
Abstract
Autosomal-dominant, nonsyndromic hearing impairment is clinically and genetically heterogeneous. We encountered a large Japanese pedigree in which nonsyndromic hearing loss was inherited in an autosomal-dominant fashion. A genome-wide linkage study indicated linkage to the DFNA2 locus on chromosome 1p34. Mutational analysis of KCNQ4 encoding a potassium channel revealed a novel one-base deletion in exon 1, c.211delC, which generated a profoundly truncated protein without transmembrane domains (p.Q71fsX138). Previously, six missense mutations and one 13-base deletion, c.211_223del, had been reported in KCNQ4. Patients with the KCNQ4 missense mutations had younger-onset and more profound hearing loss than patients with the 211_223del mutation. In our current study, 12 individuals with the c.211delC mutation manifested late-onset and pure high-frequency hearing loss. Our results support the genotype–phenotype correlation that the KCNQ4 deletions are associated with later-onset and milder hearing impairment than the missense mutations. The phenotypic difference may be caused by the difference in pathogenic mechanisms: haploinsufficiency in deletions and dominant-negative effect in missense mutations.
Similar content being viewed by others


Introduction
Hearing impairment is one of the most common communication disorders in humans and is both clinically and genetically heterogeneous. Approximately 1 in 1,000 children is affected by hearing impairment (Morton 1991), and in half of the cases genetic factors are involved (Marazita et al. 1993). Nonsyndromic hearing impairment is classified according to its mode of inheritance as DFNA, DFNB, and DFN (autosomal dominant, autosomal recessive, and X-linked, respectively). Currently, 54 autosomal dominant, 59 autosomal recessive, and 8 X-linked loci associated with nonsyndromic hearing impairment have been mapped (Hereditary Hearing Loss Homepage, http://webhost.ua.ac.be/hhh/). A total of 21 DFNA genes have been reported to date. Several of the genes are involved in both dominant and recessive deafness (GJB2, GJB6, MYO6, MYO7A, TECTA and TMC1). For example, a null GJB2 mutation, 35delG in Caucasians and 235delC in Asians, is responsible for the majority of autosomal recessive sensorineural deafness in the respective populations (Kenneson et al. 2002; Kudo et al. 2000; Usami et al. 2002). In contrast, some GJB2 mutations, including R75W, segregate with deafness in an autosomal-dominant fashion (Richard et al. 1998). The dominant-negative effect of the R75W mutation was suggested by the transgenic expression in mice (Kudo et al. 2003).
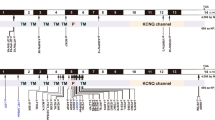
DFNA2 is a locus responsible for autosomal-dominant, nonsyndromic hearing impairment in chromosome 1p34 (Coucke et al. 1994; Van Camp et al. 1997). Two hearing impairment genes, GJB3 and KCNQ4, have been identified in the DFNA2 locus (Kubisch et al. 1999; Xia et al. 1998). The KCNQ4 gene consists of 14 exons that encode a protein of 695 amino acids (MIM*603537). The KCNQ4 protein contains six transmembrane domains and a P-loop region that forms the potassium-selective channel pore (Kubisch et al. 1999). Six missense mutations have been reported in the KCNQ4 gene to date (Akita et al. 2001; Coucke et al. 1999; Kubisch et al. 1999; Talebizadeh et al. 1999; Topsakal et al. 2005; Van Camp et al. 2002; Van Hauwe et al. 2000). Co-expression studies in Xenopus oocytes revealed that the mutant channel protein with a missense mutation exerted a strong dominant-negative effect, which may explain the autosomal-dominant inheritance of KCNQ4 deafness (Kubisch et al. 1999). Besides the missense mutations, one small deletion has also been reported (Coucke et al. 1999). Because the mutation generated a channel protein that is truncated before the first transmembrane domain, it is unlikely that the mutant protein had a dominant-negative effect. The pathogenic mechanism of this deletion in KCNQ4, therefore, remains elusive (Coucke et al. 1999). Recently, Topsakal et al. (2005) noted a phenotypic difference among eight pedigrees with six missense mutations and a single pedigree with the c.211_223del mutation. Based on these data, a hypothesis for the genotype–phenotype correlation is suggested in which younger-onset and all-frequency hearing loss is associated with missense mutations, and later-onset and pure high-frequency hearing loss with null mutations. Although it is an attractive hypothesis, more phenotypic information should be accumulated from individuals with other null KCNQ4 mutations in order to evaluate the genotype–phenotype correlation.
We identified a novel one-base deletion in KCNQ4 exon 1 in a large Japanese pedigree with hearing loss using genome-wide linkage analysis and candidate gene analysis. Individuals with the deletion manifested later-onset and pure high-frequency hearing loss, compared with reported patients with KCNQ4 missense mutations. Our observations support the phenotype–genotype correlation in KCNQ4 deafness, and suggest that haploinsufficiency is the most likely mechanism for development of hearing loss caused by the null KCNQ4 mutations.
Materials and methods
Family data
A Japanese family affected with autosomal-dominant, nonsyndromic hearing impairment was identified (Fig. 1). All affected family members had an affected and an unaffected parent, and four male-to-male transmissions were noted. Twenty-four members of the family participated in this study after giving informed consent. They were asked to complete a questionnaire to exclude other causes of hearing impairment. Special attention was paid to features that might have caused syndromic hearing impairment. All participants underwent otoscopic and audiological examinations. Pure-tone audiograms were obtained in a sound-treated room. Air conduction thresholds were measured in dB hearing level (HL) at 500, 1,000, 2,000, 4,000, and 8,000 Hz. Diagnosis of progressive sensorineural hearing impairment was based on pure-tone audiogram, questionnaire information, and medical records. Family members were considered to be affected if they had sensorineural hearing impairment of more than 25 dB at more than one frequency. Syndromic hearing impairment and environmental causes of deafness were excluded from this study. The ethics committee of Tohoku University School of Medicine approved this study.

Pedigree of the Japanese family with nonsyndromic, autosomal-dominant hearing impairment. The asterisks indicate those individuals in whom a KCNQ4 mutation was identified. The haplotype enclosed in the box shows the disease haplotype
Genetic analysis
Blood samples were obtained from 24 family members, 13 affected and 11 unaffected. Control samples were obtained from 100 Japanese subjects with normal hearing. DNA was extracted from peripheral blood leukocytes using the Genomic DNA purification kit (Promega, Madison, WI, USA). In the genome-wide linkage analysis, microsatellite markers of the Human MAPPAIRS (Invitrogen, Carlsbad, CA, USA) were used for genotyping on the ABI 373A DNA Sequencer. Detailed information for additional genetic markers used in this study can be found in the NCBI Human Map Viewer. We performed pedigree and haplotype constructions using the Cyrillic version 2.1 software. Two-point LOD scores were calculated by the MLINK program of the LINKAGE version 5.1 software package (Lathrop et al. 1984). The affected allele frequency and penetrance were set at 0.0001 and 1.0, respectively. Multipoint linkage analyses were conducted using the GENEHUNTER 2 (Kruglyak et al. 1996).
Mutation analysis was performed by genomic exon sequencing. PCR was carried out using primers flanking 1 GJB3 exon and 14 KCNQ4 exons. Reaction conditions were optimized for different primer sets. Primer sequences were based on the genomic sequence available in the Genbank (GJB3, AF099730; KCNQ4, AH007377). The amplification products from both genes were sequenced with the ABI Prism BigDye Terminator Cycle Sequencing Ready Reaction Kit (Applied Biosystems, Foster City, CA, USA) on the ABI 310 Genetic Analyzer and analyzed using the ABI DNA Sequencing Analysis version 5.1 software.
Results
Genetic analysis
In the genome-wide screening, a maximum two-point LOD score of 5.42 at θ=0 was obtained for GATA129H04 (penetrance=1.0, Table 1). In addition, multipoint linkage analysis with GATA129H04 revealed a maximum nonparametric linkage (NPL) score of 10.95 (data not shown). This marker is known to flank the DFNA2 locus on chromosome 1p34. As shown in Fig. 1, the most likely haplotypes were constructed to determine the borders of the critical region. All affected family members shared the same disease haplotype. The recombination between D1S2134 and the gene for hearing impairment was found in family member II-5. In family member II-9, recombination occurred between the gene for hearing impairment and marker D1S2892. These events localized the gene for hearing impairment between D1S2892 and D1S2134. This region is at 1p34, where the DFNA2 locus resides. These data clearly indicated linkage of the hearing impairment to the DFNA2 locus in this family.
As linkage to the DFNA2 locus was proven in this family, both GJB3 and KCNQ4 genes were examined for mutations. First, we sequenced the coding region of GJB3 in this family, but did not find any mutations. Subsequently, we analyzed all KCNQ4 exons. In exon 1 we identified a deletion of C at nucleotide position 211 of the KCNQ4 cDNA sequence (Fig. 2). All affected family members were heterozygous for this mutation (Fig. 1). The mutation was not detected in the 100 normal controls.

Sequence analysis of the KCNQ4 in a an unaffected (WT/WT) and b an affected member (WT/c.211delC) of the pedigree. The arrow indicates the c.211delC mutation, shown in c. The arrow shows the deletion of C at nucleotide position 211 of the KCNQ4 cDNA sequence. The deletion results in a frameshift after Gly70 (FS71), followed by 67 novel amino acids and a premature stop codon at amino acid position 138 (p.Q71fsX138)
Clinical features
We performed a clinical study on the Japanese family members (13 affected) with a null KCNQ4 mutation to delineate its phenotypic features (Table 2). Patients showed a nonsyndromic, postlingual, symmetric and sensorineural hearing impairment. The hearing impairment in this family was progressive, starting with high frequencies and including middle and low frequencies later in life. According to the anamnestic data, the age at onset varied from 8–50 years.
Discussion
We have identified a novel KCNQ4 mutation in a large Japanese pedigree with hearing loss by a genome-wide linkage analysis followed by the mutational analysis of candidate genes. The identified mutation was a one-base deletion in exon 1 of the KCNQ4 gene, c.211delC, which caused a profoundly truncated peptide without the transmembrane portion of the potassium channel (p.Q71fsX138). Individuals with the apparently null mutation c.211delC had late-onset and pure high-frequency hearing loss, which did not resemble the phenotypes of the patients with the KCNQ4 missense mutations. The individuals with c.211delC had phenotypes similar to those of patients with the c.211_223del (Topsakal et al. 2005). Dominant-negative mechanism has been suggested to underlie the pathogenesis of hearing loss caused by the missense KCNQ4 mutations. Concordance of the mild phenotypes in two different null mutations, c.211_223del and c.211delC, suggests the genotype–phenotype correlation in which KCNQ4 deletions are associated with later-onset and milder hearing loss than the missense mutations.
The phenotypic difference may be caused by the difference in the pathogenic mechanism. Two null mutations (c.211_223del and c.211delC) are expected to yield a KCNQ4 protein that is truncated before the first transmembrane region. Although deleterious mutations result in haploinsufficiency, the function of normal KCNQ4 protein produced from the intact allele may be spared. Fifty-percent decrease in normal KCNQ4 channels may lead to mild hearing impairment. Haploinsufficiency has been suggested to underlie the pathogenesis of both long QT syndrome and benign familial neonatal convulsions caused by KCNQ1 and KCNQ2 or KCNQ3 mutations, respectively (Gouas et al. 2004; Rogawski 2000). In contrast, the dominant-negative effect due to missense mutations significantly interferes with the normal channel subunit. Six missense mutations have been identified in the KCNQ4 gene to date. Five mutations are located in the KCNQ4 P-loop domain, and one mutation is located in the sixth transmembrane domain. Because these missense mutations are located in the critical domain of the protein, they may cause the dominant-negative effect.
The LOD score estimated at the early stage of this study was 3.0, which was far lower than the final estimation. The NPL score at the early stage was estimated to be 8.0, suggesting the possibility of incomplete penetrance. The identified KCNQ4 mutation, c.211delC, was also present in a family member III-12, who had been classified as an unaffected individual. She did not complain of any difficulty in hearing. After the identification of the causative mutation we performed the audiological examination on her. It turned out that she had mild hearing impairment only in high-frequency, in concordance with her genotype. Re-analysis using this information showed an LOD score of 5.42. Thus, the deafness caused by the c.211delC appeared to be transmitted at a level of complete penetrance. The current study illustrates the potential difficulty of linkage analysis in slowly progressive hearing loss in a limited frequency range. Thorough otological examinations may be required even in individuals who are seemingly not affected upon interview.
Although more than 30 deafness genes have been so far identified, the genotype–phenotype relation in each allelic mutation remains largely unidentified. Elucidation of the genotype–phenotype correlations would facilitate informative genetic counseling.
References
Akita J, Abe S, Shinkawa H, Kimberling WJ, Usami S (2001) Clinical and genetic features of nonsyndromic autosomal dominant sensorineural hearing loss: KCNQ4 is a gene responsible in Japanese. J Hum Genet 46:355–361
Coucke P, Van Camp G, Djoyodiharjo B, Smith SD, Frants RR, Padberg GW, Darby JK, Huizing EH, Cremers CW, Kimberling WJ et al (1994) Linkage of autosomal dominant hearing loss to the short arm of chromosome 1 in two families. N Engl J Med 331:425–431
Coucke PJ, Van Hauwe P, Kelley PM, Kunst H, Schatteman I, Van Velzen D, Meyers J, Ensink RJ, Verstreken M, Declau F, Marres H, Kastury K, Bhasin S, McGuirt WT, Smith RJ, Cremers CW, Van de Heyning P, Willems PJ, Smith SD, Van Camp G (1999) Mutations in the KCNQ4 gene are responsible for autosomal dominant deafness in four DFNA2 families. Hum Mol Genet 8:1321–1328
Gouas L, Bellocq C, Berthet M, Potet F, Demolombe S, Forhan A, Lescasse R, Simon F, Balkau B, Denjoy I, Hainque B, Baro I, Guicheney P (2004) New KCNQ1 mutations leading to haploinsufficiency in a general population: defective trafficking of a KvLQT1 mutant. Cardiovasc Res 63:60–68
Kenneson A, Van Naarden Braun K, Boyle C (2002) GJB2 (connexin 26) variants and nonsyndromic sensorineural hearing loss: a HuGE review. Genet Med 4:258–274
Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES (1996) Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet 58:1347–1363
Kubisch C, Schroeder BC, Friedrich T, Lutjohann B, El-Amraoui A, Marlin S, Petit C, Jentsch TJ (1999) KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness. Cell 96:437–446
Kudo T, Ikeda K, Kure S, Matsubara Y, Oshima T, Watanabe K, Kawase T, Narisawa K, Takasaka T (2000) Novel mutations in the connexin 26 gene (GJB2) responsible for childhood deafness in the Japanese population. Am J Med Genet 90:141–145
Kudo T, Kure S, Ikeda K, Xia AP, Katori Y, Suzuki M, Kojima K, Ichinohe A, Suzuki Y, Aoki Y, Kobayashi T, Matsubara Y (2003) Transgenic expression of a dominant-negative connexin26 causes degeneration of the organ of Corti and non-syndromic deafness. Hum Mol Genet 12:995–1004
Lathrop GM, Lalouel JM, Julier C, Ott J (1984) Strategies for multilocus linkage analysis in humans. Proc Natl Acad Sci USA 81:3443–3446
Marazita ML, Ploughman LM, Rawlings B, Remington E, Arnos KS, Nance WE (1993) Genetic epidemiological studies of early-onset deafness in the U.S. school-age population. Am J Med Genet 46:486–491
Morton NE (1991) Genetic epidemiology of hearing impairment. Ann N Y Acad Sci 630:16–31
Richard G, White TW, Smith LE, Bailey RA, Compton JG, Paul DL, Bale SJ (1998) Functional defects of Cx26 resulting from a heterozygous missense mutation in a family with dominant deaf-mutism and palmoplantar keratoderma. Hum Genet 103:393–399
Rogawski MA (2000) KCNQ2/KCNQ3 K+ channels and the molecular pathogenesis of epilepsy: implications for therapy. Trends Neurosci 23:393–398
Talebizadeh Z, Kelley PM, Askew JW, Beisel KW, Smith SD (1999) Novel mutation in the KCNQ4 gene in a large kindred with dominant progressive hearing loss. Hum Mutat 14:493–501
Topsakal V, Pennings RJ, te Brinke H, Hamel B, Huygen PL, Kremer H, Cremers CW (2005) Phenotype determination guides swift genotyping of a DFNA2/KCNQ4 family with a hot spot mutation (W276S). Otol Neurotol 26:52–58
Usami S, Koda E, Tsukamoto K, Otsuka A, Yuge I, Asamura K, Abe S, Akita J, Namba A (2002) Molecular diagnosis of deafness: impact of gene identification. Audiol Neurotol 7:185–190
Van Camp G, Coucke PJ, Kunst H, Schatteman I, Van Velzen D, Marres H, van Ewijk M, Declau F, Van Hauwe P, Meyers J, Kenyon J, Smith SD, Smith RJ, Djelantik B, Cremers CW, Van de Heyning PH, Willems PJ (1997) Linkage analysis of progressive hearing loss in five extended families maps the DFNA2 gene to a 1.25-Mb region on chromosome 1p. Genomics 41:70–74
Van Camp G, Coucke PJ, Akita J, Fransen E, Abe S, De Leenheer EM, Huygen PL, Cremers CW, Usami S (2002) A mutational hot spot in the KCNQ4 gene responsible for autosomal dominant hearing impairment. Hum Mutat 20:15–19
Van Hauwe P, Coucke PJ, Ensink RJ, Huygen P, Cremers CW, Van Camp G (2000) Mutations in the KCNQ4 K+ channel gene, responsible for autosomal dominant hearing loss, cluster in the channel pore region. Am J Med Genet 93:184–187
Xia JH, Liu CY, Tang BS, Pan Q, Huang L, Dai HP, Zhang BR, Xie W, Hu DX, Zheng D, Shi XL, Wang DA, Xia K, Yu KP, Liao XD, Feng Y, Yang YF, Xiao JY, Xie DH, Huang JZ (1998) Mutations in the gene encoding gap junction protein beta-3 associated with autosomal dominant hearing impairment. Nat Genet 20:370–373
Acknowledgements
We are grateful to the family members for participating in this study. We thank Ms. Ikuko Sato and Ms. Yasuko Murayama for excellent technical assistance. This work was supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan, and grants from the Ministry of Health, Labor, and Welfare, Japan.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kamada, F., Kure, S., Kudo, T. et al. A novel KCNQ4 one-base deletion in a large pedigree with hearing loss: implication for the genotype–phenotype correlation. J Hum Genet 51, 455–460 (2006). https://doi.org/10.1007/s10038-006-0384-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-006-0384-7
Keywords
This article is cited by
-
Genetic background in late-onset sensorineural hearing loss patients
Journal of Human Genetics (2022)
-
Novel KCNQ4 variants in different functional domains confer genotype- and mechanism-based therapeutics in patients with nonsyndromic hearing loss
Experimental & Molecular Medicine (2021)
-
Rare KCNQ4 variants found in public databases underlie impaired channel activity that may contribute to hearing impairment
Experimental & Molecular Medicine (2019)
-
Whole-exome sequencing identifies two novel mutations in KCNQ4 in individuals with nonsyndromic hearing loss
Scientific Reports (2018)
-
A novel pore-region mutation, c.887G > A (p.G296D) in KCNQ4, causing hearing loss in a Chinese family with autosomal dominant non-syndromic deafness 2
BMC Medical Genetics (2017)
