
Abstract
A wide variety of cardiac disease states can induce remodelling and lead to the functional consequence of heart failure. These complex disease states involve a plethora of parallel signal transduction events, which may be associated with tissue injury or tissue repair. Innate immunity is activated in hearts injured in different ways, evident as cytokine release from the heart, activation of toll-like receptors involved in recognizing danger, and activation of the transcription factor nuclear factor kappa B. Nuclear factor kappa B regulates gene programmes involved in inflammation as well as the resolution of inflammation. The impact of this is an enigma; while cytokines, toll-like receptors, and nuclear factor kappa B appear to elicit myocardial protection in studies of preconditioning, the literature strongly indicates a detrimental role for activation of innate immunity in studies of acute ischaemia–reperfusion injury. The impact of activation of cardiac innate immunity on the long-term outcome in in vivo models of hypertrophy and remodelling is less clear, with conflicting results as to whether it is beneficial or detrimental. More research using genetically engineered mice as tools, different models of evoking remodelling, and long-term follow-up is required for us to conclude whether activation of the innate immune system is good, bad, or unimportant in chronic injury models.
Similar content being viewed by others

Background
Heart failure (HF) is a functional consequence of a wide variety of diseases, including ischaemic heart disease, congenital disorders, hypertension, valvular heart disease, postpartum cardiomyopathy, and the complex cardiomyopathies where genetic mutations may be contributing reasons [1–3]. Regardless of etiology, the heart may respond with remodelling to maintain cardiac function despite continuous stress. In the 1960s and 1970s, HF was regarded as a mechanical pump failure and treated with inotropic and diuretic agents. In the 1980s, the neurohormonal model of HF was introduced, leading to clinical treatment with angiotensin-converting enzyme inhibitors, β-blockers, and aldosterone antagonists. However, morbidity and mortality of HF still remain high, indicating that we have not yet fully grasped the mechanisms of the underlying pathology and are not able to prevent remodelling. In the 1990s, the cardioinflammatory model of HF was introduced [3]. Since then, a large number of publications have documented the activation of the innate immune system in heart failure patients. Patients with HF have increased circulating levels of inflammatory cytokines, immune activity in the heart itself, and in circulating blood cells. Whether this is a cause or consequence of disease is currently not known. Patients with inflammatory disease (i.e. inflammatory joint disease, inflammatory bowel disease and HIV infections) have increased incidence of cardiovascular disease, indicating that the events leading to remodelling of the heart may be due to systemic effects driven by the immune system [3–6]. The current review aims at giving an overview of how the innate immune system regulates cardiac cells in the process of remodelling towards HF.
Basic biology of remodelling
Remodelling of the heart as a response to various injuries contains some specific features; at a macroscopic level, an increase of cardiac mass takes place before thinning of the ventricular wall and dilatation [1, 7, 8]. At the cellular level, many events take place; cardiomyocyte growth; cardiomyocyte death through apoptosis and autophagy; and deposition of extracellular matrix. The remodelling heart turns on foetal gene programmes originally used for heart formation, which may be responsible for some of the maladaptive events in the failing heart [1, 2]. Cardiac cell growth is an important adaptive process to an increased work load, which ultimately may contribute to HF [9]. Cardiac cell growth itself is regulated by proteasomes, regulating both proteolysis and protein synthesis in cells on their way to failure development [9, 10]. Apoptosis is the main pathway through which the organism gets rid of damaged cells, whereas autophagy is the main way of eliminating or recycling damaged organelles [9, 11]. Apoptosis and autophagy are part of normal heart maintenance but are increased in the process of remodelling [9, 11]. Evidence has existed of apoptosis in the development of heart failure for some time, although involving a minority of cells and with an unclear importance for disease development [7, 12]. Recent research pinpoints autophagy as an emerging factor controlling remodelling in the process of HF development [11, 13]. Deposition of extracellular matrix during remodelling contributes to increased cardiac size and deteriorating function [1–3]. The innate immune system may itself influence all these factors which are part of the remodelling process. The current review will focus on in vivo evidence linking the innate immune system to remodelling rather than details on life, death, and growth of different cells.
Innate immunity
Innate immunity is our first line of defence against foreign intruders. It is present from birth and is able to recognize danger “at a glance”. The innate immune system consists of phagocytosing cells such as macrophages, dendritic cells, and polymorphonuclear granulocytes, as well as cytokines, chemokines, and activation of the complement cascade [14–17]. The cells of the innate immune system recognize conserved motifs on pathogens termed pathogen-associated molecular patterns (PAMP). Recent evidence also suggests that the innate immune system recognizes endogenous danger signals, termed damage-associated molecular patterns (DAMP) [17, 18]. Polly Matzinger [19] proposed that the immune system does not discriminate between self and non-self but reacts upon danger rather than foreignness. The presence of potentially infectious PAMP would not necessarily trigger an immune response unless there is evidence of host tissue injury signified by alarm signals [18, 19]. Endogenous alarm signals may be crystalline uric acid, heat shock proteins 60, 70, and 96, fragments of hyaluronan and fibronectin [20–22]. Potentially other, yet undiscovered, molecules are released during myocardial remodelling and may act as danger molecules.
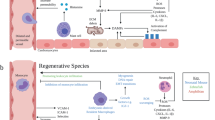
PAMP/DAMP in turn are recognized by pattern recognition receptors (PRR), which may be membrane bound or cytoplasmatic. A cartoon of the signalling downstream to PRR is shown in Fig. 1. There are currently four known families of PRR: The transmembrane protein families of toll-like receptors (TLR) and C-type lectin receptors (CLR), as well as cytoplasmatic protein families such as nucleotide oligomerization domain (NOD)- like receptors (NLR), and retinoic acid-inducible gene (RIG)-like receptors (RLR) [17]. PRR are present not only in cells of the innate immune system such as macrophages and dendritic cells, but also in “non-professional” immune cells. Very little is currently known about the expression and function of NLR, CLR, and RLR in the heart, and thus this review will summarize some of the budding knowledge of TLR below. The binding of a PAMP to a PRR leads to activation of different transcription factors through complex signalling pathways, producing substances increasing the defence against the threatening factor. Nuclear factor kappa B (NFκB) is one of the most important transcription factors regulating immune responses downstream to TLR and has been called the bridge between innate and adaptive immunity [23].

Toll-like receptors (TLR) recognize pathogen-associated molecular patterns (PAMP) or endogenous damage-associated molecular patterns (DAMP). Ligand stimulation recruits adaptor molecules TIRAP (TIR domain-containing adaptor protein) or TRAM (TRIF-related adaptor molecule). TLR signal either through the adaptor molecule MyD88 leading to activation of nuclear factor kappa B (NFκB) or independent of MyD88, leading to activation of interferon regulatory factor (IRF3). Abbreviations; AP1 transcription factor activator protein 1, IκB inhibitor of NFκB, IFN interferon gamma, IKK inhibitor of NFκB kinase type α, β, and ε, IRAK interleukin 1 receptor-associated kinases 1 and 4, MAPK mitogen-activated protein kinases, NEMO inhibitor of NFκB kinase type γ, dsRNA double-stranded RNA, RIP1 receptor-interacting protein 1, TAB TAK-binding proteins, TAK transforming growth factor beta–activated kinases, TBK1 serine-threonine protein kinase, TRAF6 tumour necrosis factor receptor–associated factor 6, TRIF TIR domain containing adaptor–inducing interferon β. Ub denotes ubiquitination, P denotes phosphorylation
Adaptive immunity is developed as a result of previous encounters with pathogens [24]. This form of immunity is specific, as it will recognize and counteract a pathogen that has been introduced earlier. The developed immunity lasts for many years and can therefore be termed to have memory. Tolerance indicates that the immune system is able to discriminate between self and non-self. The adaptive immune system works mainly through B lymphocytes and T lymphocytes and is not a subject of the present review. However, it should be mentioned that a role of adaptive immunity in remodelling is recently indicated. Patients with HF due to ischaemic heart disease or dilated cardiomyopathy have a reduced level of circulating regulatory T cells (Tregs), a cell type that suppresses the immune response [25, 26]. Tregs from HF patients had a reduced functional capacity to suppress cytokine production from CD4 + CD25- T cells [25, 26]. Transfer of Tregs to rats with hypertension-induced hypertrophy leads to reverse remodelling and improved electric remodelling [27].
Evidence supporting a role of innate immunity in myocardial remodelling
Cytokines
Cytokines are signalling molecules sent out by many different cells to attract inflammatory cells to an area under attack or needing repair. More than 200 cytokines have been cloned; they are classified into the main families of IL-2/IL-2 family, IL-6/IL-12 family, interferon-α/β family, tumour necrosis factor family, IL-10 family, IL-17 family, interleukin-1 family, TGF-β family, and chemokine family [28, 29]. Cytokines have a plethora of direct biological effects, as well as indirect effects signalling through activation of transcription factors. Patients with HF have increased circulating levels of both pro- and antiinflammatory cytokines and their receptors, as recently reviewed elsewhere [3, 12]. The remodelling heart itself has increased levels of cytokines [3, 12]. However, we do currently not know whether the increased cytokine signalling network in HF patients starts in the heart or in the periphery [12]. Furthermore, although cytokines have biological effects on cardiac contractility, extracellular matrix deposition, and apoptosis induction, their increase in remodelling could signify both beneficial attempts of tissue repair and a drive towards tissue injury. Cytokines such as TNFα, IL-1β, and TWEAK induce activation of nuclear factor kappa B [3, 12, 30]. Cytokines and their role for myocardial remodelling are reviewed more extensively elsewhere in this issue.
Nuclear factor kappa B
The transcription factor NFκB was first discovered as a B cell-specific DNA-binding protein that binds the kappa light chain enhancer region. It is implicated in the regulation of many disease states and biological phenomenon, where pathophysiologists usually regard its actions with suspicion and has researched it as the major culprit underlying injury. In research environments engaged in mechanisms of cell death, NFκB is regarded as a survival factor [7, 31]. For reviews on NFκB and the heart, see references [32–35]. The NFκB family consists of the members p50–p105, p52–p100, p65/RelA, c-Rel, and RelB. Different members can form various homo- and heterodimers, and the composition of the dimer will determine its action. In resting cells, NFκB is found in an inactive form in the cytoplasm. It is bound to a group of inhibitory proteins called IκB. IκB may be phosphorylated by different upstream kinase cascades called IKK1/IKKα and IKK2/IKKβ, after which IκB is ubiquinated and releases NFκB (Fig. 1). Ubiquitinated IκB is degraded in the proteosome [9]. When released, NFκB translocates to the nucleus where it can activate gene programs. Although more than 400 genes may be regulated by NFκB, the most relevant groups of genes in this context are cytokines, leucocyte adhesion molecules, interleukins, matrix metalloproteinases, and various genes regulating apoptosis. It should also be noted that NFκB also regulates genes associated with the resolution of inflammation, such as manganese superoxide dismutase, inducible nitric oxide synthase, and inducible cyclooxygenase [31]. Due to the diversity of the NFκB dimer and its regulatory proteins as well as the diversity in the nature of the stimuli leading to remodelling, the activities and amounts of transcribed product after NFκB activation will have many possible outcomes [31–35].
Nuclear factor kappa B and myocardial remodelling
NFκB is activated in the heart in many conditions, where the outcome for heart function and cell viability is not clarified and probably not uniform, summarized in the cartoon of Fig. 2. NFκB is activated in the heart during acute ischaemia and reperfusion [36–38]. NFκB is also activated in the heart [39] and in circulating white blood cells [40] during unstable angina. Blocking of NFκB reduces ischaemia–reperfusion injury in experimental studies with short times of observation [41–43]. NFκB is activated in the heart by preconditioning, where it is suggested to play a beneficial role for adaptation to ischaemia [34, 35, 39]. Wong et al. [44] showed for the first time that NFκB was activated in cardiomyocytes in failing hearts of patients with ischaemic heart disease, while similarly an increased NFκB expression was present in the fibrotic areas in patients with dilated cardiomyopathy. Later studies have confirmed that NFκB and its target genes are activated in cardiomyocytes of patients with failing hearts of various etiologies [45, 46] and that the same is the case in mouse and rat models of heart failure [22]. Interestingly, NFκB is also activated in peripheral white blood cells in patients with failure, except in cachectic patients [47, 48]. Furthermore, fatigued skeletal muscle in heart failure patients has increased NFκB activation [49]. Thus, although it is sure that NFκB is activated in human and animal myocardium during remodelling, the relationship between heart and periphery is far from clarified.

A summary of the main discussion in the review; Innate immunity evident as cytokine release, Toll-like receptor activation, or nuclear factor kappa activation may lead to beneficial myocardial adaptation to ischaemia. Cardiac innate immunity increases acute ischaemic injury. However, its role in long-term chronic models of remodelling and hypertrophy is not clarified
Quite a few researchers have tried to elucidate what NFκB does in the heart in the remodelling process, but with conflicting results. Mice with targeted deletion of the p50 NFκB subunit (but with normal p65 expression) subjected to myocardial infarction had an increased remodelling in the form of larger end-diastolic volume and reduced ejection fraction compared to wild type [50]. The p50 KO had an increased inflammatory response in the form of higher macrophage infiltration, higher expression of the chemoattractant MCP-1, higher TNFalpha, and IL-6 expression in the infarcted myocardium, and higher matrix metalloproteinase activity [50]. These data, indicating a beneficial effect of the p50 subunit during cardiac remodelling, fit the concept that the p50 homodimer is actually a repressor of transcription [31]. However, other groups have reported different findings in the p50 KO. Frantz et al. [51] found that postinfarct, p50 KO hearts had preserved ventricular dimensions, less matrix metalloproteinase expression, reduced collagen deposition, but higher TNFalpha expression. These findings were confirmed by Kawano et al. [52], who found reduced ventricular rupture, improved systolic and diastolic dimensions and function, reduced collagen deposition, and reduced cytokine expression postinfarction in p50 KO mice. In a model of TNF-induced cardiomyopathy through cardiac-specific TNFalpha expression, p50 KO mice had improved diastolic and systolic function, suppressed matrix metalloproteinase activity and expression, and improved survival in male mice [53].
Other approaches have been performed to suppress NFκB activation in vivo. Gupta et al. [54] used a model of hypertrophy through cardiospecific overexpression of myotrophin and delivered short hairpin RNA silencing p65 with a lentiviral vector into hearts. This resulted in a regression of hypertrophy (heart weight/body weight ratio) and reduced activation of cardiac NFκB and its target genes. The same group tested the effect of chronically inhibiting NFκB through cross-breeding the cardiac specific myotrophin mouse with a dominant-negative IκBα mouse [55]. Although the authors have only preliminary functional data in that study, it is indicated that dominant-negative IκBα inhibited the activation of NFκB and its target genes, concomitant with reducing hypertrophy and improving left ventricular systolic function [55]. In a rat model of postinfarction heart failure, chronic administration of an IκB kinase inhibitor, leading to downstream loss of NFkB activation, marginally improved left ventricular function and reduced matrix metalloproteinase activity 28 days later [56]. In contrast, Hikoso et al. [57] made a cardiospecific knock down of IKKβ, a kinase that phosphorylates the inhibitory IκBα, allowing nuclear translocation of the NFκB dimer and activation of gene programs. Mice with knock down of IKKβ had no NFκB activation after hypertrophy evoked by transaortic banding but had increased diastolic diameter, reduced fractional shortening, increased expression of atrial and brain natriuretic peptides in the left ventricle, and larger cross-sectional area of cardiomyocytes than banded wild type [57]. In agreement with that, Kratsios et al. [58] inhibited NFκB activation by cardiospecific deletion of NEMO, a regulatory subunit of the IKKγ complex. NEMO-deleted mice developed spontaneous hypertrophy progressing with age. When NEMO knockouts were subjected to transaortic banding, the maladaptive left ventricular remodelling escalated compared to wild types or NEMO KO without pressure overload [58]. In both of these papers, antioxidant status evident as lack of manganese superoxide dismutase or nitric oxide synthase appears crucial to the negative effects evoked by inhibition of NFκB activation [59, 60].
Thus, there is no doubt that NFκB is activated in hearts of patients, rats, and mice during remodelling after various forms of injury. However, the impact of this activation is not clear at the time being and warrants further research with focus on the composition of the NFκB dimer and downstream biological effects.
Toll-like receptors
In 1997, vertebrate homologues of the Drosophila spp. transmembrane PRR ‘Toll’ were identified and termed the Toll-like receptors [14–17]. To date, 11 human and 13 mouse TLR have been cloned [17, 22]. TLR recognize pathogens and possibly endogenously produced ligands as well and activate the immune system through a signalling pathway down to NFκB or interferon regulatory factor 3.
The ligands for TLR are molecular motifs associated with pathogens. TLR types 1, 2, 4, and 6 mainly recognize lipopeptides that are components of bacterial cell walls. TLR2 recognizes components of both gram-positive and gram-negative bacteria, in addition to components from fungi, parasites, and viruses. TLR4 recognizes cell wall components of gram-negative bacteria, LPS. TLR types 3, 7, 8, and 9 are named antiviral TLRs and recognize viral components or single-/double-stranded RNA/DNA. TLR5 binds protein ligands, such as flagellin from fungi [14–17]. Also endogenous activators of TLRs have been suggested (DAMP). Among these are heat shock proteins 60, 70, and 96, fibrinogen, heparan sulphate, and hyaluronan fragments [20, 22].
When activated, the TLR signalling network controls the initiation, maintenance, modulation, and termination of innate host defences by several mechanisms [14–17]. TLR activation leads to the production of cytokines and antimicrobial molecules, which in turn activate cellular immune components such as macrophages. Activated TLR on dendritic cells induce cell maturation, and the activated dendritic cells stimulate T-cell expansion and differentiation. TLR also induce the expression of costimulatory molecules necessary for sustained activation of adaptive immunity. TLR2 and TLR4 are the most well studied in the context of the heart.
The structure of TLRs and downstream signalling pathways have recently been reviewed elsewhere [17, 22, 24]. Briefly, signalling most frequently happens through the adaptor molecule MyD88 (myeloid differentiation primary response protein 88) [14–17]. Other adaptor molecules include TIRAP (TIR domain-containing adaptor protein), TRIF (TIR domain-containing adaptor inducing interferon β), and TRAM (TRIF-related adaptor molecule). MyD88-dependent signalling via TLR2 and TLR4 requires the presence of TIRAP and will end after several steps in activation of NFκB and subsequent gene transcription (Fig. 1) [14–17]. MyD88-independent signalling involves the adaptor proteins TRIF or TRAM. TLR4 can activate TRIF-dependent pathways without MyD88 association, leading to activation of the transcription factor IRF3, in turn producing interferon and costimulatory molecules.
Toll-like receptors and myocardial remodelling
TLR types 2, 3, 4, and 6 are expressed in cardiac myocytes [20, 22]. TLR are emerging as important regulators of pathophysiological processes in the heart but have been more studied in acute pathologies than in studies relevant to remodelling. For instance, in patients with acute myocardial infarction, TL4 signalling is activated, and this appears to lead to increased cytokine release [59]. TLR4 is increasingly expressed in peripheral monocytes from patients with acute myocardial infarction, a finding that was accompanied by increased cytokine release [59]. The TLR4 activation was highest in patients with acute heart failure [59]. Frantz et al. [60] demonstrated that injured human and murine myocardium exhibit focal areas of intense TLR4 expression. Other findings suggesting a role of TLR4 in myocardial ischaemia–reperfusion injury are the observations that the TLR4 ligand LPS is a possible preconditioning stimulus. Several researchers suggest that systemic administration of sublethal doses of LPS or monophosphoryl lipid A (a partial structure of the lipid A moiety of gram-negative LPS) protects the myocardium against subsequent ischaemia and reperfusion injury [61–63]. In greater doses, LPS leads to myocardial depression, and this effect is dependent of TLR4-mediated signalling [64, 65]. TLR2 has anti-apoptotic effect in stressed or injured cardiac myocytes, as inhibition of TLR2 enhances stress-induced cytotoxicity [66]. TLR2-deficient mice are also protected against postischaemic coronary endothelial dysfunction [67]. When TLR2 knockout mice were injected with doxorubin to induce remodelling, they had better left ventricular function, blunted NFkB activation, and less apoptosis than hearts of wild types 5 days after treatment [68]. Experiments carried out in TLR4-deficient mice have shown that these mice develop smaller infarcts after myocardial infarction than wild-type mice in short times of observation [69, 70].
However, when it comes to studies of in vivo myocardial remodelling with long-term follow-up, the role of TLRs for remodelling is more scarce. Ha et al. [71] subjected TLR4 knockout mice to aortic banding for 2 weeks and found that TLR4-deleted mice had less remodelling of the left ventricle. Timmers et al. [72] induced myocardial infarction in TLR4 knockout mice and found that as opposed to sham-operated mice or wild types of a different strain, TLR4 knockouts had preserved left ventricular function postinfarct 28 days later. Interstitial fibrosis and hypertrophy were reduced in the non-infarcted heart tissue of knockouts [72]. In the infarcted myocardium, the inflammatory response was reduced in the knockouts [72]. In contrast, in a model of aortic constriction in rats, activation of TLR4 signalling reduced the hypertrophy response, an effect that was reversed through administrating a TLR4 antagonist [73]. Cardiac fibrosis was reduced through inducing TLR4 signalling [73]. Shishido et al. [74] used TLR2-deficient mice with coronary artery ligation and followed them for 4 weeks. The knockouts had a higher survival after ligation than wild types, less remodelling as evaluated by echocardiography, less myocardial fibrosis, but no differences in infarct size or accumulation of polymorphonuclear granulocytes in the heart.
Thus, in order to determine more firmly whether TLR2 or 4 (or other TLR) are important for myocardial remodelling, more evidence is needed from knockout mice using different forms of cardiac injury, and longer times of observations of ventricular remodelling.
Future perspectives
A large body of evidence suggests that innate immunity is involved in the signalling underlying cardiac pathologies through cytokines such as TNFα and IL-6. Involvement of innate immunity is indicated through NFкB and TLR in models of acute myocardial injury. However, the importance of both NFkB and TLR in the chronic context of myocardial remodelling is far from determined. Use of knockout mice of different genetic backgrounds, careful experimental design with follow-up for at least 6 weeks after the index event and use of different models to evoke remodelling may give us a fuller understanding of these complex signalling cascades in the future. We still have the possibility that activation of innate immunity in acute and chronic cardiac injury is just an attempt to repair damaged tissue and is a beneficial process counteracting the viscious circle of heart failure development. The role of innate immunity in the heart versus the periphery during cardiac remodelling needs more attention.
References
Chung M-W, Tsoutsman T, Semsarian C (2003) Hypertrophic cardiomyopathy: from gene to defect to clinical disease. Cell Res 13:9–20
Francis GS, Tang WH (2003) Pathophysiology of congestive heart failure. Rev Cardiovasc Med 4:S14–S20
Yndestad A, Damås JK, Øie E, Ueland T, Gullestad L, Aukrust P (2007) Role of inflammation in the progression of heart failure. Curr Cardiol Rep 9:236–241
Sani MU (2008) Myocardial disease in human immunodeficiency virus (HIV) infection: a review. Wien Klin Wochenschr 120:77–87
Sandek A, Rauchhaus M, Anker SD, von Haehling S (2008) The emerging role of the gut in chronic heart failure. Curr Opin Clin Nutr Metab Care 11:632–639
Dhawan SS, Quyyumi AA (2008) Rheumatoid arthritis and cardiovascular disease. Curr Atheroscl Rep 10:128–133
Whelan RS, Kaplinskiy V, Kitsis RN (2010) Cell death in the pathogenesis of heart disease: mechanisms and significance. Annu Rev Physiol 72:19–44
Zelarayan L, Renger A, Noack C, Zafiriou MP, Gehrke C, van der Nagel R, Dietz R, de Windt L, Bergmann MW (2009) NFκB activation is required for adaptive cardiac hypertrophy. Cardiovasc Res 84:416–424
Hedhi N, Depre C (2010) Proteasome inhibitors and cardiac cell growth. Cardiovasc Res 85:321–329
Conraads VM, Vrints CJ, Rodrigus IE, Hoymans VY, Van Craenenbroeck EM, Bosmans J, Claeys MJ, Van Herch P, Linke A, Schuler G, Adams V (2010) Depressed expression of MuRF1 and MAFbx in areas of recent myocardial infarction: a mechanism contributing to myocardial remodelling? Basis Res Cardiol 105:219–226
Gustafsson ÅB, Gottlieb RA (2009) Autophagy in ischemic heart disease. Circ Res 104:150–158
Fildes JE, Shaw SM, Yonan N, Williams SG (2009) The immune system and chronic heart failure. Is the heart in control? J Am Coll Cardiol 53:1013–1020
Gottlieb RA, Carreira RS (2010) Autophagy in health and disease: V. mitophagy as a way of life. Am J Physiol (epub ahead of print)
Akira S, Takeda K (2004) Toll-like receptor signaling. Nat Rev Immunol 4:499–511
Palm NW, Medzhitov R (2009) Pattern recognition receptors and control of adaptive immunity. Immunol Rev 227:221–233
Yan ZQ, Hansson GK (2007) Innate immunity, macrophage activation, and atherosclerosis. Immunol Rev 219:187–203
Takeuchi O, Akira S (2010) Pattern recognition receptors and inflammation. Cell 140:805–820
Matzinger P (2002) The danger model: a renewed sense of self. Science 296:301–305
Matzinger P (2007) Friendly and dangerous signals: is the tissue in control? Nat Immunol 8:11–13
Erickson B, Sperber K, Frishman WH (2008) Toll-like receptors. New therapy strategies for the treatment of atherosclerosis, acute coronary syndroms, and myocardial failure. Cardiol Rev 16:237–279
Shi Y, Evans JE, Rode KL (2003) Molecular identification of a danger signal that alerts the immune system to dying cells. Nature 425:516–521
Frantz S, Ertl G, Bauersachs J (2007) Mechanisms of disease: toll-like receptors in cardiovascular disease. Nat Clin Pract Cardiovasc Med 4:444–454
Hayden MS, West AP, Ghosh S (2006) NF-kappaB and the immune response. Oncogene 25:6758–6780
Valeur HS, Valen G (2009) Innate immunity and myocardial adaptation to ischemia. Basic Res Cardiol 104:22–32
Tang H, Zhong Y, Zhu Y, Zhao F, Cui X, Wang Z (2010) Low responder T cell susceptibility to the suppressive function of regulatory T cells in patients with dilated cardiomyopathy. Heart 96:765–772
Tang TT, Ding YJ, Liao YH, Yu X, Xiao H, Xie JJ, Yuan J, Zhou ZH, Liao MY, Yao R, Cheng Y, Cheng X (2010) Defective circulating CD4+CD25+Foxp3+CD127low regulatory T cells in patients with chronic heart failure. Cellular Physiol Biochem 25:451–458
Kvakan H, Kleinewietfeld M, Qadri F, Park JK, Fischer R, Schwarz I, Rahn HP, Plehm R, Wellner M, Elitok S, Gratze P, Dechend R, Luft FC, Muller DN (2009) Regulatory T cells ameliorate angiotensin II induced cardiac damage. Circulation 119:2904–2912
Girn HRS, Ahilathirunayagam S, Mavor AID, Homer-Vanniasinkam S (2007) Reperfusion syndrome: cellular mechanisms of microvascular dysfunction and potential therapeutic strategies. Vasc Endovasc Surg 41:277–293
Kurdi M, Booz GW (2007) Can the protective actions of JAK-STAT in the heart be exploited therapeutically? Parsing the regulation of interleukin-6-type cytokine signalling. J Cardiovasc Pharmacol 50:126–141
Chorianopoulos E, Heger T, Lutz M, Frank D, Bea F, Katus HA, Frey N (2010) FGF-inducible 14-kDA protein (Fn14) is regulated via the RhoA/ROCK kinase pathway in cardiomyocytes and mediates nuclear factor kappa B activation by TWEAK. Basic Res Cardiol 105:301–313
Lawrence T, Fong C (2010) The resolution of inflammation: anti-inflammatory roles for NF-kB. Int J Biochem Cell Biol 42:519–523
Hall G, Hasday JD, Rogers TB (2006) Regulating the regulator: NF-kappaB signaling in the heart. J Mol Cell Cardiol 41:580–591
Jones WK, Brown M, Wilhide M, He S, Ren X (2005) NF-kappaB in cardiovascular disease: diverse and specific effects of a “general” transcription factor? Cardiovasc Toxicol 5:183–202
Valen G, Yan ZQ, Hansson GK (2001) Nuclear factor kappa-B and the heart. J Am Coll Cardiol 38:307–314
Valen G (2004) Signal transduction through nuclear factor kappa B in ischemia-reperfusion and heart failure. Basic Res Cardiol 99:1–7
Chandrasekar B, Freeman GL (1997) Induction of nuclear factor kappaB and activation protein 1 in postischemic myocardium. FEBS Lett 401:30–34
Li C, Browder W, Kao RL (1999) Early activation of transcription factor NF-kappaB during ischemia in perfused rat heart. Am J Physiol 276:H543–H552
Li C, Kao RL, Ha T, Kelley J, Browder IW, Williams DL (2001) Early activation of IKKbeta during in vivo myocardial ischemia. Am J Physiol 280:H1264–H1271
Valen G, Hansson GK, Dumitrescu A, Vaage J (2000) Unstable angina activates myocardial heat shock protein 72, endothelial nitric oxide synthase, and transcription factors NFkB and AP-1. Cardiovasc Res 47:49–56
Ritchie ME (1998) Nuclear factor-κB is selectively and markedly activated in humans with unstable angina pectoris. Circulation 98:1707–1713
Morishita R, Sugimoto T, Aoki M, Kida I, Tomita N, Moriguchi A, Maeda K, Sawa Y, Kaneda Y, Higaki J, Ogihara T (1997) In vivo transfection of cis element “decoy” against nuclear factor-kB binding site prevents myocardial infarction. Nat Med 3(8):894–899
Sawa Y, Morishita R, Suzuki K, Kagisaki K, Kaneda Y, Maeda K, Kadoba K, Matsuda H (1997) A novel strategy for myocardial protection using in vivo transfection of cis element “decoy” against NFkB binding site. Circulation 96(suppl. II):II-280–II-285
Morgan EN, Boyle EM, Yun W, Griscavage-Ennis JM, Farr A, Canty TG, Pohlman TH, Verrier ED (1999) An essential role for NFkB in the cardioadaptive response to ischemia. Ann Thorac Surg 68:377–382
Wong SCY, Fukuchi M, Melnyk P, Rodger I, Giaid A (1998) Induction of cyclooxygenase-2 and activation of nuclear factor κB in myocardium of patients with congestive heart failure. Circulation 98:100–103
Frantz S, Fracarollo D, Wagner H, Behr TM, Jung P, Angermann CE, Ertl G, Bausersachs J (2003) Sustained activation of nuclear factor kappa B and activator protein 1 in chronic heart failure. Cardiovasc Res 57:749–756
Gupta S, Sen S (2005) Role of the NFκB signalling cascade and NFκB targeted genes in failing human hearts. J Mol Med 83:993–1004
Frantz S, Stoerk S, Wagner H, Angermann CE, Ertl G, Bauersachs J (2004) Effect of chronic heart failure on nuclear factor kappa B in peripheral leukocytes. Am J Cardiol 94:671–673
Siednienko J, Jankowska EA, Banasiak W, Gorczyca WA, Ponikowski P (2007) Nuclear factor kappa B activity in peripheral blood mononuclear cells in cachectic and non-cachectic patients with chronic heart failure. Int J Cardiol 122:111–116
Adams V, Säte U, Kränkel N, Schulze PC, Linke A, Schuler G, Hambrecht R (2003) Nuclear factor kappa B activation in skeletal muscle of patients with chronic heart failure: correlation with the expression of inducible nitric oxide synthase. Eur J Cardiovasc Prev Rehabil 10:273–277
Timmers L, van Keulen K, Hoefer IE, Meijs MFL, van Middelaar B, den Ouden K, van Echteld CJA, Pasterkamp G, de Kleijn DPV (2009) Targeted deletion of nuclear factor κB p50 enhances cardiac remodelling and dysfunction following myocardial infarction. Circ Res 104:699–706
Frantz S, Hu K, Bayer B, Gerondakis S, Strotmann J, Adamek A, Ertl G, Bauersachs J (2006) Absence of NFκB subunit p50 improves heart failure after myocardial infarction. FASEB J 20:1918–1920
Kawano S, Kubota T, Monden Y, Tsutsumi T, Inoue T, Kawamura N, Tsutsui H, Sunagawa K (2006) Blockade of NFκB improves cardiac function and survival after myocardial infarction. Am J Physiol 291:H1337–H1344
Kawamura N, Kubota T, Kawano S, Monden Y, Feldman AM, Tsutsui H, Takeshita A, Sunagawa K (2005) Blockade of NFκB improves cardiac function and survival without affecting inflammation in TNFα-induced cardiomyopathy. Cardiovasc Res 66:520–529
Gupta S, Young D, Maitra RK, Gupta A, Popovic ZB, Yong SL, Mahajan A, Wang Q, Sen S (2008) Prevention of cardiac hypertrophy and heart failure by silencing of NFκB. J Mol Biol 375:637–649
Young D, Popovic ZB, Jones WK, Gupta S (2008) Blockade of NFκB using IκBα dominant negative mice ameliorates cardiac hypertrophy in myotrophin overexpressed transgenic mice. J Mol Biol 381:559–568
Wakatsuki S, Suzuki J-I, Ogawa M, Masumura M, Muto S, Shimizu T, Takayama K, Itai A, Isobe M (2008) A novel IKK inhibitor suppresses heart failure and chronic remodelling after myocardial ischemia via MMP alteration. Expert Opin Ther Targets 12:1469–1476
Hikoso S, Yamaguchi O, Nakano Y, Takeda T, Omiya S, Mizote I, Taneike M, Oka T, Tamai T, Oyabu J, Uno Y, Matsumura Y, Nishida K, Suzuki K, Kogo M, Hori M, Otsu K (2009) The IκB kinase β/nuclear factor κB signaling pathway protects the heart from hemodynamic stress mediated by the regulation of manganese superoxide dismutase expression. Circ Res 105:70–79
Kratsios P, Huth M, Temmerman L, Salimova E, Al Banchaabouchi M, Sgoifo A, Manghi M, Suzuki K, Rosenthal N, Mourkioti F (2010) Antioxidant amelioration of dilated cardiomyopathy caused by conditional deletion of NEMO/IKKγ in cardiomyocytes. Circ Res 106:133–144
Satoh M, Shimoda Y, Maesawa C, Akatsu T, Ishikawa Y, Minami Y, Hiramori K, Nakamura M (2006) Activated toll-like receptor 4 in monocytes is associated with heart failure after acute myocardial infarction. Int J Cardiol 109:226–234
Frantz S, Kelly RA, Bourcier T (2001) Role of TLR-2 in the activation of nuclear factor kappaB by oxidative stress in cardiac myocytes. J Biol Chem 276:5197–5203
Frantz S, Kobzik L, Kim YD, Fukazawa R, Medzhitov R, Lee RT, Kelly RA (1999) Toll4 (TLR4) expression in cardiac myocytes in normal and failing myocardium. J Clin Invest 104:271–280
Brown JM, Grosso MA, Terada LS, Whitman GJ, Banerjee A, White CW, Harken AH, Repine JE (1989) Endotoxin pretreatment increases endogenous myocardial catalase activity and decreases ischemia-reperfusion injury of isolated rat hearts. Proc Natl Acad Sci 86:2516–2520
Chong AJ, Shimamoto A, Hampton CR, Takayama H, Spring DJ, Rothnie CL, Yada M, Pohlman TH, Verrier ED (2004) Toll-like receptor 4 mediates ischemia/reperfusion injury of the heart. J Thorac Cardiovasc Surg 128:170–179
Song W, Furman BL, Parratt JR (1998) Monophosphoryl lipid A reduces both arrhythmia severity and infarct size in a rat model of ischaemia. Eur J Pharmacol 345:285–287
Belosjorow S, Schulz R, Dörge H, Schade FU, Heusch G (1999) Endotoxin and ischemic preconditioning: TNF-α concentration and myocardial infarct size development in rabbits. Am J Physiol 277:H2470–H2475
Nemeto S, Vallejo JG, Knuefermann P, Misra A, Defreitas G, Carabello BA, Mann DL (2002) Escherichia coli LPS-induced LV dysfunction: role of toll-like receptor 4 in the adult heart. Am J Physiol 282:H2323–H2361
Favre J, Musette P, Douin-Echinard V, Laude K, Henry JP, Arnal JF, Thuillez C, Richard V (2007) Toll-like receptors 2-deficient mice are protected against postischemic coronary endothelial dysfunction. Arterioscler Thromb Vasc Biol 27:1064–1071
Nozaki N, Shishido T, Takeishi Y, Kubota I (2004) Modulation of doxorubin-induced cardiac dysfunction in toll-like receptor-2-knockout mice. Circulation 110:2869–2874
Oyama J, Blais C, Liu X, Pu M, Kobzik L, Kelley RA, Bourcier T (2004) Reduced myocardial ischemia-reperfusion injury in Toll-like receptor 4-deficient mice. Circulation 109:784–789
Kim SC, Ghanem A, Stapel H (2007) Toll-like receptor 4 deficiency: smaller infarcts, but no gain in function. BMC Physiol 7:5–9
Ha T, Liu F, Ma J, Gao X, Kelley J, Zhao A, Haddad GE, Williams DL, William Browder I, Kao RL, Li C (2005) Reduced cardiac hypertrophy in toll-like receptor 4-deficient mice following pressure overload. Cardiovasc Res 68:224–234
Timmers L, Sluijter JP, van Keulen JK, Hoefer IE, Nederhof MG, Goumans MJ, Doevendans PA, van Echteld CJ, Joles JA, Quax PH, Piek JJ, Pasterkamp G, de Kleijn DP (2008) Toll-like receptor 4 mediates maladaptive left ventricular remodeling and impairs cardiac function after myocardial infarction. Circ Res 102:257–264
Liu YY, Cai WF, Yang HZ, Ciu B, Chen ZR, Liu HZ, Yan J, Jin W, Yan HM, Xin BM, Yuan B, Hua F, Hu ZW (2008) Bacillus Calmette-Guerin and TLR4 agonist prevent cardiovascular hypertrophy and fibrosis by regulating immune microenvironment. J Immunol 180:7349–7357
Shishido T, Nozaki N, Yamaguchi S, Shibata Y, Nitobe J, Miyamoto T, Takahashi H, Arimoto T, Maeda K, Yamakawa M, Takeuchi O, Akira S, Takeishi Y, Kubota I (2003) Toll-like receptor-2 modulates ventricular remodeling after myocardial infarction. Circulation 108:2905–2910
Acknowledgments
Financial support from The Norwegian Health Association, the Nansen Foundation, Odd Fellow, and the University of Oslo are gratefully acknowledged.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Valen, G. Innate immunity and remodelling. Heart Fail Rev 16, 71–78 (2011). https://doi.org/10.1007/s10741-010-9187-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-010-9187-1