
Abstract
Target-mediated drug disposition (TMDD) is frequently reported for therapeutic monoclonal antibodies and is linked to the high affinity and high specificity of antibody molecules for their target. Understanding TMDD of a monoclonal antibody should go beyond the empirical description of its non-linear PK since valuable insights on the antibody-target interaction itself can be gained. This makes its mechanistic understanding precious for the drug development process, in particular for the optimization of new antibody molecules, for the design and interpretation of pharmacokinetic studies, and possibly even for the evaluation of efficacy and dose selection of drug candidates. Using the observation that the molecular (microscopic) processes are usually much more rapid than the pharmacokinetic (macroscopic) processes, a series of quasi-steady-state conditions on the microscopic level is proposed to bridge the gap between simple empirical and complex mechanistic descriptions of TMDD. These considerations show the impact of parameters such as target turnover, target expression, and target accessibility on the pharmacokinetics and pharmacodynamics of monoclonal antibodies.



Similar content being viewed by others
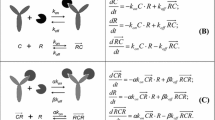

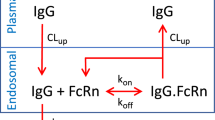
References
Mould D, Sweeney K (2007) The pharmacokinetics and pharmacodynamics of monoclonal antibodies mechanistic modeling applied to drug development. Curr Opin Drug Discov Devel 10(1):84–96
Mager DE, Jusko WJ (2001) General pharmacokinetic model for drugs exhibiting target-mediated drug disposition. J Pharmacokinet Pharmacodyn 28(6):507–532
Meno-Tetang GML, Lowe PJ (2005) On the prediction of the human response: a recycled mechanistic pharmacokinetic/pharmacodynamic approach. Basic Clin Pharmacol Toxicol 96(3):182–192
Ng CM et al (2005) Pharmacokinetic-pharmacodynamic-efficacy analysis of efalizumab in patients with moderate to severe psoriasis. Pharm Res 22(7):1088–1100
Ng CM et al (2006) Pharmacokinetics/pharmacodynamics of nondepleting anti-CD4 monoclonal antibody (TRX1) in healthy human volunteers. Pharm Res 23(1):95–103
Kovarik JM et al (2001) A population pharmacokinetic screen to identify demographic-clinical covariates of basiliximab in liver transplantation. Clin Pharmacol Ther 69(4):201–209
Lu J-F et al (2008) Clinical pharmacokinetics of bevacizumab in patients with solid tumors. Cancer Chemother Pharmacol 62(5):779–786
Mould DR et al (2007) Population pharmacokinetics-pharmacodynamics of alemtuzumab (Campath®) in patients with chronic lymphocytic leukaemia and its link to treatment response. Br J Clin Pharmacol 64(3):278–291. doi:10.1111/j.1365-2125.2007.02914.x
Ng CM et al (2005) Population pharmacokinetics of rituximab (anti-CD20 monoclonal antibody) in rheumatoid arthritis patients during a phase II clinical trial. J Clin Pharmacol 45(7):792–801. doi:10.1177/0091270005277075
Ternant D et al (2008) Infliximab pharmacokinetics in inflammatory bowel disease patients. Ther Drug Monit 30(4):523–529
Xu Z et al (2008) Population pharmacokinetics of infliximab in patients with ankylosing spondylitis. J Clin Pharmacol 48(6):681–695. doi:10.1177/0091270008316886
Nolting A, Fox FE, Kovar A (2006) Clinical drug development of cetuximab, a monoclonal antibody. In: Meibohm B (ed) Pharmacokinetics and pharmacodynamics of biotech drugs: principles and case studies in drug development. WILEY–VCH, Weinheim, pp 353–370
FDA (2006) Clinical pharmacology biopharmaceutics review(s): vectibix panitumumab injectable. FDA, Center for Drug Evaluation and Research, Rockville
Mager DE (2006) Target-mediated drug disposition and dynamics. Biochem Pharmacol 72(1):1–10
Mager D, Krzyzanski W (2005) Quasi-equilibrium pharmacokinetic model for drugs exhibiting target-mediated drug disposition. Pharm Res 22(10):1589–1596
Gibiansky L et al (2008) Approximations of the target-mediated drug disposition model and identifiability of model parameters. J Pharmacokinet Pharmacodyn 35:573–591
Holmes MH (1995) Introduction to perturbation methods. In: Marsden JE, et al (eds) Texts in applied mathematics, vol 20, 1 edn. Springer-Verlag, New York p 337
Marathe A, Krzyzanski W, Mager DE (2009) Numerical validation and properties of a rapid binding approximation of a target-mediated drug disposition pharmacokinetic model. J Pharmacokinet Pharmacodyn 36:199–219
Orditura M et al (2009) Correlation between efficacy and skin rash occurrence following treatment with the epidermal growth factor receptor inhibitor cetuximab: a single institution retrospective analysis. Oncol Rep 21(4):1023–1028
Acknowledgements
The author would like to thank Dr. Wolfgang Richter and Dr. Thierry Lavé for their valuable comments and suggestions.
Author information
Authors and Affiliations
Corresponding author
Appendix
Appendix
Derivation of CLTM(C) for the recycling case
Using \( K_{M} = {\tfrac{{k_{off} + k_{eli} }}{{k_{on} }}} = K_{d} \cdot k_{eli} \cdot \left( {{\tfrac{1}{{k_{off} }}} + {\tfrac{1}{{k_{eli} }}}} \right) \), the equations for binding equilibrium 1 and for target conservation are written as
which, after elimination of R become
At equilibrium, target-mediated clearance corresponds to the elimination of drug-target complexes. It is thus defined by
and solving 19 for RC thus yields
Furthermore, target-mediated clearance is maximal as C → 0 and thus
From this Eq. 2 follows directly.
Derivation of CLTM(C) for the target turnover case
The relevant equations are now 1 and 4, which can be written as
where 1 was used to simplify 4, \( \kappa = {{k_{eli} } \mathord{\left/ {\vphantom {{k_{eli} } {k_{de} }}} \right. \kern-\nulldelimiterspace} {k_{de} }} \), and K M being defined as previously. Eliminating R and sorting terms, this becomes:
Then, introducing a new \( \tilde{K}_{M} = {{K_{M} } \mathord{\left/ {\vphantom {{K_{M} } \kappa }} \right. \kern-\nulldelimiterspace} \kappa } = K_{d} \cdot k_{de} \cdot \left( {{\tfrac{1}{{k_{off} }}} + {\tfrac{1}{{k_{eli} }}}} \right) \) and using the definition for target-mediated clearance 20 again,
As before, target-mediated clearance is maximal as C → 0:
from which 5 ensues easily.
Derivation of CLTM(C) for the permeability limitation case
Eqs. 1, 4, and 7 can be rewritten as
with a new parameter combination \( \eta = k_{eli} /k_{per} \). Using the latter two equations to substitute C tar and R in the first equation leads to:
Similar to the previous cases, this can be solved for RC in order to calculate the target-mediated clearance directly. Alternatively, introduce
and hence, from the definition of target-mediated clearance
Substitution into 31 and after simplifying R E and C pla :
Introducing again \( \tilde{K}_{M} = {{K_{M} } \mathord{\left/ {\vphantom {{K_{M} } \kappa }} \right. \kern-\nulldelimiterspace} \kappa } = K_{d} \cdot k_{de} \cdot \left( {{\tfrac{1}{{k_{off} }}} + {\tfrac{1}{{k_{eli} }}}} \right) \), and defining \( H = {\tfrac{\eta }{\kappa }} \cdot {\tfrac{{R_{E} }}{{\tilde{K}_{M} }}} = {\tfrac{{k_{de} }}{{k_{per} }}} \cdot {\tfrac{{R_{E} }}{{\tilde{K}_{M} }}} \) this equation is
As previously, target-mediated clearance is maximal as C → 0, i.e.,
from which it follows that
This equation is equivalent to 10. It is then a matter of straight-forward (but somewhat tedious) algebra to bring 35 in the form of 8.
Derivation of saturation of CL versus saturation of target
Substituting RC from equation 29, 33 becomes
Target-mediated clearance being a continuous and monotonous function of plasma concentration, this equation can be used to express C pla as a function of CL TM :
Substituting C pla and RC in Eq. 31 by the use of Eqs. 29 and 39 and by applying relationship 37 yields (after some simplifications)
from which 12 follows easily.
Derivation of efficacious concentration
Substituting RC from Eq. 29 into 31 yields an equation which relates free receptor concentration (the closest parent to efficacy in this model framework) to C pla :
Equation 13 follows then directly, after solving for C pla . If efficacy is defined by a required ‘free fraction’ of receptors, \( {\tfrac{R}{{R_{E} }}} \), C pla can then be identified with the efficacious concentration C eff .
For a given efficacious concentration C eff , the free fraction of off-site receptors is given by solving Eq. 13 for R/R E and taking the limit H → 0, or
On the other hand, C eff is defined based on the free fraction of on-site receptors, i.e., by Eq. 13, and therefore
Multiplying both sides of the equation by R on-site /R E and after some more algebra, 14 is obtained. The equation is easily generalized for the case where R E is not the same for the on-site and the off-site targets.
Rights and permissions
About this article
Cite this article
Grimm, H.P. Gaining insights into the consequences of target-mediated drug disposition of monoclonal antibodies using quasi-steady-state approximations. J Pharmacokinet Pharmacodyn 36, 407–420 (2009). https://doi.org/10.1007/s10928-009-9129-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10928-009-9129-5