
Abstract
Molecular imaging using radioisotope- or fluorophore-labeled antibodies is increasingly becoming a critical component of modern precision medicine. Yet despite this promise, the vast majority of these immunoconjugates are synthesized via the random coupling of amine-reactive bifunctional probes to lysines within the antibody, a process that can result in heterogeneous and poorly defined constructs with suboptimal pharmacological properties. In an effort to circumvent these issues, the last 5 years have played witness to a great deal of research focused on the creation of effective strategies for the site-specific attachment of payloads to antibodies. These chemoselective modification methods yield immunoconjugates that are more homogenous and better defined than constructs created using traditional synthetic approaches. Moreover, site-specifically labeled immunoconjugates have also been shown to exhibit superior in vivo behavior compared to their randomly modified cousins. The over-arching goal of this two-part review is to provide a broad yet detailed account of the various site-specific bioconjugation approaches that have been used to create immunoconjugates for positron emission tomography (PET), single photon emission computed tomography (SPECT), and fluorescence imaging. In Part 1, we covered site-specific bioconjugation techniques based on the modification of cysteine residues and the chemoenzymatic manipulation of glycans. In Part 2, we will detail two families of bioconjugation approaches that leverage biochemical tools to achieve site-specificity. First, we will discuss modification methods that employ peptide tags either as sites for enzyme-catalyzed ligations or as radiometal coordination architectures. And second, we will examine bioconjugation strategies predicated on the incorporation of unnatural or non-canonical amino acids into antibodies via genetic engineering. Finally, we will compare the advantages and disadvantages of the modification strategies covered in both parts of the review and offer a brief discussion of the overall direction of the field.
Similar content being viewed by others


Introduction
Over the past few decades, antibodies and antibody fragments have emerged as extremely effective tumor-targeting vectors for molecular imaging (Fig. 1) [1–5]. However, the bioconjugation strategies used to synthesize these immunoconjugates leave much to be desired. The vast majority of traditional bioconjugation approaches rely on reactions between lysines in the antibody and bifunctional, amine-reactive chelators or fluorophores (e.g., benzyl isothiocyanates or N-hydroxysuccinimidyl esters; Fig. 2a–b) [6, 7]. Critically, the presence of multiple lysines distributed throughout the immunoglobulin structure makes controlling the site and frequency of these conjugation reactions impossible. This leads—inevitably and unfortunately—to the creation of constructs that are heterogeneous and poorly defined. To wit, a traditionally synthesized immunoconjugate with a degree of labeling of 3 cargoes/mAb is, in reality, a complex mixture of ~10,000 regioisomers, each with its own chemical, biological, and pharmacological properties [8–10]. Understandably, this remarkable heterogeneity can have detrimental effects on the in vitro and in vivo performance of the immunoconjugates both in the laboratory and in the clinic. In order to circumvent these issues, an increasing amount of effort has been dedicated to the development of techniques for the site-specific bioconjugation of cargoes to antibodies [8, 11–13]. While a variety of different approaches have been developed, all of them provide a route to better defined and more homogeneous immunoconjugates. It is important to note that this quest for chemoselectivity is not an academic issue, as a number of studies have shown that site-specifically modified immunoconjugates exhibit improved in vivo behavior compared to their traditionally synthesized cousins [14–17].

Detailed structural schematic of a full length IgG as well as an assortment of antibody fragments.

The basic chemical reactions underpinning the bioconjugation strategies discussed in this work.
Our over-arching goal in writing this two-part review is to provide the reader with a broad yet detailed guide to the different bioconjugation methods that have been applied to the creation of site-specifically labeled immunoconjugates for positron emission tomography (PET), single photon emission computed tomography (SPECT), and fluorescence imaging. In Part 1, we discussed site-specific bioconjugation approaches predicated on the modification of cysteine residues and the manipulation of the heavy chain glycans. In Part 2, we will shift gears and focus on two types of site-specific bioconjugation strategies that rely heavily on enzymatic transformations. First, we will address a family of modification methods that are bound by a common theme: the exploitation of peptide tags as either recognition sites for enzymatic ligations or as coordination scaffolds for the chelation of radiometals. Subsequently, we will turn our attention to modification strategies based upon the incorporation of unnatural or non-canonical amino acids (uAA and ncAA, respectively) into immunoglobulins via genetic engineering. In addition, at the end of this installment of the review, we will discuss the advantages and disadvantages of the approaches to bioconjugation covered in Parts 1 and 2 and offer our humble thoughts on the direction of the field as a whole.
Finally, before we begin, we feel compelled to make two brief logistical notes. First, as the title of the work suggests, this review is focused primarily on immunoconjugates for molecular imaging. However, we have found a number of reports in which interesting and effective site-specific bioconjugation strategies have been used to create antibody-drug conjugates (ADCs) or radioimmunotherapeutics rather than constructs for PET, SPECT, or optical imaging. Strictly speaking, these works lie outside the scope of this review. However, in these cases, we have chosen to include them here in order to increase the breadth of this work and, hopefully, facilitate the use of these approaches in the synthesis of immunoconjugates for molecular imaging. Second, given the rapid growth and burgeoning interest in this field, it is all but inevitable that we have inadvertently missed at least one of the many publications in this area, possibly more. To the authors of these works, we offer our apologies and our earnest assurance that no slight was intended.
Peptide Tags
This next section of the review marks a significant change in course. In Part 1, the site-specific conjugation strategies discussed involved no protein engineering or, at most, changes to single amino acids within the antibody structure. However, the ability to insert peptide tags or recognition sites into antibodies and antibody fragments opens up a new realm of possibilities (Fig. 3). While a few of these strategies have yet to be applied to in vivo imaging agents, we have included them here in order to encourage their application to immunoconjugates for PET, SPECT, and fluorescence imaging. Finally, a brief note to the reader: for the sake of clarity, this section has been divided into sub-sections based on the various types of modification strategies. Admittedly, this has resulted in a somewhat choppy narrative; however, we believe it best serves the educational purpose of the review.

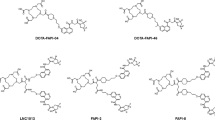
Selected chelators and cargoes used in the site-specifically labeled immunoconjugates discussed in this work.
Methods Based on Enzymatic Modifications
Transglutaminase
Transglutaminases (TGase) are enzymes that catalyze the creation of isopeptide bonds between primary amines and the acyl functionality of glutamine residues (Fig. 3). Given this ability, it is not surprising that this family of enzymes has attracted attention as tools for bioconjugation. Most notably, Schibli and coworkers at ETH Zurich have exploited TGases for chemoenzymatic antibody conjugations [18]. Working primarily with microbial transglutaminase (mTGase), the researchers have found that while antibodies possess many glutamine residues, only aglycosylated or deglycosylated antibodies can be functionalized appreciably using mTGase [19, 20]. The deglycosylation of the antibody at the N297 position increases the flexibility of the peptide backbone, exposing Q295 for reaction with the enzyme. This process was used to graft a cadaverine-bearing variant of the Cy3 fluorophore to the L1CAM-targeting antibody chCE7 for in vitro fluorescence microscopy (Fig. 4; Fig. 5a) [20]. In subsequent investigations, the laboratory used mTGase to modify variants of chCE7 and rituximab that had been enzymatically deglycosylated with PNGaseF (chCE7degl and RTXdegl). Two different chelators, desferrioxamine and a cadaverine-bearing analogue of CPTA, were employed, and chelator/mAb ratios of ~2 were obtained (Fig. 4) [19]. In an elegant twist, the authors also developed a mutant chCE7 (chCE7agl) in which the N297 residues were replaced with glutamines, abrogating glycosylation and providing two additional sites for mTGase modification in a single stroke (Fig. 5b ). This mutant antibody was then successfully modified to create immunoconjugates with degrees of labeling of ~4 chelators/mAb. Subsequent in vivo studies using mice bearing SKOV3ip xenografts revealed that site-specifically labeled 67Ga-DFO-chCE7agl exhibits significantly improved tumoral uptake and tumor-to-background activity ratios than a randomly conjugated [67Ga]DFO-chCE7agl analogue, and PET imaging data obtained using [89Zr]DFO-chCE7agl further supported these claims (Fig. 3).

Site-specific bioconjugation strategies based on the use of peptide tags.

Schematics of transglutaminase-based strategies for the site-specific modification of a deglycosylated antibodies, b aglycosylated antibodies, and c immunoglobulins bearing an LLQG transglutaminase recognition sequence.
An alternative—though admittedly more complex—strategy to the use of the endogenous Q295 site is the incorporation of a specific, engineered recognition site for enzymatic labeling. For example, Kamiya et al. engineered a GGSPLAQSHGGS tag onto the N-terminus of an scFv of the anti-hen egg-white lysozyme antibody and were thus able to use mTGase to effectively conjugate various proteins to the engineered fragment [21]. Using a similar approach, Strop et al. engineered LLQG mTGase recognition tags into several different sites in the anti-M1S1 antibody C16 in order to probe the influence of conjugation site on pharmacokinetics (Fig. 5c) [22, 23]. This method allowed them to site-specifically graft an amine-bearing MMAD construct as well as cadaverine-modified fluorophores (AlexaFluor® 350 and AlexaFluor® 488) to the engineered antibodies (Fig. 4). Ultimately, the authors found that the molecular location of the bioconjugation site exerts a very strong influence on the in vivo stability, toxicity, and efficacy of antibody-drug conjugates, though the causal relationships underlying this phenomenon remain unclear.
Sortase
Sortases (SrtA) are a family of Ca2+-dependent transpeptidases that cleave peptides at a specific LPXTG (X = D, E, A, N, Q, or K) motif and catalyze the formation of a peptidic bond between the cleaved peptide and a glycine-bearing substrate. Consequently, sortases have been employed to create a number of different fluorophore-labeled antibodies and antibody fragments [24–27]. For example, in the work of Madej et al., the enzyme cleaved between the glycine and threonine residues of an LPETGG tag sequence on an EGFR-targeting scFv and subsequently catalyzed the formation of a peptide bond between the exposed threonine and a GGG-bearing fluorescein (Fig. 4) [25]. More recently, Donnelly and coworkers employed SrtA to site-specifically append a GGG-tagged MeCOSar sarcophagine chelator for Cu-64 to an anti-LIBS scFv bearing a C-terminal LPETGG-FLAG tag (Figs. 4 and 6) [28]. In 2013, a particularly creative SrtA-based conjugation strategy was developed to reduce the number of purification steps required to isolate a final conjugate. In this approach, a fusion protein is created in which the protein of interest is attached to a (His)6-tagged variant of SrtA by a flexible linker that includes the LPXTG recognition motif [29]. After the immobilization of the protein-LPXTG-linker-SrtA-(His)6 construct on a solid support, Ca2+ and a GGG-tagged cargo are added, prompting the site-specific modification of the protein of interest and the consequent release of the protein-cargo conjugate, ultimately leaving the His6-tagged SrtA enzyme attached to the solid support.

Small animal PET/CT images collected 60 min after the injection of a site-specifically labeled scFvanti-LIBS-LPET-[64Cu]L2 (left) and a control fragment (scFvmut-LPET-[64Cu]L2; right) into a mouse model of carotid artery thrombosis. Figure adapted and reprinted with permission of [28].
Polypeptide-α-N-Acetylgalactosaminyltransferase
Somewhat ironically, peptide tags have also been used to site-specifically introduce glycosylation sites into antibodies. Along these lines, Qasba and coworkers pioneered a method in which a peptide tag containing a recognition site for a substrate-permissive glycoysyltransferase called polypeptide-α-N-acetylgalactosaminyltransferase (ppGalNAc-T2) is appended to the C-terminus of a non-glycosylated protein. This method can be used to attach the galactose derivatives GalNAz and 2-keto-Gal to threonine or serine residues of a recognition peptide tag [30]. In practice, this approach was used to site-specifically modify an anti-HER2 scFv with 2-keto-Gal and, subsequently, an aminooxy-bearing variant of AlexaFluor® 488 for in vitro fluorescence microscopy (Fig. 2c; Fig. 4) [31]. Given the presence of the heavy chain glycans, this strategy is clearly not particularly useful with intact IgGs; however, it could prove to be an attractive alternative for the creation of antibody fragment-based imaging agents.
Lysyl Endopeptidase
Proteases are most often associated with cleaving peptide bonds, but the enzymes can create them as well. Over 20 years ago, Fisch, et al. exploited this phenomenon to create a site-specifically labeled F(ab′)2-like immunoconjugate [32]. Specifically, the authors used a protease called lysyl endopeptidase, an enzyme capable of digesting antibodies by cleaving polypeptide chains on the carboxyl side of lysine residues. In this case, the enzyme was used to digest the chimeric anti-TAG72 antibody B72.3 via the cleavage of the heavy chain CH2 domain at either Lys240 or Lys242, ultimately creating a F(ab′)2-like fragment. This fragment was then coupled to a carbohydrazide moiety via the reverse proteolysis reaction catalyzed by the same enzyme. Finally, the hydrazide-bearing construct was conjugated to an aldehyde-modified 55Fe-ferrioxamine through a hydrazone linkage, and the Fe-55 labeled fragment was shown to have immunoreactivity nearly equivalent to the unmodified fragment (Fig. 2d).
Trypsiligase
In 2014, Liebscher, et al. applied a very similar concept to the development of a site-specifically labeled anti-HER2 fragment [33]. In this work, the authors designed an anti-HER2, trastuzumab-derived Fab fragment with a YRH recognition site appended to the C-terminus of the heavy chain. Using a trypsiligase enzyme designed to recognize the YRH sequence, the recombinant antibody fragment was conjugated to fluorescein and coumarin fluorophores bearing RHAK tags through the creation of peptide bonds (Fig. 4). While the authors did not report any in vitro or in vivo imaging using their fluorophore-labeled fragments, the site-specifically modified constructs exhibited almost identical binding affinity for HER2 as the unmodified fragment.
Methods Based on Self-Labeling Antibody-Enzyme Fusion Proteins
All of these peptide tag-based conjugation methodologies require modification reactions with enzymes and, consequently, purification steps after enzymatic treatment. One way to circumvent these purification steps is to genetically engineer fusion proteins in which the protein-of-interest and the modification enzyme are combined.
SNAP-tag
SNAP-tag is a widely employed 20 kDa mutant of O6-alkylguanine transferase (AGT) that catalyzes the transfer of an alkyl group from a guanidine substrate to a cysteine residue in its active site [34]. This technology was recently used to site-specifically attach a chlorin e6 (Ce6) photosensitizer to scFv-425, an anti-EGFR scFv (Fig. 4) [35]. While the authors successfully illustrated that the homogeneous scFv-425-Ce6 conjugate specifically binds EGFR-expressing cells and promotes tumor cell-specific cytotoxicity, they fail to adequately address the influence that the addition of a 20 kDa SNAP-tag moiety could have on the in vivo pharmacokinetics of the ~28 kDa scFv.
Inteins
The use of intein tags provides a way to take advantage of fusion protein conjugation approaches while avoiding the permanent attachment of a large enzyme to the immunoconjugate. Inteins, in short, are sections of proteins capable of excising themselves and ligating the remaining portions of the polypeptide to one another through a peptide bond. Generally speaking, this process is called protein splicing. In 2011, Möhlmann and colleagues creatively applied this strategy to the creation of site-specifically labeled conjugates of the anti-fibronectin antibody ED-B [26]. The authors created ED-B variants with either the GryA intein or the DnaE intein attached to the C-termini of the CH3 domains. In the ED-B-GyrA variant, an intein-catalyzed N-S acyl shift was followed by treatment with sodium 2-mercaptoethane sulfonate, incubation with a cysteine-bearing biotin derivative, and a subsequent S-N acyl shift to produce a site-specifically labeled ED-B-biotin conjugate with a degree of labeling of ~2 (Fig. 4). The antibody bearing the DnaE intein presents a slightly more complicated case (see Fig. 7 for a detailed schematic). Following a very similar approach, an anti-RhoB scFv was engineered with the GyrA intein at its C-terminus and subsequently modified with a cysteine-biotin-fluorescein construct [36]. While these strategies are elegant, their inherent complexity may ultimately limit their wide-scale use.

Diagram of the DNAE intein-based site-specific modification procedure developed by Möhlmann, et al. Figure adapted and reprinted with permission of [26].
Methods Based on In Vitro Translational Modifications
In yet another group of strategies, protein engineering methods can be harnessed to incorporate recognition and bioconjugation sites into immunoglobulins during translation.
Engineering for Intracellular Glycosylation
In one example, Leung, et al. successfully identified a natural asparagine glycosylation site with an NVT sequence in the framework-1 (FR-1) region of the VL domain of the murine anti-B cell lymphoma antibody LL-2. Subsequently, the authors genetically introduced this tripeptide glycosylation sequence into the FR-1 region of a non-glycosylated, humanized anti-carcinoembryonic antigen (CEA) antibody, hMN-14 [37]. This protein engineering resulted in the in vitro glycosylation of the antibody, as illustrated using SDS-PAGE. After isolation of the glycosylated hMN-14 antibody, a F(ab′)2 fragment was created, and then a NaIO4-based sugar oxidation strategy was used to attach an amine-bearing variant of DTPA to the fragment (Fig. 2e-f). In this way, the authors obtained degrees of labeling of ~2 DTPA per F(ab′)2 and were able to label the fragment efficiently with Y-90 and In-111 (Fig. 4). Importantly, neither the glycosylation nor the conjugation steps decreased the immunoreactivity of the F(ab′)2. However, no in vivo data for the site-specifically radiolabeled hMN-14 F(ab′)2 were presented in this initial report or subsequent publications.
Formylglycine-Generating Enzymes
Bertozzi and Rabuka have developed a related strategy based on the site-specific introduction of CxPxR recognition sites for formylglycine-generating enzyme (FGE). After the synthesis of the target protein in E. coli or mammalian cells, FGE oxidizes the cysteine residue of the recognition motif to create a formylglycine moiety on the protein of interest. The aldehyde of this formylglycine can then be exploited for conjugation with aminooxy-labeled substrates (Fig. 2 g) [38]. In related work, the same researchers developed the Pictet-Spengler and hydrazino-Pictet-Spengler (HIPS) ligations, reactions which create more stable linkages than hydrazones and oximes (Fig. 2h–i) [39]. The laboratory-engineered trastuzumab antibodies with CxPxR motifs at various locations, thereby creating a variety of immunoconjugates with aldehyde tags at specific sites [40]. Subsequently, the HIPS ligation was used to append the chemotherapeutic maytansin to the antibodies in order to investigate the role of conjugation site on the in vivo efficacy and pharmacokinetic profiles of the immunoconjugates, with the authors demonstrating that the site of bioconjugation exerts significant influence on the in vivo efficacy and pharmacokinetic behavior of antibody-drug conjugates (ADCs).
Protein Kinases
Another strategy unique to the labeling of immunoconjugates with P-32 (t 1/2 ~ 14.2 days) for therapeutic applications is the application of protein kinases. The use of these enzymes with proteins lacking native phosphorylation sites—such as antibodies and antibody fragments—requires the creation of a kinase substrate site to which the enzyme can transfer the γ-phosphate from [32P]ATP. The introduction of this recognition site, ranging from 5 to 15 amino acids depending on the specific kinase used, has been achieved using two strategies: the creation of antibody-based fusion proteins and the genetic engineering of the immunoglobulin. Both of these approaches are discussed in great detail in an excellent review dedicated to the topic [41]. In one example, Patrick et al. engineered an anti-CEA scFv with a LRRASG kinase recognition site and subsequently used protein kinase A to label the fragment with 32P-32 [42]. The authors found that the site-specifically phosphorylated construct retained the selectivity of its parent fragment and, in in vitro assays, exhibited selective cytotoxicity and a high internalization rate in CEA-expressing LS174T cells.
Metal-Coordinating Peptide Tags
Up until now, the peptide tags discussed have all served as recognition sites for enzymes. This is not, however, the only way that peptide tags can facilitate the creation of site-specifically modified conjugates. Indeed, a number of laboratories have used peptide tags not as recognition sequences to which chelators are appended but rather as the chelators themselves.
(His)6 Tag
The ubiquitious hexahistidine affinity tag provides the best example of this phenomenon. A number of laboratories have labeled antibody fragments bearing C-terminal (His)6 tags with the [99mTc(CO)3]+ synthon for SPECT imaging [43–46]. Kogelberg, et al., for example, labeled the αvβ6-targeting diabody B6.3 via the coordination of [99mTc(CO)3]+ to a C-terminal (His)6 tag, and it was found that the resultant conjugate retained nanomolar affinity for its target and demonstrated tumor-specific targeting in vivo. Kampmeier and coworkers provided a clever variation on this theme. This laboratory site-specifically labeled a PSMA-targeting, J591-derived diabody with [99mTc(CO)3]+ via a C-terminal (His)6 tag and successfully performed SPECT imaging in mice bearing subcutaneous DU145 prostate cancer xenografts. Interestingly, however, the group also incorporated a cysteine at the end of the (His)6 tag and used the thiol functionality to modify the diabody with a fluorophore, indicating that this approach could easily be applied to the synthesis of tracers for multimodal imaging.
(Gly)xCys Tag
Other laboratories have used a peptide tag composed of several glycines and a terminal cysteine to create an N3S coordination architecture for 99mTC = O species. For example, almost 20 years ago, George et al. site-specifically labeled an anti-c-erbB-2 scFv bearing a C-terminal Gly4Cys tag with 99mTc-99m [47]. To this end, the scFv was first reduced with 2-mercaptoethanol and then incubated with 99mTcO4 −1 in the presence of Sn2+ to generate an immunoconjugate with Tc-99m in an N3S coordination environment. Following a similar strategy, a different laboratory labeled an anti-ED-B fibronectin scFv bearing a (Gly)3-Cys-Ala tag [48]. This work also included a revealing comparison to the same scFv labeled with Tc-99m via a (His)6 tag. In mice bearing antigen-expressing F9 xenografts, the [99mTc]scFv-G3CA and the [99mTc]scFv-(His)6 agents showed dramatically different biodistributions: while both constructs effectively targeted tumor tissue, the authors found that the (His)6-tagged variant displayed much higher uptake in non-target tissues such as the kidney. These data serve as an effective reminder that chelators can play non-innocent roles in the pharmacokinetics of radiometal-based imaging agents.
Antibody-Metallothionein Fusion Proteins
A seldom-used but nonetheless interesting alternative to these coordinating peptide tags is the use of fusion proteins bearing metallothioneins, low molecular weight proteins capable of binding both physiological and xenobiotic metals via multiple cysteine residues. Pietersz et al. used this approach to site-specifically label a fusion protein composed of a metallothionein and an anti-CEA scFv (MET-scFv) [49]. Using a unique Zn2+ transchelation method, the authors labeled MET-scFv with Tc-99m and demonstrated that the Tc-99m labeled construct is both stable and highly immunoreactive. Further, imaging and biodistribution experiments in mice bearing LS174T xenografts revealed that despite extraordinarily rapid clearance, [99mTc]MET-scFv effectively delineated CEA-expressing tumor tissue in vivo.
Before moving on, it is important to note that a number of other chemoenzymatic protein modification approaches have recently emerged that have yet to be applied to antibodies or antibody fragments [50]. This knowledge, coupled with the tremendous variety of peptide-based bioconjugation strategies discussed in this section, underscores that the next few years will be an exceptionally exciting time for the development of site-specifically labeled immunoconjugates of all kinds.
Unnatural Amino Acids
Arguably the most sophisticated method of facilitating site-specific conjugation is the incorporation of orthogonally reactive functional groups during the translation of recombinant antibodies using non-canonical (ncAA) or unnatural amino acids (uAA). This approach relies on expanding the genetic code of cells to reinterpret nonsense codons—amber (UAG), ochre (UAA), or opal (UGA)—as sequences coding for uAA, leading to the ribosomal incorporation of the uAA into the protein in question [51, 52]. At present, the number of uAAs that have been incorporated into recombinant proteins hovers around 70, with p-aceto-l-phenylalanine (pAcF) and p-azido-l-phenylalinine (pAzF) among the most often employed (Fig. 8) [52]. While this technology has been used to create ADCs based on recombinant antibodies and antibody fragments, there are curiously few examples of uAA-based immunoconjugates for imaging [53]. Therefore, in this section, we will primarily provide examples of how this technology has been used to create immunoconjugates for other applications with a view toward inspiring the future application of these methods to molecular imaging.

Structures of selected unnatural and non-canonical amino acids.
Incorporation of uAA by engineered cells
The Schultz laboratory has pioneered the genetic engineering of cells for the integration of uAAs and has used this technology for the production of site-specifically labeled immunoconjugates [54, 55]. In one example, the laboratory used engineered E. coli to incorporate the ketone-bearing uAA pAcF into an anti-HER2 Fab based on trastuzumab, which could then be modified with an aminooxy-bearing AlexaFluor® 488 fluorophore. In an alternate strategy, this pAcF-Fab was modified with an aminooxy-bearing variant of biotin and then conjugated to neutravidin to form multimers which were shown to be more potent inhibitors of HER2 phosphorylation than their unmodified, monomeric parents [54]. In an expansion of this methodology, it is now possible to introduce aminoacyl tRNA synthetase (aaRS)/tRNA pairs corresponding to two different uAAs and two different nonsense codons, thus enabling the incorporation of two different uAAs into a single recombinant protein. Using this strategy, Xiao et al. inserted pAcF and azido-lysine (AzK) into the sequence of trastuzumab [56]. These two functional groups were then used to modify the antibody with an aminooxy-bearing variant of auristatin and a cyclooctyne-bearing Alexa Fluor® 488 fluorophore, and the resulting immunoconjugate was subsequently employed in in vitro assays (Fig. 2c and j).
Incorporation of uAA in cell-free system
Despite the promise of systems based on genetically engineered cells, these methods are hampered by the expense and complexity of creating stable cell clones as well as the need for the uAA in question to cross the cell membrane rapidly in order to join the translational machinery. Cell-free protein expression systems circumvent these drawbacks. These systems contain all the machinery needed for protein expression and provide a powerful tool for the incorporation of uAAs after the introduction of the suitable aaRS/tRNA pair. The principal advantages of cell-free protein expression lie in addition by subtraction: in the absence of a cell, all the available resources are dedicated to the production of the protein of interest, and the aaRSs have unobstructed access to the uAAs. Unfortunately, the folding of the recombinant proteins and the incorporation of post-translational modifications (like glycosylation) are significant challenges in cell-free systems, as these functions are normally handled by organelles such as the endoplasmic reticulum and Golgi apparatus. Recently, however, avenues around these issues have emerged [57, 58]. Using these methods, Zimmerman, et al. created a pAzF-containing variant of trastuzumab, which was subsequently coupled to a DBCO-bearing MMAF drug moiety (Fig. 4) [59]. In addition, Patel and coworkers created both a homopropargyl glycine-containing variant of luciferase and a pAzF-bearing fusion construct of an scFv and the IM9 protein [60]. The investigators then coupled the IM9-scFv fusion protein to the luciferase and employed the resulting conjugate for the in vivo detection of B cell lymphoma cells.
Non-canonical amino acids
The effort and expense of using uAAs have prompted some research groups to revisit the use of natural, though non-canonical, amino acids for the construction of site-specifically modified proteins. In this regard, selenocysteine (Sec; U) and pyrrolysine (Pyl; O)—the 21st and 22nd naturally occurring, genetically encoded amino acids—have been the center of attention (Fig. 8). While pyrrolysine has been found exclusively in prokaryotic organisms, selenocysteine has been observed in all kingdoms of life. From a protein modification standpoint, pyrrolysine boasts unique orthogonal reactivity, while selenocysteine can be coupled to maleimide analogues even in the presence of cysteine due to the difference in the pK a of the two residues (5.2 for selenocysteine vs. 8.3 for cysteine; Fig. 2k–m)) [61, 62]. Along these lines, Li et al. have recently produced a scFv-Fc fragment of trastuzumab bearing two engineered cysteines and one selenocysteine in the Fc region [63]. After reduction, the selenocysteine residue of the fragment was selectively coupled to a methyl sulfone-bearing variant of biotin at pH 5.2, while the cysteine residues were conjugated to a methyl sulfone-containing fluorescein moiety at pH 7.4. The resulting immunoconjugate was then successfully employed for the in vitro targeting of HER2 (Fig. 2n–o).
Over the last ten years, great strides have been made in the production of recombinant proteins bearing unnatural or non-canonical amino acids. Yet reports of using these methods to create site-specifically modified immunoconjugates are few and far between. Indeed, in assembling this review, we were wholly unable to find any examples of site-specifically modified immunoconjugates for in vivo imaging synthesized using these methods. This is almost certainly due to the intrinsic complexity of these approaches. However, we are optimistic that the molecular imaging community will be able to leverage this technology in the near future.
Moving Forward
The development of site-specifically labeled immunoconjugates is a rapidly evolving field, as illustrated by the emergence of a number of new studies during the writing of this work [64–66]. In the two installments of this review, we have covered a wide variety of approaches, each with distinct advantages and limitations (Fig. 9). For example, while strategies based on unnatural amino acids offer exquisite control over the molecular location and stoichiometry of the conjugation reaction, they also require specialized and complex genetic engineering. On the other hand, methodologies based on the reaction of native cysteines with maleimide-bearing probes are simple and straightforward but admittedly do not readily produce the degree of homogeneity offered by other approaches. Specifics aside, the benefits of all of these strategies over traditional synthetic methods are clear: the reproducible creation of better defined and more homogeneous immunoconjugates with in vivo performance comparable to—if not superior to—their randomly modified cousins.

The advantages and limitations of selected approaches to site-specific bioconjugation.
Ultimately, it is our belief that four of these bioconjugation approaches—two that require genetic manipulation and two that do not—hold particular promise for the future. First, the incorporation of engineered cysteines into antibodies and fragments offers complete control over the site of conjugation while leaving the rest of the immunoglobulin structure untouched [67]. However, the development of thiol-reactive alternatives to maleimides is essential to this technology’s ongoing success [68]. Second, the use of sortases to modify C-terminal recognition sequences on antibody fragments requires genetic manipulation as well, yet it offers an operationally simple, highly specific, and all-but-traceless bioconjugation strategy [69]. While the genetic engineering methods required for these two approaches are relatively straightforward, site-specific bioconjugation strategies that employ native, unmodified antibodies offer modularity and flexibility that can be tremendously beneficial in both the laboratory and the clinic. In this regard, both the use of bioorthogonal click chemistry to modify enzymatically tagged heavy chain glycans and the use of transglutaminase to modify deglycosylated antibodies are exciting approaches [19, 70]. Both methods provide facile and modular ways to site-specifically modify antibodies without any genetic manipulation and, equally importantly, both routes ensure that the site of conjugation is within the Fc region, far from the sensitive antigen-binding domains of the antibody. Critically, however, both of these approaches dramatically alter the heavy chain glycans of the antibody: the former caps the glycans with an azide-modified sugar and a dibenzocyclooctyne-bearing cargo, while the latter removes the sugar chains entirely. Going forward, further study into the pharmacokinetic and pharmacodynamic effects of these glycans modifications is urgently needed.
We are writing this review at a critical time for the field. Over the next few years, growth in a number of directions is needed in order to fully harness the potential of site-specifically labeled immunoconjugates for molecular imaging. First, the field will surely benefit from the further preclinical development, refinement, and optimization of site-specific bioconjugation methods. Second, as we have discussed, a number of innovative approaches—for example, the use of bridging dibromomaleimide reagents (Fig. 2p)—have been applied to the synthesis of ADCs but not immunoconjugates for imaging. This represents an important missed opportunity for the development of imaging agents that should be remedied in the near future. And third, the dissemination of these bioconjugation technologies to laboratories that would otherwise not have access to them is critical to widen the application of site-specifically labeled immunoconjugates.
Yet above all, we think it is time for priorities to shift to the clinic. Indeed, the ability to create site-specifically modified immunoconjugates that are well-defined, homogeneous, highly stable, and highly immunoreactive could have a transformational impact on the way imaging agents are synthesized for the clinic. More than anything else, the clinical demonstration of the safety and efficacy of site-specifically radiolabeled immunoconjugates for PET, SPECT, and optical imaging will convince clinicians and regulatory agencies of the value of this technology. Hopefully, the next few years will be a tipping point during which an increasing number of first-in-human clinical trials act as the vanguard for a new era in which site-specifically labeled immunoconjugates become the standard of care.
Abbreviations
- 2-keto-Gal:
-
2-Acetyl-2-deoxy-galactose
- aaRS:
-
Aminoacyl tRNA synthase
- ADC:
-
Antibody-drug conjugate
- AGT:
-
O6-Alkylguanine transferase
- AzK:
-
Azido-lysine
- CEA:
-
Carcinoembryonic antigen
- Db:
-
Diabody
- DBCO:
-
Dibenzocyclooctyne
- DFO:
-
Desferrioxamine
- DOTA:
-
1,4,7,10-Tetraazacyclododecane-1,4,7,10-tetraacetic acid
- DTPA:
-
Diethylenetriaminepentaacetic acid
- EGFR:
-
Epidermal growth factor receptor
- FGE:
-
Formylglycine-generating enzyme
- FR-1:
-
Framework-1 region
- HER2:
-
Human epidermal growth factor receptor 2
- HIPS:
-
hydrazino-Pictet-Spengler
- mAb:
-
Monoclonal antibody
- Mb:
-
Minibody
- MET:
-
Metallothionein
- MMAD:
-
Monomethyl auristatin D
- MMAF:
-
Monomethyl auristatin F
- ncAA:
-
Non-canonical amino acid
- NCS:
-
Isothiocyanate
- NHS:
-
N-hydroxysuccinimide
- NIRF:
-
Near-infrared fluorescence
- OI:
-
Optical imaging
- pAcF:
-
p-Aceto-l-phenylalanine
- pAzF:
-
p-Azido-l-phenylalinine
- PET:
-
Positron emission tomography
- PSMA:
-
Prostate specific membrane antigen
- scFv:
-
Single-chain variable fragment
- sdAb:
-
Single-domain antibody
- SDS-PAGE:
-
Sodium dodecyl sulfate polyacrylamide gel electrophoresis
- SPECT:
-
Single photon computed tomography
- SrtA:
-
Sortase
- TGase:
-
Transglutaminase
- uAA:
-
Unnatural amino acid
References
Freise AC, Wu AM (2015) In vivo imaging with antibodies and engineered fragments. Mol Immunol 67:142–152
Wu AM (2009) Antibodies and antimatter: the resurgence of immuno-PET. J Nucl Med 50:2–5
Deri MA, Zeglis BM, Francesconi LC, Lewis JS (2013) PET imaging with 89Zr: from radiochemistry to the clinic. Nucl Med Biol 40:3–14
Frangioni JV (2003) In vivo near-infrared fluorescence imaging. Curr Opin Chem Biol 7:626–634
van Dongen GAMS, Visser GWM, Lub-de Hooge MN et al (2007) Immuno-PET: a navigator in monoclonal antibody development and applications. Oncologist 12:1379–1389
Vosjan M, Perk LR, Visser GWM et al (2010) Conjugation and radiolabeling of monoclonal antibodies with zirconium-89 for PET imaging using the bifunctional chelate p-isothiocyanatobenzyl-desferrioxamine. Nat Protoc 5:739–743
Zeglis BM, Lewis JS (2011) A practical guide to the construction of radiometallated bioconjugates for positron emission tomography. Dalton Trans 40:6168–6195
Agarwal P, Bertozzi CR (2015) Site-specific antibody-drug conjugates: the nexus of bioorthogonal chemistry, protein engineering, and drug development. Bioconj Chem 26:176–192
Harris LJ, Skaletsky E, McPherson A (1998) Crystallographic structure of an intact IgG1 monoclonal antibody. J Mol Biol 275:861–872
Wang LT, Amphlett G, Blattler WA et al (2005) Structural characterization of the maytansinoid—monoclonal antibody immunoconjugate, huN901-DM1, by mass spectrometry. Protein Sci 14:2436–2446
Behrens CR, Liu B (2014) Methods for site-specific drug conjugation to antibodies. mAbs 6:46–53
Li X, Patterson JT, Sarkar M, et al. (2015) Site-specific dual antibody conjugation via engineered cysteine and selenocysteine residues. Bioconjugate Chem
Zhou Q, Stefano JE, Manning C et al (2014) Site-specific antibody-drug conjugation through glycoengineering. Bioconj Chem 25:510–520
Alvarez VL, Wen ML, Lee C et al (1986) Site-specifically modified In-111 labeled antibodies give low liver backgrounds and improved radioimmunoscintigraphy. Nucl Med Biol 13:347–352
Ding H, Carlton MM, Povoski SP et al (2013) Site specific discrete PEGylation of 124I-labeled mCC49 Fab′ fragments improves tumor microPET/CT imaging in mice. Bioconj Chem 24:1945–1954
Junutula JR, Raab H, Clark S et al (2008) Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat Biotechnol 26:925–932
Pillow TH, Tien J, Parsons-Reponte KL et al (2014) Site-specific trastuzumab maytansinoid antibody-drug conjugates with improved therapeutic activity through linker and antibody engineering. J Med Chem 57:7890–7899
Grünberg J, Jeger S, Sarko D et al (2013) DOTA-functionalized polylysine: A high mumber of DOTA chelates positively influences the biodistribution of enzymatic conjugated anti-tumor antibody chCE7agl. PLoS One 8:e60350
Jeger S, Zimmermann K, Blanc A et al (2010) Site-specific and stoichiometric modification of antibodies by bacterial transglutaminase. Angew Chem Int Ed 49:9995–9997
Mindt TL, Jungi V, Wyss S et al (2008) Modification of different IgG1 antibodies via glutamine and lysine using bacterial and human tissue transglutaminase. Bioconj Chem 19:271–278
Kamiya N, Takazawa T, Tanaka T et al (2003) Site-specific cross-linking of functional proteins by transglutamination. Enzyme Microb Technol 33:492–496
Farias SE, Strop P, Delaria K et al (2014) Mass spectrometric characterization of transglutaminase based site-specific antibody-drug conjugates. Bioconj Chem 25:240–250
Strop P, Liu S-H, Dorywalska M et al (2013) Location matters: site of conjugation modulates stability and pharmacokinetics of antibody drug conjugates. Chem Biol 20:161–167
Swee LK, Guimaraes CP, Sehrawat S et al (2013) Sortase-mediated modification of alphaDEC205 affords optimization of antigen presentation and immunization against a set of viral epitopes. Proc Natl Acad Sci U S A 110:1428–1433
Madej MP, Coia G, Williams CC et al (2012) Engineering of an anti-epidermal growth factor receptor antibody to single chain format and labeling by sortase A-mediated protein ligation. Biotechnol Bioeng 109:1461–1470
Mohlmann S, Bringmann P, Greven S, Harrenga A (2011) Site-specific modification of ED-B-targeting antibody using intein-fusion technology. BMC Biotechnol 11:76
Möhlmann S, Mahlert C, Greven S et al (2011) In vitro sortagging of an antibody Fab fragment: overcoming unproductive reactions of sortase with water and lysine side chains. ChemBioChem 12:1774–1780
Paterson BM, Alt K, Jeffery CM et al (2014) Enzyme-mediated site-specific bioconjugation of metal complexes to proteins: Sortase-mediated coupling of copper-64 to a single-chain antibody. Angew Chem Int Ed 53:6115–6119
Warden-Rothman R, Caturegli I, Popik V, Tsourkas A (2013) Sortase-tag expressed protein ligation: combining protein purification and site-specific bioconjugation into a single step. Anal Chem 85:11090–11097
Ramakrishnan B, Boeggeman E, Qasba PK (2007) Novel method for in vitro O-glycosylation of proteins: application for bioconjugation. Bioconj Chem 18:1912–1918
Ramakrishnan B, Boeggeman E, Manzoni M et al (2009) Multiple site-specific in vitro labeling of single-chain antibody. Bioconj Chem 20:1383–1389
Fisch I, Kunzi G, Rose K, Offord RE (1992) Site-specific modification of a fragment of a chimeric monoclonal antibody using reverse proteolysis. Bioconj Chem 3:147–153
Liebscher S, Kornberger P, Fink G et al (2014) Derivatization of antibody Fab fragments: a designer enzyme for native protein modification. ChemBioChem 15:1096–1100
Gautier A, Juillerat A, Heinis C et al (2008) An engineered protein tag for multiprotein labeling in living cells. Chem Biol 15:128–136
Hussain AF, Kampmeier F, von Felbert V et al (2011) SNAP-Tag technology mediates site-specific conjugation of antibody fragments with a photosensitizer and imprives target specific phototoxicity in tumor cells. Bioconj Chem 22:2487–2495
Chaisemartin L, Chinestra P, Favre G et al (2009) Synthesis and application of a N-1′ fluorescent biotinyl derivative inducing the specific carboxy-terminal dual labeling of a novel RhoB-selective scFv. Bioconj Chem 20:847–855
Leung S, Losman MJ, Govindan SV et al (1995) Engineering a unique glycosylation site for site-specific conjugation of haptens to antibody fragments. J Immunol 154:5919–5926
Hudak JE, Barfield RM, de Hart GW et al (2012) Synthesis of heterobifunctional protein fusions using copper-free click chemistry and the aldehyde tag. Angew Chem Int Ed 51:4161–4165
Agarwal P, van der Weijden J, Sletten EM et al (2013) A Pictet-Spengler ligation for protein chemical modification. Proc Natl Acad Sci U S A 110:46–51
Drake PM, Albers AE, Baker J et al (2014) Aldehyde tag coupled with HIPS chemistry enables the production of ADCs conjugate site-specifically to different antibody regions with distinct in vivo efficacy and PK outcomes. Bioconj Chem 25:1331–1341
Clark WA, Izotova L, Philipova D et al (2002) Site-specific 32P-labeling of cytokines, monoclonal antibodies, and other protein substrates for quantitative assays and therapeutic application. Biotechniques Suppl 76–78:80–77
Patrick MR, Chester KA, Pietersz GA (1998) In vitro characterization of a recombinant 32P-phosphorylated anti-(carcinoembryonic antigen) single-chain antibody. Cancer Immunol Immunother 46:229–237
Kogelberg H, Miranda E, Burnet J et al (2013) Generation and characterization of a diabody targeting the alphavbeta6 integrin. PLoS One 8:e73260
Vaneycken I, Devoogdt N, Van Gassen N et al (2011) Preclinical screening of anti-HER2 nanobodies for molecular imaging of breast cancer. FASEB J 25:2433–2446
Kampmeier F, Williams JD, Maher J et al (2014) Design and preclinical evaluation of a 99mTc-labelled diabody of mAb J591 for SPECT imaging of prostate-specific membrane antigen (PSMA). Eur J Nucl Med Mol Imaging 4:13
Badar A, Williams J, de Rosales R et al (2014) Optimising the radiolabelling properties of technetium tricarbonyl and His-tagged proteins. EJNMMI Res 4:14
George AJ, Jamar F, Tai MS et al (1995) Radiometal labeling of recombinant proteins by a genetically engineered minimal chelation site: technetium-99m coordination by single-chain Fv antibody fusion proteins through a C-terminal cysteinyl peptide. Proc Natl Acad Sci U S A 92:8358–8362
Berndorff D, Borkowski S, Moosmayer D et al (2006) Imaging of tumor angiogenesis using 99mTc-labeled human recombinant anti-ED-B fibronectin antibody fragments. J Nucl Med 47:1707–1716
Pietersz GA, Patrick MR, Chester KA (1998) Preclinical characterization and in vivo imaging studies of an engineered recombinant technetium-99m-labeled metallothionein-containing anti-carcinoembryonic antigen single-chain antibody. J Nucl Med 39:47–56
Rabuka D (2010) Chemoenzymatic methods for site-specific protein modification. Curr Opin Chem Biol 14:790–796
Liu CC, Schultz PG (2010) Adding new chemistries to the genetic code. Annu Rev Biochem 79:413–444
Lang K, Chin JW (2014) Cellular incorporation of unnatural amino acids and bioorthogonal labeling of proteins. Chem Rev 114:4764–4806
Hallam TJ, Wold E, Wahl A, Smider VV (2015) Antibody conjugates with unnatural amino acids. Mol Pharm 12:1848–1862
Hutchins BM, Kazane SA, Staflin K et al (2011) Site-specific coupling and sterically controlled formation of multimeric antibody Fab fragments with unnatural amino acids. J Mol Biol 406:595–603
Kazane SA, Sok D, Cho EH et al (2012) Site-specific DNA-antibody conjugates for specific and sensitive immuno-PCR. Proc Natl Acad Sci U S A 109:3731–3736
Xiao H, Chatterjee A, S-h C et al (2013) Genetic incorporation of multiple unnatural amino acids into proteins in mammalian cells. Angew Chem Int Ed 52:14080–14083
Groff D, Armstrong S, Rivers PJ et al (2014) Engineering toward a bacterial “endoplasmic reticulum” for the rapid expression of immunoglobulin proteins. mAbs 6:671–678
Guarino C, DeLisa MP (2012) A prokaryote-based cell-free translation system that efficiently synthesizes glycoproteins. Glycobiology 22:596–601
Zimmerman ES, Heibeck TH, Gill A et al (2014) Production of site-specific antibody-drug conjugates using optimized non-natural amino acids in a cell-free system. Bioconj Chem 25:351–361
Patel KG, Ng PP, Kuo C-C et al (2009) Cell-free production of Gaussia princeps luciferase—antibody fragment bioconjugates for ex vivo detection of tumor cells. Biochem Biophys Res Commun 390:971–976
Ou W, Uno T, Chiu H-P et al (2011) Site-specific protein modifications through pyrroline-carboxy-lysine residues. Proc Natl Acad Sci U S A 108:10437–10442
Hofer T, Skeffington LR, Chapman CM, Rader C (2009) Molecularly defined antibody conjugation through a selenocysteine interface. Biochemistry 48:12047–12057
Li X, Patterson JT, Sarkar M, et al. (2015) Site-specific dual antibody conjugation via engineered cysteine and selenocysteine residues. Bioconjugate Chem:Online Ahead of Print
VanBrunt MP, Shanebeck K, Caldwell Z, et al. (2015) Genetically encoded azide containing amino acid in mammalian cells enables site-specific antibody-drug conjugates using click cycloaddition chemistry. Bioconjugate Chem:Online Ahead of Print
Thompson P, Bezabeh B, Fleming R et al (2015) Hydrolytically stable site-specific conjugation at the N-terminus of an engineered antibody. Bioconj Chem 26:2085–2096
Rashidian M, Keliher EJ, Dougan M et al (2015) Use of 18F-2-fluorodeoxyglucose to label antibody fragments for immuno-positron emission tomography of pancreatic cancer. ACS Cent Sci 1:142–147
Tinianow JN, Gill HS, Ogasawara A et al (2010) Site-specifically 89Zr-labeled monoclonal antibodies for ImmunoPET. Nucl Med Biol 37:289–297
Patterson JT, Asano S, Li X et al (2014) Improving the serum stability of site-specific antibody conjugates with sulfone linkers. Bioconj Chem 25:1402–1407
Mao HY, Hart SA, Schink A, Pollok BA (2004) Sortase-mediated protein ligation: a new method for protein engineering. J Am Chem Soc 126:2670–2671
Zeglis BM, Davis CB, Abdel-Atti D et al (2014) Chemoenzymatic strategy for the synthesis of site-specifically labeled immunoconjugates for multimodal PET and optical imaging. Bioconj Chem 25:2123–2128
Acknowledgments
The authors would like to thank all of those researchers whose work has contributed to creating the immunoconjugates discussed in this review. We are also very grateful to the Hunter College, the Research Foundation of the City University of New York, and the NIH (4R00CA178205-02) for their generous funding.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of Interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Rights and permissions
About this article

Cite this article
Adumeau, P., Sharma, S.K., Brent, C. et al. Site-Specifically Labeled Immunoconjugates for Molecular Imaging—Part 2: Peptide Tags and Unnatural Amino Acids. Mol Imaging Biol 18, 153–165 (2016). https://doi.org/10.1007/s11307-015-0920-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11307-015-0920-y
Key words
- Positron emission tomography
- PET
- Single photon emission tomography
- SPECT
- Fluorescence imaging
- Near-infrared fluorescence imaging
- Optical imaging
- Click chemistry
- Site-specific conjugation
- Site-selective conjugation
- Bioconjugation
- Bioorthogonal chemistry
- Glycoengineering
- Protein engineering, antibody
- Antibody fragment
- Immunoglobulins
- Peptide tags
- Unnatural amino acids
- Non-canonical amino acids