gp64 protein. Hum Gene Ther 18:665–672. doi:10.1089/hum.2007.020
Zheng DP, Ando T, Fankhauser RL et al (2006) Norovirus classification and proposed strain nomenclature. Virology 346:312–323. doi:10.1016/j.virol.2005.11.015
Acknowledgments
We thank Eeva Jokela at the laboratory of the Vaccine Research Center for excellent technical assistance during the study. We are also grateful to Helena Vihinen for her guidance and assistance with transmission electron microscopy.
Conflict of interest
None of the authors have any conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Huhti, L., Tamminen, K., Vesikari, T. et al. Characterization and immunogenicity of norovirus capsid-derived virus-like particles purified by anion exchange chromatography. Arch Virol 158, 933–942 (2013). https://doi.org/10.1007/s00705-012-1565-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-012-1565-7