
Abstract
The objectives of the following investigation were (1) development of a physiologically based pharmacokinetic (PBPK) model capable of characterizing the plasma and tissue pharmacokinetics (PK) of nonspecific or antigen specific monoclonal antibodies (mAbs) in wild type, FcRn knockout, tumor bearing and non tumor bearing mice and (2) evaluation of the scale up potential of the model by characterizing the mouse, rat, monkey and human plasma PK of mAbs, simultaneously. A PBPK model containing 15 tissues, a carcass and a tumor compartment was developed by modifying/augmenting previously published PBPK models. Each tissue compartment was subdivided into plasma, blood cell, endothelial, interstitial and cellular sub-compartments. Each tissue was connected through blood and lymph flow to the systemic circulation. Lymph flow was set to a value 500 times lower than plasma flow and vascular reflection coefficients for each tissue were adjusted according to their vascular pore size. In each tissue endothelial space, mAb entered via pinocytosis and the interaction of FcRn with mAb was described by on and off rates. FcRn bound mAb was recycled and unbound mAb was eliminated by a first order process (K deg ). The PBPK model was simultaneously fit to the following datasets to estimate four system parameters: (1) plasma and tissue PK of nonspecific mAb in wild type mouse with or without simultaneous intravenous immunoglobulin (IVIG) administration, (2) plasma and tissue PK of nonspecific mAb in FcRn knockout mouse, (3) plasma and tissue PK of nonspecific mAb in tumor bearing mouse, (4) plasma and tissue PK of tumor antigen specific mAb in tumor bearing mouse, and (5) plasma PK of mAb in rat, monkey and human. The model was able to characterize all the datasets reasonably well with a common set of parameters. The estimated value of the four system parameters i.e. FcRn concentration (FcRn), rate of pinocytosis per unit endosomal space (CL up ), K deg and the proportionality constant (C_LNLF) between the rate at which antibody transfers from the lymph node compartment to the blood compartment and the plasma flow of the given species, were found to be 4.98E−05 M (CV% = 11.1), 3.66E−02 l/h/l (%CV = 3.48), 42.9 1/h (%CV = 15.7) and 9.1 (CV% > 50). Thus, a platform PBPK model has been developed that can not only simultaneously characterize mAb disposition data obtained from various previously published mouse PBPK models but is also capable of characterizing mAb disposition in various preclinical species and human.
Similar content being viewed by others
Introduction
Traditionally plasma pharmacokinetics of a small molecule drug has been used to infer its tissue pharmacokinetics and interpret its pharmacodynamic or toxicodynamic effects [1]. However, unlike small molecules, for many mAbs plasma concentration may not accurately reflect concentration at the site of action [2] (e.g., tumor), necessitating a modeling approach capable of accurately characterizing and predicting not only plasma but tissue concentration as well [3]. Additionally, mAbs often demonstrate complex nonlinear plasma pharmacokinetics [4] (e.g., target mediated drug disposition [5]), which is difficult to scale up using conventional allometric methods [6] and mammillary-type compartmental models [7], necessitating a modeling approach capable of accurately scaling up mAb disposition from preclinical species to human.
Physiologically based pharmacokinetic (PBPK) models have gained wide spread use as a mechanistic and more realistic modeling approach than empirical compartmental models to describe the disposition of drugs [8–10]. These models are highly complex in nature and integrate drug-specific parameters, like intrinsic clearance and tissue partition coefficients, with a drug-independent structural model consisting of anatomical compartments (e.g., organs and tissues) connected via physiological processes, e.g., blood flow and lymph flow [11, 12]. Because of their intricacy PBPK models are able to achieve detailed quantitative assessment of the plasma and tissue disposition of mAb drugs [13] along with the ease of scaling up the model to different species since the structural model is relatively common to most mammalian species [14]. These models are also flexible enough to adapt to changes in pathological conditions and can help in investigation and prediction of mAb targeting, correlation of target expression and turnover with mAb disposition and prediction of non-linear process involved in mAb disposition across species [15, 16].
One of the first reports characterizing the biodistribution, catabolism, and excretion of a nonspecific immunoglobulin G (IgG) and its fragments (F(ab′)2 and Fab′) in mice, using a physiologically based organ-specific model, was published by Covell et al. [17]. Their work established a framework for the future mAb PBPK models, which included the processes related to (a) circulation of plasma (b) exchange of mAb across the capillary wall (c) return of mAb from the interstitial space to the bloodstream via lymph and (d) interaction of antibody with cell-associated material and formation of metabolic products. However, for each organ they estimated three parameters (including the apparent distribution volume of mAb in the non-plasma and non-interstitial space) by fitting a three-compartment linear model to the data for each organ, rendering the model less ideal and more empirical. Subsequently Baxter et al. [13] published a PBPK model characterizing the biodistribution of a specific mAb and its fragments and a nonspecific mAb in a human colon carcinoma xenograft in nude mice, which was later shown to be able to predict the biodistribution of mAb in humans based on data obtained from rodents and known human physiological parameters [14]. Their work was not only the first to characterize the biodistribution of a specific binding mAb but also created a framework for subsequent PBPK models incorporating a two-pore formalism for trans-capillary mAb exchange [18, 19]. Following the increased understanding about the role of neonatal Fc receptor (FcRn) in the disposition of mAb, the next generation of PBPK models included IgG–FcRn interaction [15, 16, 18, 19]. While the models from Ferl et al. [18] and Davda et al. [19] used the two-pore formalism for drug extravasation, with IgG–FcRn interaction incorporated in few chosen tissue compartments, the model from Garg and Balthasar [15, 16] incorporated IgG–FcRn interaction in all the tissue compartments and used a simplified approach for drug extravasation with considerably fewer model parameters to estimate.
Although each of the aforementioned PBPK models for mAb [13–16, 18] were able to characterize the respective experimental data reasonably well, each of them has some limitations regarding either number of tissues involved in the analysis or number of species used to validate the model, and this manuscript attempts to overcome those limitations by developing a “platform” PBPK model by modifying/augmenting previously published PBPK models [15, 16]. The two main objectives of the present investigation were (1) development of a PBPK model capable of characterizing the plasma and tissue PK of nonspecific or antigen specific mAbs in wild type, FcRn knockout, tumor bearing and non tumor bearing mice and (2) evaluation of the scale up potential of the model by characterizing the mouse, rat, monkey and human plasma PK of mAbs, simultaneously. The analysis presented in this manuscript demonstrates following distinct improvements in the features of the platform PBPK model compared to previously published PBPK models [15, 16]: (1) presence of more pertinent tissues in the model structure and complete mass balance, (2) use of four different species to build the model, (3) different values for the physiological parameters, (4) mechanistically more detailed and divided tissue compartment that includes blood cell sub-compartment into tissue vascular space and cellular space sub-compartment in each tissue, (5) use of different vascular reflection coefficients for different tissues, which were fixed based on the vascular pore size of the given tissue, compared to the use of same vascular reflection coefficients for all the tissues, (6) removal of the characterization of endogenous mAb concentrations in all the compartments and its competition with exogenous mAb for FcRn in the endosomal compartment, (7) no assumption of equilibrium between mAb and FcRn in the endosomal space and hence the use of association and dissociation rate constants instead of equilibrium dissociation constant, (8) degradation of FcRn unbound mAb is characterized using a first order rate constant that was estimated from the data and kept the same for all tissues in all species compared to the use of tissue normalized clearance, which was fixed, (9) use of pinocytosis clearance to describe the entry and exit of mAb from the endosomal space, which was estimated from the data and kept the same for all tissue in all species, compared to the use of a rate constant that was kept the same for all the tissues, and (10) concentration of accessible FcRn in the endosomal space was estimated from the data and kept the same for all tissue in all species compared to fixing the tissue normalized value for each tissue based on the estimates of total body capacity of FcRn.
Methods
Datasets for modeling
The datasets that are used for the development of the PBPK model not only cover a diverse set of antibodies and animal species but also cover data from several previously published PBPK models for mAbs. All the datasets were digitized from the literature (using the software ‘Grab It! XP’) and mean data were used for the analysis. The data for plasma and tissue PK of control IgG in wild-type and FcRn knockout mouse were obtained from Garg and Balthasar [15]. This study analyzes PK of a nonspecific mouse IgG1 antibody, 7E3, in plasma, heart, lung, liver, spleen, muscle, skin, gut, and kidneys after intravenous administration of 8 mg/kg dose. In order to account for the effect of saturation of FcRn on the disposition of mAbs, plasma and tissue PK of control IgG in wild-type mouse after excess intravenous IgG (IVIG) co-administration was used [20]. This study analyzes PK of 7E3 in plasma, heart, lung, liver, spleen, muscle, skin, gut, and kidneys after intravenous co-administration of 8 mg/kg 7E3 with 2 g/kg IVIG. Plasma and tissue PK of a control IgG in tumor bearing mouse were obtained from Baxter et al. [13]. Their study analyzes PK of a nonspecific mouse IgG1 antibody, MOPC21, in plasma, bone, heart, kidney, liver, lung, muscle, skin, spleen, tumor, and GI tract after intravenous administration of 3.8 μg dose. Plasma and tissue PK of a specific IgG in tumor bearing mouse were obtained from Ferl et al. [18, 21]. This study analyzes the PK of a chimeric anti-CEA IgG1 mAb (cT84.66) in blood, liver, spleen, kidney, lung, stomach, bowel, bone, carcass and tumor of a human colorectal tumor (LS174T) mouse xenograft after intravenous administration of ~4.5 μg/kg dose. Representative PK of mAbs in rat, monkey and human were collected from the literature. Plasma PK of a nonspecific rat IgG1 in wild-type rat after intravenous administration of 0.7 mg/kg dose were obtained from Bazin-Redureau et al. [22]. Plasma PK of a nonspecific human IgG2, OST577, in rhesus monkey after intravenous administration of 1 mg/kg dose was obtained from Hinton et al. [23]. Plasma PK of a fully human anti-TNF-alpha IgG1 mAb, adalimumab, in human after intravenous administration of 5 mg/kg dose was obtained from Weisman et al. [24].
Model structure
A schematic diagram for the structure of the platform PBPK model is shown in the Fig. 1. The PBPK model includes 16 tissue compartments (blood, lung, heart, kidney, muscle, skin, liver, brain, adipose, thymus, bone, small intestine, large intestine, spleen, pancreas and other), a tumor compartment and a lymph node compartment connected to each other in an anatomical manner using blood and lymph flow. The “other” compartment represents carcass tissues e.g., thyroid, gonads etc., not specifically accounted for in the model structure. Arterial blood to each tissue compartment is delivered by the efferent blood supply from the lung and venous return from most tissues except small intestine, large intestine, spleen, pancreas, is delivered to the blood compartment, which represents a venous pool. Venous return from small intestine, large intestine, spleen and pancreas is delivered to the liver. Lymph flow from all the tissues is delivered to the blood compartment via a lymph node compartment. The flow circuitry is completed by delivering the blood from the blood (venous pool) compartment to the lung compartment.

Structure of the whole body platform PBPK model for mAb disposition. All organs are represented by a rectangular compartment and connected in an anatomical manner with blood flow (solid arrows) and lymphatic flow (dashed arrows). Arrows represent the direction of the flow. Each tissue within this model, except blood and lymph node, is divided into sub-compartments as shown in Fig. 2. The central blood compartment is divided into plasma and blood cell (B. Cells) compartments. Small intestine (S.I.) refers to duodenum, jejunum and ileum; whereas large intestine (L.I.) refers to cecum and colon
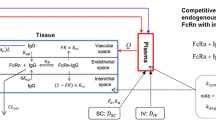
Each tissue compartment is further divided into vascular, endosomal, interstitial and cellular sub-compartments as shown in Fig. 2. The vascular sub-compartment is further divided into plasma and blood cell compartments whereas the cellular compartment is further divided into cell membrane and intracellular compartments. MAb enters each tissue vascular compartment using arterial blood flow (Q) and exits using venous blood flow (i.e. arterial blood flow reduced by the lymphatic flow) (Q-L). From the vascular compartment, mAb distributes to the plasma sub-compartment and, if required by the property of the drug, the mAb in plasma interacts with the cells present in the blood cell sub-compartment. MAb in plasma distributes to the interstitial sub-compartment via paracellular pores using the convective lymph flow (L) where the vascular reflection coefficient (σ v ) represents the level of resistance provided to the mAb convection by the vascular endothelial cells. MAb in plasma space is also taken up by the vascular endothelial cells via pinocytosis (CL up ). It is assumed that the intact mAb in the vascular endothelial cells stays within the endosomal space and once within the endosomal space mAb interacts with FcRn molecules via association (K on_FcRn ) and dissociation (K off_FcRn ) rate constants. Unbound mAb within the endosomal space degrades with a first order rate constant (K deg ). MAb bound to FcRn recycles either to the plasma space (fraction recycled to plasma space is FR) or to the interstitial space (fraction recycled to interstitial space is 1-FR) via exocytosis. MAb present in interstitial space is taken up by the vascular endothelial cells via pinocytosis. Interstitial mAb exits the tissue compartment using the convective lymph flow (L) where the interstitial reflection coefficient (σ I ) represents the level of resistance provided to the mAb convection by the lymphatic openings. For mAb drugs that have specific affinity against antigens on the cell surface of certain tissues (e.g., tumor), interstitial molecules of mAb are allowed to interact with the antigen using association (K on_Ag ) and dissociation (K off_Ag ) rate constants. For antigens capable of internalization, the mAb-antigen complex internalizes using the first order ‘net’ internalization rate constant (K int_Ag-IgG ), and the internalized molecules are assumed to be metabolized.

Structure of the tissue level PBPK model for mAb disposition. Each tissue compartment, except blood and lymph node, is further divided into vascular, endosomal, interstitial and cellular sub-compartments. The vascular sub-compartment is further divided into plasma and blood cell compartments whereas the cellular compartment is further divided into cell membrane and intracellular space compartments. For detailed description of the symbols and drug disposition process please refer to the “model structure” sub-section in the “methods” section
Model equations
Please refer to the glossary in the Appendix section for detailed descriptions and units of the symbols used in the following equations.
Blood compartment
Plasma
Blood Cells
Lymph node compartment
Typical tissue compartment
Vascular space plasma
Vascular space blood cells
Endosomal space mAb unbound to FcRn
Endosomal space mAb bound to FcRn
Endosomal FcRn
Interstitial
Cell membrane
Equations 4–10 describe a typical tissue compartment and the subscript “i” refers to the individual tissue being considered for the analysis. For all the tissues except lung and liver the subscript “j” in EqS. 4 and 5 refers to the lung concentration, whereas for the lung subscript “j” refers to plasma/blood cell concentrations. Due to the unique anatomical arrangement of the liver the vascular space in this tissue compartment is described differently using the following equations.
Liver compartment
Vascular space plasma
Vascular space blood cells
For a given tissue the observed total antibody concentration in tissue homogenate is described using following equation:
Model parameters
The model structure and equations for the PBPK model remain the same for the different species considered in this manuscript i.e., mouse, rat, monkey and human. However, the physiological parameters for each species change and the values were obtained from a diverse set of literature and from BioDMET documents (http://pdsl.research.ge.com/BioDMET/userdocumentation.html) (GE Corp.) [13, 16, 20, 25, 26]. Values of the physiological parameters used for the PBPK modeling are compiled and presented in Tables 1, 2, 3 and 4. Lymph flow for all the tissues across the species was set 500 times lower than the plasma flow of the given tissue [27]. Endosomal volume for all the tissues across the species was assumed to be 0.5% of the total tissue volume based on Garg and Balthasar [15]. Based on the literature reviewing physiologic upper limits of pore size of different blood capillary types [28], the vascular reflection coefficient (\( \sigma_{i}^{V} \)) for lung, heart, muscle, skin, adipose, large intestine and others compartments was set to a value of 0.95; the value for kidney, thymus, small intestine and pancreas was set to 0.9; the value for spleen, liver and bone was set to 0.85 and the value for brain was set to 0.99 a priori. The vascular reflection coefficient value for tumor was set to a value of 0.84 based on the estimates from Urva et al. [16]. Consistent with previously published literature the values of lymphatic reflection coefficient (\( \sigma_{i}^{IS} \)) and fraction of FcRn bound antibody that recycles to the vascular space (FR) were set to 0.2 and 0.715 [15, 16]. The association (\( K_{on}^{FcRn} \)) and dissociation rate constants (\( K_{off}^{FcRn} \)) between IgG and FcRn for each species were obtained from literature [23, 29–33]. Values for the association rate constant were 8.06E + 07, 8.00E + 08, 7.92E + 08 and 5.59E + 08 1/M/h for mouse, rat, rhesus monkey and human. And, values for the dissociation rate constant were 6.55, 144, 46.8 and 23.9 1/h for mouse, rat, rhesus monkey and human.
Four parameters, for which a certain literature reported values were not available, were estimated by fitting the PBPK model to the data. Each parameter was assumed constant in all tissues and species. These parameters were: (1) FcRn concentration (FcRn) in the endosomal space, (2) the rate of pinocytosis and exocytosis per unit endosomal space of vascular endothelia (CL up ) (this rate was scaled by multiplying it with the endosomal volume of the given tissue), (3) the rate at which FcRn unbound antibody molecules are degraded in the endosomal space (K deg ), and (4) the proportionally constant (C_LNLF) between the rate at which antibody transfers from the lymph node compartment to the plasma/blood compartment (L LymphNode ) and the plasma flow of the given species.
For the two datasets (i.e. Baxter et al. [13] and Ferl et al. [18]) consisting of antibody plasma and tissue PK in tumor bearing mice, extra parameterization for tumor was required. Initial tumor volumes for both the studies and the growth characteristics for tumor (i.e. constant volume vs. growing) were kept the same as in original publications [13, 18]. The vascular volume for tumor was set to 7%, the interstitial volume was set to 55% and the endosomal volume was set to 0.5% of the total tumor volume [16]. The lymph reflection coefficient was fixed at 0.2 and the vascular reflection coefficient was fixed at the value of 0.842 [16]. Tumor plasma flow was set to 12.7 l/h/l [18]. The lymph flow was set to a value of 0.2% of plasma flow, for the dataset obtained from Baxter et al. Consistent with the publication of Ferl et al. [18] their dataset was fit by adjusting the perfusable tumor mass, which was set as a function of total tumor size in mice. For the specific antibody against tumor antigen (cT84.66), the antibody-antigen interaction parameters (i.e., association and dissociation rate constants) and tumor antigen concentrations were obtained from literature [18]. Antigen–antibody complex internalization rate was estimated by fitting the model to the data. During individual optimization of the dataset from Ferl. et al. [18] the estimated internalization rate constant for tumor antigen (K int ) was found to be 2.47E−02 1/h and the tumor lymph flow was found to be 0.29% of plasma flow.
Parameter estimation and model fitting evaluation
Fifty two different plasma and tissue concentration versus time datasets from 8 different literature studies were fitted simultaneously with the PBPK model using a total of 820 equations to estimate 4 unknown parameters (i.e. FcRn, CL up , K deg and C_LNLF), while keeping all other parameters fixed. The model was fitted to the data using the maximum likelihood (ML) estimation method in ADAPT-5 software (BMSR, CA) with the following variance model:
where Y(t) is the model output at a given time t and Var(t) is the variance associated with the output. σIntercept and σSlope are the variance parameters representing a linear relationship between the standard deviation of the model output and Y(t).
Final model fittings and parameter estimates were confirmed using standard model fitting criteria e.g., visual inspection, observed versus predicted plot, predicted versus residual plot and CV% of the estimated parameters. For quantitative comparison of observed and model generated data, the median percent predictive error \( \left( {\% PE = \frac{{\left| {C_{Pred} - C_{Obs} } \right|}}{{C_{Obs} }} \bullet 100} \right) \) was calculated for all the plasma and tissue datasets. In the equation above, C Pred is the model generated total concentrations of antibody obtained with the optimized set of parameters and C Obs is the observed total antibody concentrations.
Sensitivity analysis
A local sensitivity analysis was performed on the final model and parameter set to assess the sensitivity of the model output to model parameters. All the parameters except literature derived physiological parameters were included in the sensitivity analysis. Area under the plasma concentration curve (AUC) was chosen as the relevant model output to represent drug exposure. The analysis was conducted by evaluating the percentage change in AUCs with ±50% alteration in the model parameters [16, 34]:
AUC SIM refers to the AUC obtained with the optimized set of parameters and AUC ±50% is the AUC obtained following a 50% increase or decrease in the parameter value. The analysis was performed on all the datasets from all species at the same dose levels as the ones used for parameter estimation.
Results
All the analyzed plasma and tissue PK datasets for mAb were reasonably well characterized by the PBPK model, simultaneously (Figs. 3, 4, 5, 6, 7, 8). The model was able to recapitulate the salient features of each of the datasets and the quantitative comparison between the observed and model generated concentrations, in the form of median %PE, is summarized in Table 5. Figure 3 displays the observed and model generated plasma and tissue PK of a nonspecific mAb in wild-type mouse after the dose of 8 mg/kg, superimposed on each other. As evident from the figure the model was able to characterize all the data reasonably well except for the concentrations in heart, which were under-predicted at all time points. The best characterized tissue was lung (%PE = 8.27) and the worst characterized tissue was spleen (%PE = 58.2). Figure 4 displays the observed and model generated plasma and tissue PK of a nonspecific mAb in FcRn knockout mouse after the dose of 8 mg/kg. The model adequately accounted for the removal of FcRn mediated recycling of mAb, leading to prediction of enhanced elimination of mAb in FcRn knockout mouse relative to wild-type mouse (Figs. 4, 8d). The best characterized tissue was again lung (%PE = 18) and the worst characterized tissue was heart (%PE = 63.2), which was consistently under-predicted. Figure 5 displays superimposed observed and model generated plasma and tissue PK of a nonspecific mAb in wild-type mouse, which was collected after co-administration of 8 mg/kg mAb with 2 g/kg dose of IVIG. The model was adequately able to account for the saturation of FcRn by the high dose of IVIG, leading to prediction of enhanced elimination of mAb compared to its elimination in wild-type mouse at the same dose when IVIG is not co-administered (Figs. 5, 8d). The best characterized tissue was liver (%PE = 10.6) and the worst characterized tissue was kidney (%PE = 50.4). Figure 6 displays the observed and model generated plasma and tissue PK of a nonspecific mAb in tumor bearing mouse, superimposed on each other. The model was not only able to characterize the PK of mAb in plasma and normal tissues but also in xenograft tumor reasonably well, capturing the delayed increase in tumor mAb concentrations compared to many other tissues. The best characterized tissue was liver (%PE = 11.2) and the worst characterized tissue was gut/small intestine (%PE = 121). Figure 7 displays the observed and model generated plasma and tissue PK of a tumor antigen specific mAb in human xenograft tumor bearing mouse, demonstrating the model was able to adequately capture the typical target mediated drug disposition signature profile [5] observed for the antibody at given dose. The model was also able to capture the accumulation of mAb in tumor, characterized by the delayed increase in tumor mAb concentrations and relatively high mAb concentrations in tumor compared to any other tissues. The best characterized tissue was plasma (%PE = 5.66) and the worst characterized tissue was spleen (%PE = 90.3). Figure 8a–c, shows the observed and model generated plasma PK of mAb in rat, rhesus monkey and human. The model was able to characterize the plasma PK of mAb in all three species reasonably well with the median %PE value of 39.6, 36.8 and 16.0 for rat, rhesus monkey and human. As such, %PE values for all but one dataset were less than 100%, suggesting model generated concentration values were on average within two fold of the observed concentration values.

PK of nonspecific mAb in wild type mouse. Figure displays the observed (solid dots) and model generated (solid lines) plasma and tissue concentration versus time profiles of mAb in wild-type mouse after intravenous administration of 8 mg/kg dose

PK of nonspecific mAb in FcRn knockout mouse. Figure displays the observed (solid dots) and model generated (solid lines) plasma and tissue concentration versus time profiles of mAb in FcRn knockout mouse after intravenous administration of 8 mg/kg dose

PK of nonspecific mAb in wild type mouse after saturation of FcRn. Figure displays the observed (solid dots) and model generated (solid lines) plasma and tissue concentration versus time profiles of mAb in wild type mouse after intravenous co-administration of 8 mg/kg dose with 2 g/kg IVIG

PK of nonspecific mAb in tumor bearing mouse. Figure displays the observed (solid dots) and model generated (solid lines) plasma and tissue concentration versus time profiles of mAb in human xenograft tumor bearing mouse after intravenous administration of 3.8 μg dose

PK of a specific mAb in tumor bearing mouse. Figure displays the observed (solid dots) and model generated (solid lines) plasma and tissue concentration versus time profiles of a tumor antigen specific mAb in human xenograft tumor bearing mouse

PK of nonspecific mAbs in different species. Figure displays the observed (solid dots) and model generated (solid lines) plasma concentration versus time profiles a Plasma PK of a nonspecific rat IgG1 in wild type rat after intravenous administration of 0.7 mg/kg dose, b Plasma PK of a nonspecific IgG2 in rhesus monkey after intravenous administration of 1 mg/kg dose, c Plasma PK of a fully human IgG1 mAb in human after intravenous administration of 5 mg/kg dose, d Plasma PK of a nonspecific mAb after the dose of 8 mg/kg in wild-type mouse (solid line, solid circles), in FcRn knockout mouse (dashed line, solid squares) and in wild-type mouse that is co-administered with 2 g/kg IVIG (dotted line, open circles)
The overall quality of the fit for all the datasets with the optimized set of parameters is illustrated in Fig. 9. Figure 9a displays the plot of individual model generated versus observed concentration along with the identity line. Figure 9B displays the plot of predicted concentration versus residuals, demonstrating the uniform spread of the residuals around the x-axis without any obvious patterns. The parameter characterizing the rate of pinocytosis and exocytosis per unit endosomal space (CL up ) was estimated with good precision (%CV = 3.48) and the optimized value was found to be 3.66E−02 l/h/l. The parameter characterizing the rate at which antibody molecules unbound to the FcRn is degraded in the endosomal space (K deg ) was also estimated with good precision (%CV = 15.7) and the optimized value was found to be 42.9 1/h. The concentration of FcRn (FcRn) necessary to simultaneously characterize all the datasets was estimated to be 4.98E−05 M, which was estimated with good precision and CV% value of 11.1. The proportionality constant between the rate at which antibody transfers from the lymph node compartment to the plasma/blood compartment and the plasma flow of the given species (C_LNLF) was estimated to be 9.1, however, the confidence in the estimated value was found to be low (CV% > 50). System parameters estimated from the PBPK model are summarized in Table 6.

Evaluation of the platform PBPK model performance. a The plot of individual model generated versus observed concentration along with the identity line. b The plot of weighted residuals versus observed concentration, demonstrating the spread of the residuals around the x-axis
To investigate the relative importance of the parameters and to determine the parameters to which the mAb plasma concentrations are most sensitive to, a local sensitivity analysis was conducted [16, 34]. The results from the sensitivity analysis based on percentage change in plasma AUCs with ±50% alteration in the model parameters are displayed in Fig. 10. The higher the absolute value of percent change in AUC higher the sensitivity of the parameter and a negative value for percent change indicates a decrease in AUC with an increase in the parameter value. The sensitivity analysis showed that the direction and extent of changes in plasma AUCs with ±50% alteration in the model parameters were similar for mouse, rat, monkey and human (Fig. 10). The most sensitive parameters were found to be the rate at which FcRn unbound antibody molecules degraded (K deg ) and the dissociation rate constants between IgG and FcRn (\( K_{off}^{FcRn} \)). The model was also notably sensitive to the FcRn concentration (FcRn), the association rate constants between IgG and FcRn (\( K_{on}^{FcRn} \)), vascular reflection coefficient (\( \sigma_{i}^{V} \)) and the rate of pinocytosis per unit endosomal space (CL up ). It is important to note that due to the upper limit of 1 for the vascular reflection coefficient it was not possible to change the value to +50% for sensitivity analysis. The model output was also moderately sensitive to the fraction of FcRn bound antibody that recycles to the vascular space (FR) and the significance of this parameter seemed to increase as the size of species increased. The models output was least sensitive to the lymphatic reflection coefficient (\( \sigma_{i}^{IS} \)) and the rate at which antibody transfers from the lymph node compartment to the blood compartment. Keeping the dissociation constant (ratio of dissociation and association rate constant) the same and changing the absolute value of the dissociation and association rate constants simultaneously by ±50% led to a slight change in the model output and the effect was more prominent in mouse compared to other species. The sensitivity analysis performed on the output from the FcRn knockout mouse revealed that the vascular reflection coefficient and the rate of pinocytosis per unit endosomal space were the most significant parameters and other parameters did not influence the output as much as they did in wild-type mouse (data not shown). The sensitivity analysis performed for the nonspecific antibody in tumor bearing mouse provided results that were similar to the ones found with wild-type mouse. However, the sensitivity analysis performed for the specific antibody in tumor bearing mouse revealed that the model output was significantly sensitive to tumor specific parameters and the vascular reflection coefficient was the only system parameter the model output was notably sensitive to. Tumor specific parameters the model output was most sensitive to were the tumor vascular reflection coefficient, tumor plasma and lymph flow, the tumor antigen concentration and binding parameters between tumor antigen and antibody.

Local sensitivity analysis of the parameters used in the platform PBPK model. The bar chart shows percentage change in plasma AUCs with ±50% alteration in the model parameter values. The higher the absolute value of percent change in AUC, the higher the sensitivity of the parameter and a negative value for percent change indicates a decrease in AUC with an increase in the parameter value. The sensitivity analysis showed that the direction and extent of changes in plasma AUCs with ±50% alteration in the model parameters were similar for mouse, rat, monkey and human. Key: (K deg ) first order degradation rate constant of FcRn unbound mAb within the endosomal space; (K off ) dissociation rate constant between mAb and FcRn; (FcRn) concentration of FcRn in the endosomal space; (K on ) association rate constant between mAb and FcRn; (V_RC) vascular reflection coefficient representing the level of resistance provided to the mAb convection by the vascular endothelial cells; (CL_UP) rate of pinocytosis and exocytosis per unit endosomal space of vascular endothelia; (FR) fraction of FcRn bound mAb that recycles to the vascular space; (IS_RC) lymphatic reflection coefficient representing the level of resistance provided to the mAb convection by the lymph vessels; (C_LNLF) proportionally constant between the rate at which antibody transfers from the lymph node compartment to the plasma/blood compartment and plasma flow of the given species
Discussion
Translational research conducted during the drug development process relies on the efficacy and toxicity data obtained from animals to make informed decisions and predictions about human efficacy and toxicity. One of the main reasons this strategy has been widely accepted is the fact that many of the anatomical structures and physiological processes have been conserved between mammalian species. PBPK models rely on the same principle of conserved inter-relationships between anatomical, physiological, biochemical and physicochemical processes [11, 35–37] to not only provide quantitative descriptions of the drug disposition process in a biological systems but also to scale drug ADME (absorption, distribution, metabolism and, elimination) processes between species [12, 38]. In this report efforts have been made to accommodate many such essential processes involved in the disposition of mAb in a single platform PBPK model, which can be used to characterize the plasma and tissue concentrations of mAb in preclinical species and human.
Since it is imperative for a mass balance approach like the PBPK model to have all the distribution space accounted for, one of the main goals of the platform PBPK model was to incorporate all the possible tissues along with the carcass “other” compartment in the model structure (Fig. 1) to account for all the volume of distribution available to mAb. This is in contrast to previously published PBPK models that incorporated only few of the tissues in the model, which were considered to represent the major distribution sites for mAb, accounting for more than 90–95% of the injected dose in mice a short time after administration [13–16, 18]. Having more tissues also makes the platform PBPK model better than other previously published PBPK models in terms of its ability to incorporate wider range of mAb targets and sites of actions for treating and diagnosing various disease conditions e.g., anti-DR5 and anti-CTLA4 mAbs for pancreatic cancer, anti-CD133 + and anti-EGFRvIII + mAbs for brain tumor, anti-adipocyte mAbs to alter lipid metabolism in adipose, anti-sclerostin antibody for osteoporosis etc.
It is well known that FcRn is the homeostatic receptor responsible for extending the serum half-life of mAbs [39–42]. Although the biological importance of this receptor is still being investigated [39, 43] and the exact site(s) at which it protects mAb from degradation is still being delineated [43, 44], characterizing this interaction using a mathematical model becomes very valuable to take the full advantage of strategies, which design drug molecules with desired PK properties by altering the FcRn–mAb interaction [45]. Among the previously published mAb PBPK models, earlier models did not account for this interaction [13, 14], some of the models account for the FcRn–mAb interaction only in few chosen tissues [18, 19] and other models account for this interaction in all the tissues considered in the model [15, 16]. FcRn protein is found to be expressed by a wide variety of tissues e.g., the renal proximal tubules [46, 47], endothelial cells of the muscle vasculature [48], keratinocytes [49], hepatocytes [50], mammary epithelium [51], monocytes, intestinal macrophages, dendritic cells [52] aortic endothelial cells, and spleen tissue [53]. Additionally, the gene encoding FcRn receptor (FCGRT) [54] is found to exist in all the tissues analyzed in mouse, rat and human [55]. Considering the aforementioned ubiquitous presence of FcRn, the present model incorporates FcRn–mAb interaction in all the tissues considered in the model. The site where FcRn is protecting IgG from catabolism has been believed to exist mainly within the vascular endothelial cells where FcRn has a large contact area with the blood [39, 48, 56], and consistent with that observation the platform PBPK model allows the interaction between mAb and FcRn in the endosomal space of each tissue’s vascular endothelium (Fig. 2) [15, 16, 19]. Assuming that the concentration of FcRn in the vascular endothelial cells is conserved between different species a single value of FcRn concentration was estimated by fitting the model to all the datasets simultaneously. The fitted value of FcRn concentration was found to be 4.98E−05 M, which is similar to the value of 4.00E−05 M reported by Ferl et al. [18] for mouse and 3.30E−05 M reported by Garg and Balthasar [20] for human.
It is hypothesized that mAbs enter the endothelial cells via pinocytosis (fluid phase endocytosis) and upon acidification of the endosomes they bind to FcRn [39, 43]. Subsequently, unbound mAbs are subjected to lysosomal degradation and FcRn–bound mAbs are recycled to the cell surface, where they are exposed to the physiological pH and get released into the extracellular fluid [20, 39, 43]. In order to mimic this mechanism the platform PBPK model allows mAb to enter endosomal compartment (either from vascular or from interstitial compartments) via a pinocytosis clearance uptake term (CL up ). Assuming that the rate of pinocytosis into the vascular endothelial cells is conserved between different species a single value of this uptake clearance was estimated by fitting the model to all the datasets simultaneously. The fitted value of pinocytosis uptake clearance was found to be 3.66E−02 l/h/l, which is very close to the value of 3.75E−02 l/h/l reported by Haigler et al. [57]. Once inside the endosomal space the interaction between the FcRn and mAb molecules was characterized using association and dissociation rate constants, in contrast to previously published literature that assume equilibrium is achieved within the endosomal space and use the equilibrium dissociation constant to characterize this interaction [15, 16]. One of the main reason for adopting this approach is the relatively short endosomal recycling time compared to the dissociation half life of mAbs from FcRn, suggesting the equilibrium cannot be achieved between mAb and FcRn during their stay in the endosomal phase. Of note, the model output is highly sensitive to the association and dissociation rate constant parameters and before using these values it is important for a PBPK modeling scientist to understand the validity of the analytical approach used to determine these parameters, as the value of these parameters highly depends on the analytical approach taken to measure them [23, 29–32]. FcRn bound mAb was allow to recycle in a polarized manner where the fraction recycling to the vascular space was fixed to the value of 0.715 obtained from Garg and Balthasar [15]. Degradation of mAb molecules not bound to FcRn was described with a first order rate constant (K deg , which mimics the process of lysosomal transition [43]) assumed to be the same throughout all the tissues and species. The fitted rate of degradation of unbound antibody from the endosomal space was 42.9 1/h, which yields a half life of ~1 min. Although there is no literature value to compare to the estimated value of this parameter, the relative time frame of the endocytic recycling process and some evidential videos about this sorting process [43] support the estimated short half life.
Consistent with the anatomy and a previously published PBPK model [16], the current version of the platform PBPK model uses a lymph node compartment to collect the effluent lymph from all the interstitial compartments and then transfer the amount present in this compartment to the central circulation. However, through the estimation process and sensitivity analysis it was found that the parameter related to the rate of transfer from lymph node to central circulation was not estimated with great confidence and the output was not sensitive to this parameter (Fig. 10). In the future, one needs to measure the physiological value of this process and fix this parameter or, for the purpose of parsimony, remove the lymph node compartment. However, considering the model has been developed to use as a platform model capable of incorporating many pathophysiological scenarios and some recent data suggesting the possibility of lymphatic catabolism of macromolecules, there is a strong case for keeping this lymph node compartment. Sensitivity analysis also demonstrated that vascular reflection coefficient is one of the parameters the model output is very sensitive to. In the present model this value was fixed to different values for different tissues, based on the literature reviewing pore diameter for each tissue vasculature [28] and the fact that mAb have a diameter between 9 and 13 nm [58]. Although these fixed values performed reasonably well for fitting most of the datasets (Figs. 3, 4, 5, 6, 7, 8) tissues like heart were consistently under-predicted in many cases and there were subtle systematic deviations in few tissues, demanding a better non-subjective approach to determined these parameter values. Ideally this values should be estimated and, rich datasets for mAb tissue biodistribution where drug concentrations in tissues are determined after removing the residual blood content (e.g., Vugmeyster et al. [59]) should be employed to estimate this parameter for each tissue with confidence.
One of the motives behind developing this platform model was to make it applicable to different macromolecular drug modalities e.g., antibody drug conjugates [60], CovX-bodies [61], IgNARs [62], Fc fusion proteins, F(ab), F(ab)2, ScFv, bi-functional mAbs, albumin etc. Although the current manuscript only deals with the characterization of simple mAb molecules, the reflection coefficient parameters can be adjusted (or fitted) so that different size of macromolecules can be characterized with the same model structure. Additionally, macromolecules that contain the Fc region (e.g., Fc fusion proteins) can also be well characterized with the same model owing to the incorporation of the FcRn recycling component in the model. It is also important to note that some tissues like kidney, where FcRn has been shown to reabsorb mAb [46] and whose contribution to elimination of macromolecules becomes significant as the molecular weight becomes close to or less than 70 kDa, may deserve special attention and modification when applying the present model to a wide array of macromolecular modalities. The platform PBPK model is also amenable to disease related anatomical changes that would be necessary to characterize the disposition of mAb in a diseased animal or subject. As is demonstrated in the manuscript, the model is easily able to incorporate a xenograft tumor model and is also able to characterize the plasma and tissue disposition of nonspecific or tumor antigen specific mAbs in this diseased model (Figs. 6, 7). Since the model incorporates most of the relevant tissues in the structure it would be also relatively easy to incorporate pathophysiological changes in any given tissue by just altering the tissue specific parameters and to incorporate any desired target into given tissues.
Another objective behind developing this platform model was to have a model structure and parameter set that is able to characterize the plasma and tissue disposition of mAb across different species. In order to achieve this it was assumed that many parameters remain conserved between species and these parameters were estimated by fitting the same model structure to plasma and tissue PK data of mAb in four different species. As shown in Fig. 8 the model was able to characterize the data from different species simultaneously, delivering a common set of parameters that remain constant across four different species. The model can further be expanded to additional species (e.g., dog, guinea pig etc.) if the necessary physiological parameters are available.
Ideally, using the presented PBPK model and estimated parameter sets, one should be able to predict the plasma and tissue PK of mAb, just based on the knowledge of a few drug specific parameters measured in vitro (e.g., the affinity of the molecule with FcRn). Additionally, if one is able to characterize the PK of a mAb in one species, using the present model and a given set of parameters, just by changing the physiological parameters from one species to the other species the PK of mAb in the other species should also be predicted a priori. However, as our experience with mAb is increasing and our understanding of the molecular construction and interactions of mAb is increasing we are coming across examples that are difficult to explain using our past body of knowledge. The notion that a mAb has a typical half-life of 3 weeks in human is no longer true for all the molecules and there are many parameters that have been identified to be responsible for the deviation of mAb half-life from this typically assumed value. Going forward, it is very important for a model like this platform PBPK model to keep up to date with the current state of knowledge and have ability to adapt to the newly discovered mechanisms. Many a times the target for the mAb itself is responsible for eliminating the drug faster after the interaction, leading to a target mediated drug disposition [5] of mAb and a shorter plasma half-life. The present model is able to take this phenomenon into account and in fact for the plasma and tissue PK data of a mAb that shows target mediated drug disposition (from Ferl et al. [18]) the model was successfully able to characterize the data (Fig. 7). In the literature there are some reports suggesting that the isoelectric point (pI) of mAb can influence its plasma and tissue disposition. Results from Igawa et al. [63] suggest that lowering the pI by engineering the variable region could reduce the elimination of mAbs and results from Boswell et al. [64] suggest that (1) shifts in pI of approximately one unit or more can produce measurable changes in tissue distribution kinetics, (2) increases in net positive charge generally result in increased tissue retention and blood clearance and (3) decreases in net positive charge generally result in decreased tissue retention and increased whole body clearance [64]. If this relationship between pI and the disposition of mAb is confirmed, the present model can account for this relationship by establishing a correlation between the pI value for the mAb and CL up in the model, which according to the work from Boswell et al. [64] would be the main parameter influenced by changes in the charge of the mAb molecule. There are tremendous molecular biology efforts going on to produce different variants of mAbs by site directed mutagenesis to alter its PK behavior and effector function. These efforts produce a variety of mAb molecules with very different scaffolds and properties that can help us evaluate different hypothesis. In one such effort it was found out that improved affinity of mAb for FcRn at pH 6.0 by inducing mutations in the Fc region of mAb leads to an increased in vivo half-life [65], which was mainly attributed to changes observed in the dissociation rate constant between mAb and FcRn. This result has been further supported by many studies that suggest a correlation between mAb affinity for FcRn and its serum half-life. The present model structure is able to incorporate this phenomenon by simply altering the association and dissociation rate constants between a mAb and FcRn to alter the half-life of the mAb in plasma. As shown by the sensitivity analysis (Fig. 10) the model output is highly sensitive to the changes made in association and dissociation rate constants and the direction of the changes predicted in plasma exposure is consistent with the aforementioned experimental results. There are also some reports that failed to establish a correlation between affinity of mAb at pH 6.0 and serum half life [66], which according to some studies could be explained by simultaneously looking at the affinity of the mAb–FcRn at pH 7.4 [67–69]. The present model structure, however, is not capable of incorporating two different pHs in the endosomal compartment and in order to consider different affinities of mAb with FcRn at two different pH one will need to split the endosomal compartment into two different pH compartments by incorporating different affinity parameters between mAb and FcRn in each pH compartment. It is also important to note that several recent publications support the expression and function of FcRn in hematopoietic cells of mouse and human, and within this cell type, it has been shown to be involved in mAb homeostasis together with endothelial cells [44, 52, 53, 70]. If the aforementioned information is confirmed in other species (i.e., rat and monkey) and literature reports support the quantitative significance and distinction for the mAb homeostasis phenomenon occurring via hematopoietic cells compared to the endothelial cells, one would have to incorporate the mAb homeostasis by hematopoietic cells into the PBPK model, by characterizing mAb–FcRn interaction between plasma and blood cell compartments.
In summary, a platform PBPK model has been developed that is able to characterize the plasma and tissue PK of different mAbs in wild-type, FcRn knockout and tumor bearing mouse simultaneously. The model was also able to characterize the plasma PK of mAb in mouse, rat, monkey and human with a common set of estimated parameters, whose values were close to some of the literature reported values. Apart from mAbs the model is also capable of characterizing other macromolecular modalities without any major structural changes. The model is also capable of handling different disease (e.g., tumor) and pathophysiological conditions by altering the relevant parameter values. The platform model was able to characterize the data from many previously published mAb PBPK models simultaneously, and it provides a better alternative to over-parameterized two-pore models [13, 14, 18, 19] and an augmentation to previously published single-pore models [15, 16].
References
Levy G (1966) Kinetics of pharmacologic effects. Clin Pharmacol Ther 7:362–372
Lobo ED, Hansen RJ, Balthasar JP (2004) Antibody pharmacokinetics and pharmacodynamics. J Pharm Sci 93:2645–2668
Wang W, Wang EQ, Balthasar JP (2008) Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther 84:548–558
Mould DR, Sweeney KR (2007) The pharmacokinetics and pharmacodynamics of monoclonal antibodies-mechanistic modeling applied to drug development. Curr Opin Drug Discov Devel 10:84–96
Mager DE, Jusko WJ (2001) General pharmacokinetic model for drugs exhibiting target-mediated drug disposition. J Pharmacokinet Pharmacodyn 28:507–532
Mahmood I (2009) Pharmacokinetic allometric scaling of antibodies: application to the first-in-human dose estimation. J Pharm Sci 98:3850–3861
Dong JQ, Salinger DH, Endres CJ, Gibbs JP, Hsu CP, Stouch BJ, Hurh E, Gibbs MA (2011) Quantitative prediction of human pharmacokinetics for monoclonal antibodies: retrospective analysis of monkey as a single species for first-in-human prediction. Clin Pharmacokinet 50:131–142
De Buck SS, Sinha VK, Fenu LA, Nijsen MJ, Mackie CE, Gilissen RA (2007) Prediction of human pharmacokinetics using physiologically based modeling: a retrospective analysis of 26 clinically tested drugs. Drug Metab Dispos 35:1766–1780
Grime KH, Bird J, Ferguson D, Riley RJ (2009) Mechanism-based inhibition of cytochrome P450 enzymes: an evaluation of early decision making in vitro approaches and drug–drug interaction prediction methods. Eur J Pharm Sci 36:175–191
Thygesen P, Macheras P, Van Peer A (2009) Physiologically-based PK/PD modelling of therapeutic macromolecules. Pharm Res 26:2543–2550
Dedrick RL (1973) Animal scale-up. J Pharmacokinet Biopharm 1:435–461
Bischoff KB, Dedrick RL, Zaharko DS, Longstreth JA (1971) Methotrexate pharmacokinetics. J Pharm Sci 60:1128–1133
Baxter LT, Zhu H, Mackensen DG, Jain RK (1994) Physiologically based pharmacokinetic model for specific and nonspecific monoclonal antibodies and fragments in normal tissues and human tumor xenografts in nude mice. Cancer Res 54:1517–1528
Baxter LT, Zhu H, Mackensen DG, Butler WF, Jain RK (1995) Biodistribution of monoclonal antibodies: scale-up from mouse to human using a physiologically based pharmacokinetic model. Cancer Res 55:4611–4622
Garg A, Balthasar JP (2007) Physiologically-based pharmacokinetic (PBPK) model to predict IgG tissue kinetics in wild-type and FcRn-knockout mice. J Pharmacokinet Pharmacodyn 34:687–709
Urva SR, Yang VC, Balthasar JP (2010) Physiologically based pharmacokinetic model for T84.66: a monoclonal anti-CEA antibody. J Pharm Sci 99:1582–1600
Covell DG, Barbet J, Holton OD, Black CD, Parker RJ, Weinstein JN (1986) Pharmacokinetics of monoclonal immunoglobulin G1, F(ab’)2, and Fab’ in mice. Cancer Res 46:3969–3978
Ferl GZ, Wu AM, DiStefano JJ 3rd (2005) A predictive model of therapeutic monoclonal antibody dynamics and regulation by the neonatal Fc receptor (FcRn). Ann Biomed Eng 33:1640–1652
Davda JP, Jain M, Batra SK, Gwilt PR, Robinson DH (2008) A physiologically based pharmacokinetic (PBPK) model to characterize and predict the disposition of monoclonal antibody CC49 and its single chain Fv constructs. Int Immunopharmacol 8:401–413
Garg A (2007). Investigation of the role of FcRn in the absorption, distribution, and elimination of monoclonal antibodies, Chap. 3. PhD Thesis, Department of Pharmaceutical Sciences. 71–111
Williams LE, Wu AM, Yazaki PJ, Liu A, Raubitschek AA, Shively JE, Wong JY (2001) Numerical selection of optimal tumor imaging agents with application to engineered antibodies. Cancer Biother Radiopharm 16:25–35
Bazin-Redureau MI, Renard CB, Scherrmann JM (1997) Pharmacokinetics of heterologous and homologous immunoglobulin G, F(ab’)2 and Fab after intravenous administration in the rat. J Pharm Pharmacol 49:277–281
Hinton PR, Johlfs MG, Xiong JM, Hanestad K, Ong KC, Bullock C, Keller S, Tang MT, Tso JY, Vasquez M, Tsurushita N (2004) Engineered human IgG antibodies with longer serum half-lives in primates. J Biol Chem 279:6213–6216
Weisman MH, Moreland LW, Furst DE, Weinblatt ME, Keystone EC, Paulus HE, Teoh LS, Velagapudi RB, Noertersheuser PA, Granneman GR, Fischkoff SA, Chartash EK (2003) Efficacy, pharmacokinetic, and safety assessment of adalimumab, a fully human anti-tumor necrosis factor-alpha monoclonal antibody, in adults with rheumatoid arthritis receiving concomitant methotrexate: a pilot study. Clin Ther 25:1700–1721
Brown RP, Delp MD, Lindstedt SL, Rhomberg LR, Beliles RP (1997) Physiological parameter values for physiologically based pharmacokinetic models. Toxicol Ind Health 13:407–484
Davies B, Morris T (1993) Physiological parameters in laboratory animals and humans. Pharm Res 10:1093–1095
Swartz MA (2001) The physiology of the lymphatic system. Adv Drug Deliv Rev 50:3–20
Sarin H (2010) Physiologic upper limits of pore size of different blood capillary types and another perspective on the dual pore theory of microvascular permeability. J Angiogenes Res 2:14
Vaughn DE, Bjorkman PJ (1997) High-affinity binding of the neonatal Fc receptor to its IgG ligand requires receptor immobilization. Biochemistry 36:9374–9380
Hinton PR, Xiong JM, Johlfs MG, Tang MT, Keller S, Tsurushita N (2006) An engineered human IgG1 antibody with longer serum half-life. J Immunol 176:346–356
Datta-Mannan A, Witcher DR, Tang Y, Watkins J, Jiang W, Wroblewski VJ (2007) Humanized IgG1 variants with differential binding properties to the neonatal Fc receptor: relationship to pharmacokinetics in mice and primates. Drug Metab Dispos 35:86–94
Datta-Mannan A, Witcher DR, Tang Y, Watkins J, Wroblewski VJ (2007) Monoclonal antibody clearance. Impact of modulating the interaction of IgG with the neonatal Fc receptor. J Biol Chem 282:1709–1717
Chen Y, Balthasar JP (2010) A physiologically-based pharmacokinetic (PBPK) model for disposition of IgG with altered FcRn binding kinetics, AAPS Annual Meeting, New Orleans, p R6397
Emond C, Birnbaum LS, DeVito MJ (2006) Use of a physiologically based pharmacokinetic model for rats to study the influence of body fat mass and induction of CYP1A2 on the pharmacokinetics of TCDD. Environ Health Perspect 114:1394–1400
Dedrick RL, Bischoff KB (1980) Species similarities in pharmacokinetics. Fed Proc. 39:54–59
Johnson TN (2005) Modelling approaches to dose estimation in children. Br J Clin Pharmacol 59:663–669
Parrott N, Lave T (2002) Prediction of intestinal absorption: comparative assessment of GASTROPLUS and IDEA. Eur J Pharm Sci 17:51–61
Bischoff KB, Dedrick RL (1968) Thiopental pharmacokinetics. J Pharm Sci 57:1346–1351
Roopenian DC, Akilesh S (2007) FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol 7:715–725
Ghetie V, Hubbard JG, Kim JK, Tsen MF, Lee Y, Ward ES (1996) Abnormally short serum half-lives of IgG in beta 2-microglobulin-deficient mice. Eur J Immunol 26:690–696
Junghans RP, Anderson CL (1996) The protection receptor for IgG catabolism is the beta2-microglobulin-containing neonatal intestinal transport receptor. Proc Natl Acad Sci USA 93:5512–5516
Brambell FW (1969) The transmission of immune globulins from the mother to the foetal and newborn young. Proc Nutr Soc 28:35–41
Ober RJ, Martinez C, Vaccaro C, Zhou J, Ward ES (2004) Visualizing the site and dynamics of IgG salvage by the MHC class I-related receptor, FcRn. J Immunol 172:2021–2029
Montoyo HP, Vaccaro C, Hafner M, Ober RJ, Mueller W, Ward ES (2009) Conditional deletion of the MHC class I-related receptor FcRn reveals the sites of IgG homeostasis in mice. Proc Natl Acad Sci USA 106:2788–2793
Zhou J, Johnson JE, Ghetie V, Ober RJ, Ward ES (2003) Generation of mutated variants of the human form of the MHC class I-related receptor, FcRn, with increased affinity for mouse immunoglobulin G. J Mol Biol 332:901–913
Haymann JP, Levraud JP, Bouet S, Kappes V, Hagege J, Nguyen G, Xu Y, Rondeau E, Sraer JD (2000) Characterization and localization of the neonatal Fc receptor in adult human kidney. J Am Soc Nephrol 11:632–639
Kobayashi N, Suzuki Y, Tsuge T, Okumura K, Ra C, Tomino Y (2002) FcRn-mediated transcytosis of immunoglobulin G in human renal proximal tubular epithelial cells. Am J Physiol Renal Physiol 282:F358–F365
Borvak J, Richardson J, Medesan C, Antohe F, Radu C, Simionescu M, Ghetie V, Ward ES (1998) Functional expression of the MHC class I-related receptor, FcRn, in endothelial cells of mice. Int Immunol 10:1289–1298
Cauza K, Hinterhuber G, Dingelmaier-Hovorka R, Brugger K, Klosner G, Horvat R, Wolff K, Foedinger D (2005) Expression of FcRn, the MHC class I-related receptor for IgG, in human keratinocytes. J Invest Dermatol 124:132–139
Blumberg RS, Koss T, Story CM, Barisani D, Polischuk J, Lipin A, Pablo L, Green R, Simister NE (1995) A major histocompatibility complex class I-related Fc receptor for IgG on rat hepatocytes. J Clin Invest 95:2397–2402
Cianga P, Cianga C, Cozma L, Ward ES, Carasevici E (2003) The MHC class I related Fc receptor, FcRn, is expressed in the epithelial cells of the human mammary gland. Hum Immunol 64:1152–1159
Zhu X, Meng G, Dickinson BL, Li X, Mizoguchi E, Miao L, Wang Y, Robert C, Wu B, Smith PD, Lencer WI, Blumberg RS (2001) MHC class I-related neonatal Fc receptor for IgG is functionally expressed in monocytes, intestinal macrophages, and dendritic cells. J Immunol 166:3266–3276
Akilesh S, Christianson GJ, Roopenian DC, Shaw AS (2007) Neonatal FcR expression in bone marrow-derived cells functions to protect serum IgG from catabolism. J Immunol 179:4580–4588
Kandil E, Egashira M, Miyoshi O, Niikawa N, Ishibashi T, Kasahara M (1996) The human gene encoding the heavy chain of the major histocompatibility complex class I-like Fc receptor (FCGRT) maps to 19q13.3. Cytogenet Cell Genet 73:97–98
Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, Zhang J, Soden R, Hayakawa M, Kreiman G, Cooke MP, Walker JR, Hogenesch JB (2004) A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci USA 101:6062–6067
Israel EJ, Taylor S, Wu Z, Mizoguchi E, Blumberg RS, Bhan A, Simister NE (1997) Expression of the neonatal Fc receptor, FcRn, on human intestinal epithelial cells. Immunology 92:69–74
Haigler HT, McKanna JA, Cohen S (1979) Rapid stimulation of pinocytosis in human carcinoma cells A-431 by epidermal growth factor. J Cell Biol 83:82–90
Pease LF III, Elliott JT, Tsai DH, Zachariah MR, Tarlov MJ (2008) Determination of protein aggregation with differential mobility analysis: application to IgG antibody. Biotechnol Bioeng 101:1214–1222
Vugmeyster Y, DeFranco D, Szklut P, Wang Q, Xu X (2010) Biodistribution of [125I]-labeled therapeutic proteins: application in protein drug development beyond oncology. J Pharm Sci 99:1028–1045
Alley SC, Okeley NM, Senter PD (2010) Antibody-drug conjugates: targeted drug delivery for cancer. Curr Opin Chem Biol 14:529–537
Doppalapudi VR, Tryder N, Li L, Aja T, Griffith D, Liao FF, Roxas G, Ramprasad MP, Bradshaw C, Barbas CF 3rd (2007) Chemically programmed antibodies: endothelin receptor targeting CovX-Bodies. Bioorg Med Chem Lett 17:501–506
Stanfield RL, Dooley H, Verdino P, Flajnik MF, Wilson IA (2007) Maturation of shark single-domain (IgNAR) antibodies: evidence for induced-fit binding. J Mol Biol 367:358–372
Igawa T, Tsunoda H, Tachibana T, Maeda A, Mimoto F, Moriyama C, Nanami M, Sekimori Y, Nabuchi Y, Aso Y, Hattori K (2010) Reduced elimination of IgG antibodies by engineering the variable region. Protein Eng Des Sel 23:385–392
Boswell CA, Tesar DB, Mukhyala K, Theil FP, Fielder PJ, Khawli LA (2010) Effects of charge on antibody tissue distribution and pharmacokinetics. Bioconjug Chem 21:2153–2163
Ghetie V, Popov S, Borvak J, Radu C, Matesoi D, Medesan C, Ober RJ, Ward ES (1997) Increasing the serum persistence of an IgG fragment by random mutagenesis. Nat Biotechnol 15:637–640
Gurbaxani B, Dela Cruz LL, Chintalacharuvu K, Morrison SL (2006) Analysis of a family of antibodies with different half-lives in mice fails to find a correlation between affinity for FcRn and serum half-life. Mol Immunol 43:1462–1473
Yeung YA, Leabman MK, Marvin JS, Qiu J, Adams CW, Lien S, Starovasnik MA, Lowman HB (2009) Engineering human IgG1 affinity to human neonatal Fc receptor: impact of affinity improvement on pharmacokinetics in primates. J Immunol 182:7663–7671
Deng R, Loyet KM, Lien S, Iyer S, DeForge LE, Theil FP, Lowman HB, Fielder PJ, Prabhu S (2010) Pharmacokinetics of humanized monoclonal anti-tumor necrosis factor-{alpha} antibody and its neonatal Fc receptor variants in mice and cynomolgus monkeys. Drug Metab Dispos 38:600–605
Wang W, Lu P, Fang P, Hamuro L, Pittman T, Carr B, Hochman J, Prueksaritanont T (2011) Monoclonal antibodies with identical Fc sequences can bind to FcRn differentially with pharmacokinetic consequences. Drug Metab Dispos 39(9):1469–1477
Qiao SW, Kobayashi K, Johansen FE, Sollid LM, Andersen JT, Milford E, Roopenian DC, Lencer WI, Blumberg RS (2008) Dependence of antibody-mediated presentation of antigen on FcRn. Proc Natl Acad Sci USA 105:9337–9342
Acknowledgments
The author would like to acknowledge the scientific support from the State University of New York at Buffalo under the UB-Pfizer strategic alliance. Author would also like to thank Prof. Joseph P. Balthasar and his past and present lab members for the scientific discussions and impact on the presented work. The authors would also like to thank Hugh Barton and Craig Giragossian for critical review of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Appendix
Rights and permissions
About this article
Cite this article
Shah, D.K., Betts, A.M. Towards a platform PBPK model to characterize the plasma and tissue disposition of monoclonal antibodies in preclinical species and human. J Pharmacokinet Pharmacodyn 39, 67–86 (2012). https://doi.org/10.1007/s10928-011-9232-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10928-011-9232-2