
Abstract
Respiratory muscle contractile inactivity during mechanical ventilation (MV) induces diaphragm muscle weakness, a condition referred to as ventilator-induced diaphragmatic dysfunction (VIDD). Although VIDD pathophysiological mechanisms are still not fully understood, it has been recently suggested that remodeling of the sarcoplasmic reticulum (SR) calcium release channel/ryanodine receptors (RyR1) in the diaphragm is a proximal mechanism of VIDD. Here, we used piglets, a large animal model of VIDD that is more relevant to human pathophysiology, to determine whether RyR1 alterations are observed in the presence of diaphragm weakness. In piglets, diaphragm weakness induced by 72 h of respiratory muscle unloading was associated with SR RyR1 remodeling and abnormal resting SR Ca2+ leak in the diaphragm. Specifically, following controlled mechanical ventilation, diaphragm contractile function was reduced. Moreover, RyR1 macromolecular complexes were more oxidized, S-nitrosylated and phosphorylated at Ser-2844 and depleted of the stabilizing subunit calstabin1 compared with controls on adaptive support ventilation that maintains diaphragmatic contractile activity. Our study strongly supports the hypothesis that RyR1 is a potential therapeutic target in VIDD and the interest of using small molecule drugs to prevent RyR1-mediated SR Ca2+ leak induced by respiratory muscle unloading in patients who require controlled mechanical ventilation.
Similar content being viewed by others


Introduction
Mechanical ventilation (MV) is the most efficient treatment for respiratory failure in intensive care units (ICU). However, the respiratory muscle contractile inactivity induced by MV (i.e., the unloading of the respiratory muscle) has various adverse cellular effects that lead, in a time-dependent way, to diaphragm weakness, atrophy and injury (Powers et al. 2009; Levine et al. 2008; Jaber et al. 2011), a pathological condition known as ventilator-induced diaphragmatic dysfunction (VIDD) (Vassilakopoulos and Petrof 2004). The identification of the different cellular pathways involved in diaphragm weakness is important because VIDD increases the difficulties of MV weaning (Tobin et al. 2009), with a major impact on healthcare costs (Esteban et al. 2008).
Oxidative stress is considered to be a major pathophysiological mechanism of VIDD (Shanely et al. 2002; Falk et al. 2006; Zergeroglu et al. 2003; Betters et al. 2004), and mitochondria are an essential source of reactive oxygen species (ROS) (Betters et al. 2004; Picard et al. 2012; Kavazis et al. 2009). It is not clear why mitochondrial ROS production is increased following respiratory muscle unloading induced by MV. However, this energetic homeostasis imbalance with acute substrate oversupply is a potential source of mitochondrial dysfunction (Picard et al. 2012).
Recently, we showed that Ca2+ homeostasis impairment is involved in the structural and functional impairment of type 1 ryanodine receptors (RyR1) induced by oxidative stress (Matecki et al. 2016). RyRs are located in the sarcoplasmic reticulum (SR) and are responsible for the release of Ca2+ from this intracellular store during excitation–contraction (EC) coupling. RyR1-mediated Ca2+ release is modulated by regulatory proteins (for instance, the FK506 binding protein 1A, also known as calstabin1) that bind to its large cytoplasmic scaffold domain (Brillantes et al. 1994). Following protein kinase A (PKA)-mediated RyR1 phosphorylation and oxidation, the association between calstabin1 and the rest of the RyR1 complex is lost (Ward et al. 2003; Reiken et al. 2003), leading to leaky RyR1 and consequently to impaired Ca2+ handling and contractile dysfunction in many different conditions, such as VIDD (Matecki et al. 2016), heart failure (Ward et al. 2003), aging (2011), chronic muscle fatigue (Bellinger et al. 2008) and muscular dystrophy (Bellinger et al. 2009). This important cellular pathway could be considered as a proximal mechanism of the Ca2+-dependent proteolysis and muscle atrophy observed in different animal models of VIDD (Shanely et al. 2002; Maes et al. 2007; Kavazis et al. 2007; McClung et al. 2009; Jung et al. 2010) and in human patients (Levine et al. 2008; Jaber et al. 2011). This suggests that RyR1 is a potential therapeutic target in VIDD.
We recently reported that in patients hospitalized in ICU and mechanically ventilated for a mean period of 4 days, post-translational RyR1 modifications in the diaphragm are a proximal mechanism of VIDD (Matecki et al. 2016). However, the mechanisms involved in such modifications remain unclear. Indeed, several causes could be involved, such as catecholamine stress, sepsis, mechanical stress induced by patient-ventilator asynchrony, drugs used for sedation or respiratory muscle unloading. Using a murine model of VIDD, we established the link between respiratory muscle unloading and diaphragm force deficit (Matecki et al. 2016). However, mouse models of VIDD with the simple device used for mechanical ventilation (a syringe that pumps a constant level of air in the airways) are physiologically not comparable to human patients. Moreover, this model does not allow monitoring the diaphragm activity to confirm the lack of contraction at each breathing cycle.
In view of future clinical trials, it is mandatory to establish the link between respiratory muscle unloading and RyR1 dysfunction in an animal model that is physiologically similar to humans. Indeed, respiratory muscle unloading is the only putative cause of RyR1 dysfunction that cannot be removed when MV is used to efficiently ventilate lungs. Thus, a larger animal model of VIDD, in which diaphragm contractile activity can be monitored during MV with devices similar to those used in patients admitted in ICU, is required to determine the direct consequence of diaphragm contractile inactivity on RyR1 function. Using a piglet model of VIDD, we previously reported that total unloading of respiratory muscles during controlled mechanical ventilation (CMV) induces diaphragm weakness. This could be prevented by maintaining a certain level of diaphragmatic contractile activity using adaptive support ventilation (ASV) (Jung et al. 2010). Therefore, the aim of the present study was to evaluate in the same animal model, whether CMV for 3 days induced RyR1 remodeling and dysfunction. To specifically link RyR1 alteration to respiratory muscle unloading we tested whether RyR1 dysfunction could be prevented by ASV.
Methods
Piglet model
As previously described (Jung et al. 2010; Jaber et al. 2008), ten piglets (15–20 kg) were separated in two groups of five animals/each. In both groups, oxygenation was maintained with a fraction of inspired oxygen (FIO2) ranging from 25 to 35%, tidal volume between 10 and 12 ml kg−1 body weight and breathing frequency from 15 to 30 cycles/min to maintain normocapnia. In the first group (MV group), piglets were mechanically ventilated using CMV, with a positive end-expiratory pressure level at 5 cm H2O. The absence of spontaneous breathing was verified on the ventilator trend graphs, and the diaphragm electromyographic activity was measured to ensure the absence of diaphragm electrical activity. In the second group (control group), piglets were ventilated using ASV, with settings in phase with the piglet body weight (inspiratory flow trigger at 0.3 L min−1, percentage of mechanical ventilation between 100 and 150%, positive end-expiratory pressure level at 5 cm H2O, and expiratory trigger at 25% of the peak inspiratory flow). The diaphragm electromyographic activity was assessed to verify that piglets could breathe spontaneously. Both groups received the same care, except for the MV mode.
Briefly, piglets were anesthetized by intravenous injection of 5–6 mg kg−1 pentobarbital sodium, intubated with a cuffed endotracheal tube and mechanically ventilated (Galileo®; Hamilton Medical AG, Rhazuns, Switzerland). Anesthesia was maintained with continuous intravenous infusion of 15–20 mg kg−1 propofol, 0.1–0.3 mg kg−1 midazolam and 3–4 mg kg−1 h−1 ketamine. The level of sedation was monitored by using a bispectral index system (BIS®; Aspect, Norwood, MA). Heating pads were employed, as needed, to maintain the normal body temperature of 38.5–39.5 °C. A carotidal arterial catheter (PiCCO®; Pulsion, Munich, Germany) was inserted for monitoring the heart rate, arterial blood pressure and cardiac output. The absence of spontaneous breathing in the MV group was verified using the ventilator trend graphs and the diaphragm electromyographic activity measurement.
Arterial carbon dioxide pressure was monitored using a capnograph (Deltatrac®; Datex-Ohmeda, Helsinki, Finland) and verified by arterial blood gas analysis (iSTAT®; Abbott, Abbott Park, IL). Parenteral nutrition was given from the first day (10% glucose, 20% amino acids solution and Hyperamine 20®; Braun, Boulogne Billancourt, France) to provide 30–35 kcal kg−1 day−1. All procedures were performed aseptically. All animals received prophylactic intravenous injections of antibiotics three times per day (100 mg kg−1 day−1 amoxicillin–clavulanate).
The diaphragm contractile function was assessed in vivo by measuring the transdiaphragmatic pressure (Pdi) (Jung et al. 2010). A double-lumen air-filled balloon-tipped catheter was placed transorally in the stomach and the distal third of the esophagus. Bipolar transvenous pacing catheters were introduced through each internal jugular vein and adjusted to stimulate the phrenic nerve and subsequently induce the diaphragm contraction by supramaximal stimulation at frequencies ranging from 20 to 120 Hz with trains of stimulation of 2 s and 150 ms.
RyR1 biochemical analysis
Diaphragm muscle biopsies were homogenized in 150 µl of buffer containing 5% SDS, 5% beta-mercaptoethanol, 10% glycerol, 10mM EDTA and 50mM Tris/HCl buffer (pH 8.0). Each sample was immediately denatured at 90 °C for 4 min. After centrifugation (5000 rpm) at 4 °C, protein concentration in supernatants was measured in duplicate using the BCA protein assay. Samples were then diluted to the same concentration (2 µg/µl) with loading buffer and aliquoted. RyR1 was immunoprecipitated from 250 µg of protein homogenates using 4 µg of home-made anti-RyR antibody (RyR1-1327, see below for description) in 0.5 ml of modified RIPA buffer (50mM Tris–HCl pH 7.4, 0.9% NaCl, 5.0 mM NaF, 1.0 mM Na3VO4, 1% Triton-X100, and protease inhibitors) at 4 °C for 1h. Immune complexes were incubated with protein A Sepharose beads (Amersham Pharmacia) at 4 °C for 1h and beads were washed three times with buffer. Proteins were separated on 4–20% SDS–PAGE gradient gels and transferred onto nitrocellulose membranes at 200 mA for 2 h (SemiDry transfer blot, Bio-Rad). To prevent non-specific antibody binding, membranes were incubated with blocking solution (LICOR Biosciences) and washed with Tris-buffered saline/0.1% Tween-20.
Blots were incubated with the primary anti-RyR1 antibody RyR1-1327. This is an affinity-purified rabbit polyclonal antibody that was raised against a KLH-conjugated peptide with the amino acid sequence CAEPDTDYENLRRS that corresponds to residues 1327–1339 of mouse skeletal RyR1 and an additional cysteine residue added to the amino terminus and then affinity-purified with the unconjugated peptide. Antibodies against calstabin1 (1:2500 in blocking buffer, LICOR Biosciences), human RyR2 phosphorylated on Ser-2808 (1:5000), PKA-phosphorylated mouse RyR1 (on Ser-2844), and S-nitrosylated cysteine residues (1:1000, Sigma) were also used. To determine RyR1 oxidation, immunoprecipitates were incubated with 2, 4-dinitrophenyl hydrazine and the derivatized carbonyls were detected using the OxyBlot Protein Oxidation Detection Kit (catalog S7150, Chemicon International Inc.). After three washes, membranes were incubated with infrared-labeled secondary antibodies. Samples from MV and control piglets were added on each gel and after normalization, RyR1 total levels were not different between groups.
Sarcoplasmic reticulum vesicle preparation
Piglet diaphragm muscle biopsies were homogenized on ice in 300 mM sucrose, 20 mM PIPES (pH 7.0) in the presence of protease inhibitors (Roche) and centrifuged at 8000 rpm (5900g) at 4 °C for 20 min. Supernatants were then ultracentrifuged at 32,000 rpm (100,000g) at 4 °C for 1 h. The final pellets that contain microsomal fractions enriched in SR vesicles were resuspended and aliquoted in 300 mM sucrose, 5 mM PIPES (pH 7.0) with protease inhibitors. Samples were frozen in liquid nitrogen and stored at −80 °C.
Single channel data from planar lipid bilayer measurements
Planar lipid bilayers were formed from a 3:1 mixture of phosphatidylethanolamine and phosphatidylcholine (Avanti Polar Lipids, Alabaster, AL) suspended (30 mg/ml) in decane by painting the lipid/decane solution across a 200-µm aperture on one side of a polysulfonate cup (Warner Instruments) that separates two chambers. The trans chamber (1 ml), which represents the intra-SR (luminal) compartment, was connected to the headstage input of a bilayer voltage clamp amplifier (BC-525D, Warner Instruments) and the cis chamber (1 ml), which represents the cytoplasmic compartment, was held at virtual ground. Solutions in both chambers were as follows: 1mM EGTA, 250/125 mM HEPES/Tris, 50 mM KCl, 0.64 mM CaCl2, pH 7.35 (cis solution), and 53 mM Ca(OH)2, 50 mM KCl, 250mM HEPES, pH 7.35 (trans solution). The concentration of free Ca2+ in the cis chamber was calculated with the WinMaxC program (version 2.50; http://www.stanford.edu/~cpatton/maxc.html). SR vesicles were added to the cis side and fusion with the lipid bilayer was induced by making the cis side hyperosmotic by addition of 400–500 mM KCl. After the appearance of potassium and chloride channels, the cis compartment was perfused with the cis solution. Single-channel currents were recorded at 0 mV using a Bilayer Clamp BC-535 amplifier (Warner Instruments), filtered at 1 kHz and digitized at 4 kHz. All experiments were performed at room temperature. Data acquisition was performed with the Digidata 1440A and Axoscope 10.2 software programs and recordings were analyzed using Clampfit 10.2 (Molecular Devices). The open probability (Po) was calculated by 50% threshold analysis using a least 2 min of continuous recording, as previously described (Matecki et al. 2016, 2017; Bellinger et al. 2008). At the end of each experiment, 5 µM ryanodine was added to the cis chamber to confirm that the channels were RyRs.
Statistical analysis
Data are presented as the mean ± SEM. Differences between groups were analyzed with the ANOVA test. Statistical significance was set at p < 0.05.
Results
One limitation of using respiratory muscle biopsies from patient in ICU to investigate the pathophysiological mechanisms of VIDD is the potential influence of comorbid conditions and confounding factors associated with their critical condition. Therefore, to specifically determine the effect of respiratory muscle unloading on RyR1 properties and diaphragm function, we used diaphragm biopsies from healthy piglets that were anesthetized and ventilated using a controlled ventilation mode (MV group) or ASV (control group) for 72 h. In the MV group, as previously described (Jung et al. 2010), spontaneous diaphragm activity was observed for no more than 5% of all delivered breaths. Conversely, in the control group, 80% of breaths delivered by the ventilator were triggered by spontaneous contractile activity of the diaphragm. Throughout the study (3 days of CMV), animals were afebrile with normal intestinal transit and diuresis as well as normal fluid balance and stable hemodynamic parameters.
At the beginning of the ventilation (baseline), the maximal diaphragm strength, evaluated by measuring the maximal transdiaphragmatic pressure, was not significantly different in the MV and control group (49 ± 6 vs 43 ± 7 cm H20). In the control group (ASV mode), Pdi did not decrease at all tested frequencies between baseline and the end of the 3 days of ventilation (Fig. 1). This suggests that the ASV mode does not affect diaphragm function over 3 days. Conversely, in the MV group, Pdi was significantly lower after 3 days of MV compared with baseline (30 ± 5 versus 43 ± 6 cm H20, respectively; p < 0.05) (Fig. 1).

Diaphragmatic contractile function was assessed by in vivo measurements of transdiaphragmatic pressure (Pdi) at t = 0 (baseline) and t = 72 (72 h of anesthesia) in control (control_0 and control_72) and mechanically ventilated piglets (MV_0 and MV_72), produced by supramaximal stimulation and expressed as a function of the stimulation frequency. MV_0 Piglets on controlled mechanical ventilation for less than 3 h. MV_72 Piglets on controlled mechanical ventilation for 3 days. Control_0 Piglets on assisted support ventilation for less than 3 h. Control_72 Piglets on assisted support ventilation during 3 days (*p < 0.05, MV_72 vs. MV_0)
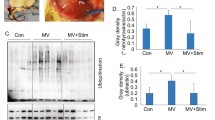
Then, to analyze the biochemical properties of the RyR1 macromolecular complex, SR fractions were purified from diaphragm muscle biopsies obtained after 72 h of ventilation in both groups. Western blot analysis after RyR1 immunoprecipitation showed that RyR1 S-nitrosylation, oxidation and phosphorylation on Ser-2844 as well as calstabin1 dissociation from RyR1 were significantly higher in MV samples compared with controls (ASV) (Fig. 2). This biochemical remodeling of RyR1 channels is known as the biochemical signature of “leaky” RyR1 channels (Bellinger et al. 2008) and is associated with a significant increase in RyR1 open probability (Po) measured in channels incorporated in planar lipid bilayers. Accordingly, compared with non-remodeled RyR1 channels (control group) that were tightly closed (Po = ~ 0) (Fig. 3), Po in the MV group increased to 0.021 ± 0.007 after 3 days of MV, suggesting increased SR Ca2+ leak.

Representative immunoblots of immunoprecipitated RyR1 from diaphragm muscle samples collected from control group (ventilated with ASV mode) and MV group (ventilated with CMV mode) after 72 h of ventilation (each blot corresponds to adjacent wells of the same gel) (left). Graph (right) shows the quantification relative to total RyR1 (mean ± SEM, n = 5 in both groups, *p < 0.05, MV vs. control). CysNO, thio-nitrosylation; DNP, 2,4-dinitrophenylhydrazone; P∗RyR1, RyR1 phosphorylated at serine 2844. Control and MV refer to samples collected at t = 72. Data are expressed as the mean ± SEM

Single channel traces of single RyR1 incorporated in planar lipid bilayers, corresponding to representative experiments performed with porcine diaphragm muscle biopsies from Control and MV animals after 72 h of ventilation. Respiratory muscle unloading increased RyR1 open probability (Po) value in MV group (n = 20 biopsies) compared with control group (n = 12 biopsies) (0.02130 ± 0.007519 and 0.00115 ± 0.00069, respectively; *p < 0.05 MV vs controls)
Discussion
This study shows that, in piglets mechanically ventilated with CMV, for 72 h, the diaphragm exhibit the biochemical signature of leaky RyR1 channels, suggesting intracellular Ca2+ leak, as shown by the increased mean channel RyR1 open probability, associated with a reduced diaphragm force production. By using piglets, an animal model physiologically similar to humans, and the same ventilation mode and anesthesia care routinely employed in ICU for patients with acute respiratory distress syndrome, we show here for the first time, that diaphragm inactivity, induced by CMV and associated with passive muscle fiber length changes, contributes to the RyR1 defect associated with diaphragm weakness. This demonstrates the link between respiratory muscle unloading after 3 days of CMV and RyR1 alteration associated with SR Ca2+ leak, which is considered to be the proximal mechanism of VIDD in ICU patients (Matecki et al. 2016).
Similar results were previously reported using mouse models of VIDD. Specifically, oxidative stress and catecholamine stress during MV have been recently involved in RyR1 dysfunction, and linked to diaphragm weakness in a murine model (Matecki et al. 2016). However, several limitations precluded the translation of this finding to the clinic, thus emphasizing the necessity to confirm, using a model closer to humans, that the RyR1 defect is linked to diaphragm unloading. Indeed, ventilators for mice are rudimentary, without any possibility to prove the lack of diaphragm contractile activity during mechanical ventilation. Moreover, in mice, the occurrence of asynchrony, defined as the mismatching between neural inspiratory and mechanical inspiratory time that induces episodic diaphragm contractions without air delivery, cannot be excluded (Sassoon and Foster 2001). As a consequence, the RyR1 defect observed in mechanically ventilated mice could have been caused mainly by the diaphragm mechanical stress caused by asynchrony, rather than unloading. Indeed, the presence of mechano-transduction elements that activates NADPH- oxidase 2 (Nox2)-dependent ROS generation during mechanical stress may also induce RyR1 defects (Ward et al. 2014). Moreover, this hypothesis is also supported by the beneficial effect of apocynin, a NADPH oxidase inhibitor, on diaphragm function in in rats undergoing MV (McClung et al. 2009). Finally, in the rodent model of VIDD, 18 h of MV is considered as protracted (Powers et al. 2009), and very few data are available on the effects of longer MV periods on the diaphragm due to the rudimentary ventilation techniques used and the few possibilities to monitor vital functions and nutritional status. These limitations may introduce significant confounding factors when evaluating the effects of MV.
In a previous study using a similar piglet model, 5 days of MV induced loss of diaphragm fiber stiffness, suggesting an increased number of “non-functional” myosin or actin molecules due to myofibrillar disassembly and myofiber injury (Ochala et al. 2011). This could be caused by activation of calpains or caspases, two Ca2+-dependent proteolysys systems that are upregulated in rat diaphragm after mechanical ventilation (Shanely et al. 2002; Maes et al. 2007; McClung et al. 2007). These and our present results strongly suggest that the SR Ca2+ leak observed in the MV group could be involved in myofibrillar remodeling linked to Ca2+-dependent proteolysis, ultimately leading to the muscle weakness observed in mechanically ventilated patients.
The finding by Ochala et al. (2011a, b) that in rats, calpain-1 activation is not increased before 9 days of MV could be in contradiction with our hypothesis on the early involvement of SR Ca2+ leakage and Ca2+-stimulated myofibrillar remodeling in VIDD. However, this late calpain-1 activation was observed in hindlimb muscle, a different model of muscle unloading compared with the diaphragm. Indeed, in hindlimbs, muscle ‘disuse’ is mainly induced by contractile inactivity during MV. On the other hand, the diaphragm is concomitantly mechanically unloaded, electrically quiescent and undergoing changes in myofiber length caused by the cyclical lung inflation or positive end-expiratory pressure. Moreover, the diaphragm is the only muscle exposed to a negative-pressure environment along its pleural surface that can potentially serve as a stretch-like hypertrophic stimulus (Yang et al. 1998), which is removed by the application of positive-pressure ventilation. In addition, the diaphragm is more active than most other skeletal muscles (30–40% of the time, 24 h/day). All these factors could explain the very rapid diaphragmatic muscle atrophy and force loss observed during MV. Indeed, it has been estimated that 12 h of MV in rats is approximately equivalent to 96 h of locomotor muscle unloading in terms of induced muscle wasting responses (Powers et al. 2007). In line with these considerations, in humans, atrophy, injury and activation of Ca2+-dependent proteolysys have been observed after a MV duration similar to the one used in the present study (Levine et al. 2008; Jaber et al. 2011).
The main objective of MV is to unload the respiratory muscle when the breathing work required to efficiently ventilate the lungs overcomes the performance of respiratory muscles. This unloading may be total (CMV) or partial (ASV). Previous studies have reported the beneficial effect of ventilation modes that only partially unload the diaphragm to prevent VIDD in small animal models (Gayan-Ramirez et al. 2005; Sassoon et al. 2004; Futier et al. 2008) and in a large animal model (Jung et al. 2010). However, the cellular mechanisms accounting for these beneficial effects are still poorly understood. We propose here, that during MV, the partial spontaneous activity of the diaphragm preserved in ASV might prevent RyR1 post-translational modifications induced by the total unloading of respiratory muscle. By only partially unloading the respiratory muscles, ASV might attenuate the energy homeostasis rupture and substrate oversupply to the mitochondria, described in VIDD (Picard et al. 2012). This beneficial effect could prevent mitochondrial ROS production that then leads to RyR1 modification, Ca2+ homeostasis rupture and activation of Ca2+-dependent proteolysis. This hypothesis needs now to be demonstrated experimentally.
Since VIDD description, clinicians have tried to develop alternative forms of MV, such as assist-control ventilation or pressure support ventilation, where a partial neuronal activation and mechanical activity of the diaphragm still exists. Although these new modes of ventilation support are now frequently used in the clinical practice (Esteban et al. 2000), few studies have evaluated their impact on the cellular pathways involved in VIDD. Our study brings new insights that might help understanding the beneficial effect on diaphragm function of these new MV modes that should be employed in ICU as often as possible to prevent VIDD. However, CMV is still commonly used in patients with acute respiratory distress syndrome in ICU, and cannot be efficiently replaced by ASV (Putensen et al. 2001). Thus, it is clearly in this particular context that clinical trials are needed to test small molecule drugs to stabilize RyR1 and counteract Ca2+ homeostasis defect.
We previously observed in humans after a similar period of CMV, the presence of diaphragm weakness associated with RyR1 defects (Jaber et al. 2011; Matecki et al. 2016). However, other mechanisms, including anesthetics, pain which could induce an overstimulation of the adrenergic response and consequently elevated levels of circulating endogenous catecholamines (Dunser and Hasibeder 2009; Boldt et al. 1995), have been associated with generalized myopathy in animal models (Ng et al. 2002) and could also explain these observations in patients on MV. The main advantage of the piglet model is the possibility to have a control group (ASV group) that only differs from the experimental model (MV group) by the level of diaphragm contractile activity. This was the only way to single out the unloading effect, in a similar context as observed in ICU. Thus, our study emphasizes the necessity of clinical trials to target specifically the unloading effect, the only parameters that cannot be removed, to prevent VIDD when CMV is necessary.
The main weakness of our study is the absence of any evidence on the causal link between diaphragm weakness and RyR1 defect. To make this link, the effect of small molecule drugs that stabilize RyR1 defect should be evaluated in this model, as previously done in the mouse model (Matecki et al. 2016).
In patients, diaphragm weakness induced by MV could have several other causes that might interfere with the unloading effect, such as metabolic disorders, sepsis, drug used for sedation, type of ventilation mode or comorbidity (Jung et al. 2010; Jaber et al. 2008; Sassoon et al. 2004; Divangahi et al. 2007; Testelmans et al. 2006). The relative contributions of each of these mechanisms of diaphragm weakness, which could cause failure to wean from the ventilator in ICU, are extremely difficult to evaluate in humans. Mechanically ventilated piglets represent a unique model that allows separating all these VIDD contributors, in a context very similar to clinical reality. Mechanically ventilated mice are an interesting tool to test easily new pharmacological treatments, or to better explore a particular cellular pathway involved in VIDD; however, larger animal models should be developed in parallel for pre-clinical studies of VIDD treatments. Indeed, RyR dysfunction and abnormal resting SR Ca2+ leak have been observed in different animal models and in mechanically ventilated human patients (Fig. 4). This abnormal pathway seems to be present since the early onset of VIDD with our without comorbidity and may participate to the mechanisms involved in muscular weakness.

RyR1 dysfunction is observed in the different animal models of VIDD and also in patients, independently of the mechanical ventilation (MV) duration and the presence of confounding factors
A previous study demonstrated in a mouse model, that preventing RyR1 leak with S107, a small molecule drug known to stabilize RyR1-calstabin1 interaction, can prevent MV-induced muscle weakness and muscle fiber atrophy in mice (Matecki et al. 2016). Our study strongly supports the interest of a clinical trial in patients who require artificial respiratory support, using similar a compound to prevent the RyR1-mediated SR Ca2+ leak induced by MV.
References
Andersson DC, Betzenhauser MJ, Reiken S, Meli AC, Umanskaya A, Xie W, Shiomi T, Zalk R, Lacampagne A, Marks AR (2011) Ryanodine receptor oxidation causes intracellular calcium leak and muscle weakness in aging. Cell Metab 14:196–207
Bellinger AM, Reiken S, Dura M, Murphy PW, Deng SX, Landry DW, Nieman D, Lehnart SE, Samaru M, LaCampagne A, Marks AR (2008) Remodeling of ryanodine receptor complex causes “leaky” channels: a molecular mechanism for decreased exercise capacity. Proc Natl Acad Sci USA 105:2198–2202
Bellinger AM, Reiken S, Carlson C, Mongillo M, Liu X, Rothman L, Matecki S, Lacampagne A, Marks AR (2009) Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat Med 15:325–330
Betters JL, Criswell DS, Shanely RA, Van Gammeren D, Falk D, Deruisseau KC, Deering M, Yimlamai T, Powers SK (2004) Trolox attenuates mechanical ventilation-induced diaphragmatic dysfunction and proteolysis. Am J Respir Crit Care Med 170:1179–1184
Boldt J, Menges T, Kuhn D, Diridis C, Hempelmann G (1995) Alterations in circulating vasoactive substances in the critically ill—a comparison between survivors and non-survivors. Intensive Care Med 21:218–225
Brillantes AB, Ondrias K, Scott A, Kobrinsky E, Ondriasova E, Moschella MC, Jayaraman T, Landers M, Ehrlich BE, Marks AR (1994) Stabilization of calcium release channel (ryanodine receptor) function by FK506-binding protein. Cell 77:513–523
Divangahi M, Demoule A, Danialou G, Yahiaoui L, Bao W, Xing Z, Petrof BJ (2007) Impact of IL-10 on diaphragmatic cytokine expression and contractility during Pseudomonas Infection. Am J Respir Cell Mol Biol 36:504–512
Dunser MW, Hasibeder WR (2009) Sympathetic overstimulation during critical illness: adverse effects of adrenergic stress. J Intensive Care Med 24:293–316
Esteban A, Anzueto A, Alia I, Gordo F, Apezteguia C, Palizas F, Cide D, Goldwaser R, Soto L, Bugedo G, Rodrigo C, Pimentel J, Raimondi G, Tobin MJ (2000) How is mechanical ventilation employed in the intensive care unit? An international utilization review. Am J Respir Crit Care Med 161:1450–1458
Esteban A, Ferguson ND, Meade MO, Frutos-Vivar F, Apezteguia C, Brochard L, Raymondos K, Nin N, Hurtado J, Tomicic V, Gonzalez M, Elizalde J, Nightingale P, Abroug F, Pelosi P, Arabi Y, Moreno R, Jibaja M, D’Empaire G, Sandi F, Matamis D, Montanez AM, Anzueto A (2008) Evolution of mechanical ventilation in response to clinical research. Am J Respir Crit Care Med 177:170–177
Falk DJ, Deruisseau KC, Van Gammeren DL, Deering MA, Kavazis AN, Powers SK (2006) Mechanical ventilation promotes redox status alterations in the diaphragm. J Appl Physiol 101:1017–1024
Futier E, Constantin JM, Combaret L, Mosoni L, Roszyk L, Sapin V, Attaix D, Jung B, Jaber S, Bazin JE (2008) Pressure support ventilation attenuates ventilator-induced protein modifications in the diaphragm. Crit Care 12:R116
Gayan-Ramirez G, Testelmans D, Maes K, Racz GZ, Cadot P, Zador E, Wuytack F, Decramer M (2005) Intermittent spontaneous breathing protects the rat diaphragm from mechanical ventilation effects. Crit Care Med 33:2804–2809
Jaber S, Jung B, Sebbane M, Ramonatxo M, Capdevila X, Mercier J, Eledjam JJ, Matecki S (2008) Alteration of the piglet diaphragm contractility in vivo and its recovery after acute hypercapnia. Anesthesiology 108:651–658
Jaber S, Petrof BJ, Jung B, Chanques G, Berthet JP, Rabuel C, Bouyabrine H, Courouble P, Koechlin-Ramonatxo C, Sebbane M, Similowski T, Scheuermann V, Mebazaa A, Capdevila X, Mornet D, Mercier J, Lacampagne A, Philips A, Matecki S (2011) Rapidly progressive diaphragmatic weakness and injury during mechanical ventilation in humans. Am J Respir Crit Care Med 183:364–371
Jung B, Constantin JM, Rossel N, Le Goff C, Sebbane M, Coisel Y, Chanques G, Futier E, Hugon G, Capdevila X, Petrof B, Matecki S, Jaber S (2010) Adaptive support ventilation prevents ventilator-induced diaphragmatic dysfunction in piglet: an in vivo and in vitro study. Anesthesiology 112:1435–1443
Kavazis AN, DeRuisseau KC, McClung JM, Whidden MA, Falk DJ, Smuder AJ, Sugiura T, Powers SK (2007) Diaphragmatic proteasome function is maintained in the ageing Fisher 344 rat. Exp Physiol 92:895–901
Kavazis AN, Talbert EE, Smuder AJ, Hudson MB, Nelson WB, Powers SK (2009) Mechanical ventilation induces diaphragmatic mitochondrial dysfunction and increased oxidant production. Free Radic Biol Med 46:842–850
Levine S, Nguyen T, Taylor N, Friscia ME, Budak MT, Rothenberg P, Zhu J, Sachdeva R, Sonnad S, Kaiser LR, Rubinstein NA, Powers SK, Shrager JB (2008) Rapid disuse atrophy of diaphragm fibers in mechanically ventilated humans. N Engl J Med 358:1327–1335
Maes K, Testelmans D, Powers S, Decramer M, Gayan-Ramirez G (2007) Leupeptin inhibits ventilator-induced diaphragm dysfunction in rats. Am J Respir Crit Care Med 175:1134–1138
Matecki S, Dridi H, Jung B, Saint N, Reiken SR, Scheuermann V, Mrozek S, Santulli G, Umanskaya A, Petrof BJ, Jaber S, Marks AR, Lacampagne A (2016) Leaky ryanodine receptors contribute to diaphragmatic weakness during mechanical ventilation. Proc Natl Acad Sci USA 113:9069–9074
McClung JM, Kavazis AN, DeRuisseau KC, Falk DJ, Deering MA, Lee Y, Sugiura T, Powers SK (2007) Caspase-3 regulation of diaphragm myonuclear domain during mechanical ventilation-induced atrophy. Am J Respir Crit Care Med 175:150–159
McClung JM, Van Gammeren D, Whidden MA, Falk DJ, Kavazis AN, Hudson MB, Gayan-Ramirez G, Decramer M, DeRuisseau KC, Powers SK (2009) Apocynin attenuates diaphragm oxidative stress and protease activation during prolonged mechanical ventilation. Crit Care Med 37:1373–1379
Ng Y, Goldspink DF, Burniston JG, Clark WA, Colyer J, Tan LB (2002) Characterisation of isoprenaline myotoxicity on slow-twitch skeletal versus cardiac muscle. Int J Cardiol 86:299–309
Ochala J, Gustafson AM, Diez ML, Renaud G, Li M, Aare S, Qaisar R, Banduseela VC, Hedstrom Y, Tang X, Dworkin B, Ford GC, Nair KS, Perera S, Gautel M, Larsson L (2011a) Preferential skeletal muscle myosin loss in response to mechanical silencing in a novel rat intensive care unit model: underlying mechanisms. J Physiol 589:2007–2026
Ochala J, Renaud G, Llano Diez M, Banduseela VC, Aare S, Ahlbeck K, Radell PJ, Eriksson LI, Larsson L (2011b) Diaphragm muscle weakness in an experimental porcine intensive care unit model. PLoS One 6:e20558
Picard M, Jung B, Liang F, Azuelos I, Hussain S, Goldberg P, Godin R, Danialou G, Chaturvedi R, Rygiel K, Matecki S, Jaber S, Des Rosiers C, Karpati G, Ferri L, Burelle Y, Turnbull DM, Taivassalo T, Petrof BJ (2012) Mitochondrial dysfunction and lipid accumulation in the human diaphragm during mechanical ventilation. Am J Respir Crit Care Med 186:1140–1149
Powers SK, Kavazis AN, McClung JM (2007) Oxidative stress and disuse muscle atrophy. J Appl Physiol (1985) 102:2389–2397
Powers SK, Kavazis AN, Levine S (2009) Prolonged mechanical ventilation alters diaphragmatic structure and function. Crit Care Med 37:S347–S353
Putensen C, Zech S, Wrigge H, Zinserling J, Stuber F, Von Spiegel T, Mutz N (2001) Long-term effects of spontaneous breathing during ventilatory support in patients with acute lung injury. Am J Respir Crit Care Med 164:43–49
Reiken S, Lacampagne A, Zhou H, Kherani A, Lehnart SE, Ward C, Huang F, Gaburjakova M, Gaburjakova J, Rosemblit N, Warren MS, He KL, Yi GH, Wang J, Burkhoff D, Vassort G, Marks AR (2003) PKA phosphorylation activates the calcium release channel (ryanodine receptor) in skeletal muscle: defective regulation in heart failure. J Cell Biol 160:919–928
Sassoon CS, Foster GT (2001) Patient-ventilator asynchrony. Curr Opin Crit Care 7:28–33
Sassoon CS, Zhu E, Caiozzo VJ (2004) Assist-control mechanical ventilation attenuates ventilator-induced diaphragmatic dysfunction. Am J Respir Crit Care Med 170:626–632
Shanely RA, Zergeroglu MA, Lennon SL, Sugiura T, Yimlamai T, Enns D, Belcastro A, Powers SK (2002) Mechanical ventilation-induced diaphragmatic atrophy is associated with oxidative injury and increased proteolytic activity. Am J Respir Crit Care Med 166:1369–1374
Testelmans D, Maes K, Wouters P, Gosselin N, Deruisseau K, Powers S, Sciot R, Decramer M, Gayan-Ramirez G (2006) Rocuronium exacerbates mechanical ventilation-induced diaphragm dysfunction in rats. Crit Care Med 34:3018–3023
Tobin MJ, Laghi F, Brochard L (2009) Role of the respiratory muscles in acute respiratory failure of COPD: lessons from weaning failure. J Appl Physiol 107:962–970
Vassilakopoulos T, Petrof BJ (2004) Ventilator-induced diaphragmatic dysfunction. Am J Respir Crit Care Med 169:336–341
Ward CW, Reiken S, Marks AR, Marty I, Vassort G, Lacampagne A (2003) Defects in ryanodine receptor calcium release in skeletal muscle from post-myocardial infarct rats. Faseb J 17:1517–1519
Ward CW, Prosser BL, Lederer WJ (2014) Mechanical stretch-induced activation of ROS/RNS signaling in striated muscle. Antioxid Redox Signal 20:929–936
Yang L, Bourdon J, Gottfried SB, Zin WA, Petrof BJ (1998) Regulation of myosin heavy chain gene expression after short-term diaphragm inactivation. Am J Physiol 274:L980–L989
Zergeroglu MA, McKenzie MJ, Shanely RA, Van Gammeren D, DeRuisseau KC, Powers SK (2003) Mechanical ventilation-induced oxidative stress in the diaphragm. J Appl Physiol 95:1116–1124
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Matecki, S., Jung, B., Saint, N. et al. Respiratory muscle contractile inactivity induced by mechanical ventilation in piglets leads to leaky ryanodine receptors and diaphragm weakness. J Muscle Res Cell Motil 38, 17–24 (2017). https://doi.org/10.1007/s10974-017-9464-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10974-017-9464-x