
Abstract
Since adult neurogenesis became a widely accepted phenomenon, much effort has been put in trying to understand the mechanisms involved in its regulation. In addition, the pathophysiology of several neuropsychiatric disorders, such as depression, has been associated with imbalances in adult hippocampal neurogenesis. These imbalances may ultimately reflect alterations at the cell cycle level, as a common mechanism through which intrinsic and extrinsic stimuli interact with the neurogenic niche properties. Thus, the comprehension of these regulatory mechanisms has become of major importance to disclose novel therapeutic targets. In this review, we first present a comprehensive view on the cell cycle components and mechanisms that were identified in the context of the homeostatic adult hippocampal neurogenic niche. Then, we focus on recent work regarding the cell cycle changes and signaling pathways that are responsible for the neurogenesis imbalances observed in neuropathological conditions, with a particular emphasis on depression.
Similar content being viewed by others


Introduction
The view that no new neurons can be added to the adult brain was deconstructed over the past years. Neurogenesis in the adult smammalian brain is now a widely accepted neuroplastic event [1–3] that enables the brain to adapt to intrinsic and extrinsic stimuli. In fact, during the past few years, a large amount of data has provided evidence that the production, differentiation and survival of neurons in the adult brain have significant implications for several physiological processes, such as memory and learning [4–6]. Moreover, many studies have linked neurogenesis deregulation with the emergence of several pathological features in neuropsychiatric disorders. However, the role of neurogenesis in these disorders is yet to be completely established. Presently, much effort is devoted to the generation of behavioral and molecular data that establish a mechanistic link between neurogenesis and disease-state, so that ultimately directed therapeutic interventions can be designed.
Adult stem cells are responsible for tissue integrity, by adding new cells to the networks or by promoting the capacity to repopulate mature differentiated tissues as their constituting cells die due to damage or degeneration. The complex process of producing new cells throughout an organism’s lifespan may rely on a common denominator—the cell cycle regulatory machinery (Fig. 1). However, the specificity of this transversal phenomenon in each tissue or cell type offers a wide spectrum of responses to a particular stem cell niche. In this review we aim to provide a comprehensive and integrated view of the cell cycle regulation in the adult hippocampal neurogenic niche both in basal conditions and in disease (namely, in depression). With this, we offer a perspective on how the cell cycle machinery may constitute an interesting and still largely unrecognized link between alterations in postnatal hippocampal neurogenesis and disease, highlighting its relevance for the discovery of new molecular targets for the treatment of neurobiological disorders.

Cell cycle regulation in the adult hippocampal neurogenic niche. Some niche-specific cell cycle regulators in the adult hippocampus have been identified. Cdk6-cyclin D2 and Cdk4-cyclin D1 complexes promote the expansion of the neural progenitor pool. P21 and p27 Cdk inhibitors have a role in proliferation arrest, both at the G1 and G2 phase. E2F1 has an important role in the neurogenic process by inducing the expression of genes involved in cell proliferation and differentiation. Cdk5 activity is associated with cell cycle reentry inhibition in postmitotic neurons. Signaling pathways, such as Notch, BMP, Shh and Wnt, are also involved in proliferation regulation, and in the balance between proliferation induction and stem cell quiescence maintenance. Nevertheless, several key molecules remain to be identified in this process in the context of adult hippocampal neurogenic niche (represented by a question mark). Rb retinoblastoma protein
Neurogenesis in the Adult Mammalian Brain
Adult neurogenesis is the process by which neural progenitors divide mitotically to produce new neurons in the adult brain. This complex process involves several steps beyond cell division; namely, the commitment of the new cell to a neuronal phenotype, the migration and maturation of the cells, and the establishment of appropriate synaptic contacts that culminate with a full integration on the pre-existent network. Well described, adult neurogenesis is known to occur at least in two different regions of the mammalian brain: the subependymal zone (SEZ) of the lateral ventricles, and the subgranular zone (SGZ) of the hippocampal dentate gyrus (DG) [7]. In both regions, astroglial cells act as the source of adult progenitor cells [8, 9]. The neuroblasts born in the SEZ migrate along the rostral migratory stream (RMS) becoming mostly mature GABAergic granule and periglomerular interneurons in the olfactory bulb (OB). The cells born in the adult SGZ migrate into the granular cell layer (GCL) of the DG and differentiate into glutamatergic granule cells [7]. Additionally, although disputable [10, 11], several reports describe the generation of new neurons in other regions of the adult brain, including the cortex [12, 13], the amygdala [14–16], the hypothalamus [17, 18], the striatum [19, 20] and the substantia nigra [21, 22]. However, in all these areas, neurogenesis appears to occur at very low levels or under non-physiological conditions [23].
Adult Hippocampal Neurogenesis
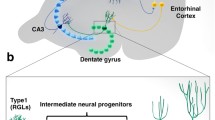
Neurogenesis in the adult DG occurs from a progenitor population residing in a narrow layer of about three nuclei wide, the SGZ. The first type of progenitor cells, defined as multipotent, are the neural stem cells (NSCs or type_1 cells). These cells express nestin and glial fibrillary acidic protein (GFAP), among other markers, and can be divided in two subtypes based on their orientation in the SGZ: radial astrocytes/NSCs (rA) and horizontal astrocytes/NSCs (hA). Radial NSCs, morphologically characterized by having a single radial process, are slow dividing cells, whereas horizontal NSCs have a short horizontal process and divide faster than rA [24, 25]. Either one or both of these cell subtypes will then divide asymmetrically into one daughter cell and one progenitor cell, both of which already committed to a neuronal lineage. These latter cells, designated by transit amplifying neural progenitors (tANPs, or type_2 cells), are mitotically active and divide to give rise to neuroblasts (also known as type_3 cells). This last stage corresponds to a transition from a slow proliferating neuroblast, which is exiting the cell cycle, to a postmitotic immature neuron, that will migrate a short distance into the GCL. Neuroblasts transiently express markers of the neuronal lineage, such as the calcium-binding protein calretinin, doublecortin (DCX) and polysialylated-neural cell adhesion molecule (PSA-NCAM) [10, 26]. The newborn cells will then fully maturate into granule neurons, elongating their axons towards the hippocampal Cornus Ammonis 3 (CA3) area [23] and making the appropriate axonal connections [26]. These adult-born neurons become integrated in the pre-existing neuronal network 4 to 8 weeks after their birth [3, 27–29] (Fig. 2a).

a Neurogenesis in the hippocampus comprises several steps, including proliferation of neural stem cells and transit amplifying neural progenitors in the subgranular zone (SGZ), cell cycle exit, neuroblasts migration throughout the granule cell layer (GCL), and maturation of the newborn neurons. b,_c Cell cycle regulators implicated in neurogenesis imbalances observed in animal models of depression and in the pro-neurogenic effects of antidepressant drugs and other stimuli. b Neurogenesis imbalances have been observed in animal models of depression. These imbalances are attributed to an increased expression of p27 Cdk inhibitor (green arrow) in the DGs of animal models of depression. P27 inhibits neural progenitor cells proliferation in this neurogenic niche. Cdk5 is involved in the development of depressive-like signs in an animal model of depression. The increased activity of Cdk5 (green arrow), together with the translocation of p35 activator to the membrane, was observed in chronic mild stress (CMS) exposed animals. c The pro-neurogenic actions of antidepressant drugs and stimuli, such as physical exercise, have also been correlated with alterations in the “expanded cell cycle.”Antidepressants are able to specifically inhibit p21 expression (red arrow) in the DG, while increasing neurogenesis. Additionally, signaling pathways with recognized effects over the cell cycle regulation, such as Wnt, Notch and BMP, were implicated in the modulation of adult hippocampal neurogenesis in the context of depression and antidepressant stimuli
Importantly, not all cells expressing immature neuronal markers develop into fully mature neurons [30]. In fact, if not recruited to perform any function, the great majority of these newly-born cells are eliminated by apoptosis once they exit cell cycle [31, 32]; a mechanism that until recently was largely undescribed. However, a recent report by Lu et al. [33], has very elegantly shown that DCX-positive neuronal progenitors present a phagocytic activity in the DG as well as in the SEZ, with important implications for the neurogenic process [33].
The generation of new neurons in the hippocampal niche of the adult brain, depends on the harmonization of several processes and cellular activities, which include proliferation, cell cycle exit, activation of survival/death pathways, migration through the GCL and differentiation/maturation of the newborn neurons [34]. These processes are regulated by both intrinsic and extrinsic factors that are ultimately responsible for the modulation of the neurogenic phenomenon. While there are numerous focused studies on several of these steps, little is known about the cell cycle regulation in the context of adult hippocampal neurogenesis and its repercussions for disease states. Here, to provide an integrated view, we will consider the “expanded cell cycle” [35], taking into consideration some of the most well-described mitogenic signals and the interacting signaling pathways.
Cell Cycle Regulation in the Adult Hippocampal Neurogenic Niche
Providing important clues on the regulation of the adult neurogenic process, the expression of cell cycle proteins and their regulation have been extensively explored in the context of embryonic development [36–40]. On one hand, some of the mechanisms are common to both embryonic and adult brain; however, there are essential differences between them especially regarding niche properties. Whereas during development, the cellular environment is highly specialized to support proliferation, in the adult hippocampus the environmental context includes a population of fully mature and functionally active neurons [7, 37], thus providing a different set of both intrinsic and extrinsic signals. In fact, in the adult mammalian brain, the vast majority of neuronal cells are in a quiescent differentiated state (G0 phase of the cell cycle), which is probably promoted by an increase in the expression of region-specific Cdk inhibitors [34, 41, 42]. Nonetheless, the expression of cell cycle proteins in the postnatal brain and their definite role in this neurogenic niche are still being unveiled [36].
Contrary to the traditional concept that postmitotic mature neurons are stably maintained in a quiescent differentiated state, recent, albeit still controversial, evidence has demonstrated that in some disease conditions, such as Alzheimer’s disease [43, 44], traumatic brain injury [45] and cerebral hypoxia-ischemia [46], these cells are capable of responding to mitogenic signals and reenter the cell cycle [35, 47]. However, apparently these neurons neither finish dividing nor revert to their G0 quiescent state, ultimately undergoing apoptotic cell death and suggesting that they lack the factors needed for cell cycle progression [35].
It is important to mention, at this point, that the expression of cell cycle proteins in neuronal populations is not always associated with cell proliferation or cell cycle reentry. Indeed, a small number of studies have demonstrated that the expression of key cell cycle components may be associated with other neuronal processes, such as neuronal migration, dendrite morphogenesis, synaptic maturation and plasticity [48, 49]. Nonetheless, a few cell cycle molecules and signaling pathways have been implicated in the regulation of adult hippocampal neurogenesis. Moreover, their deregulation is often the cause for neurogenesis imbalances observed in several disorders, such as depression. As such, we will first provide a brief overview on these molecules and its functions in the context of adult hippocampal neurogenesis.
Cell Cycle Regulators
The cell cycle consists of a succession of events that lead to cell division. It comprises four distinct consecutive phases: the first gap (G1) phase, during which cells prepare for DNA replication in the synthetic (S) phase, followed by a second gap (G2) phase and mitosis (M). A highly coordinated network of molecules mediates progression through these four phases. There are two major classes of cell cycle regulators that cooperate in order to promote cell cycle progression: cyclins and cyclin-dependent kinases (Cdks). Cdks are serine/threonine kinases stably expressed during cell cycle progression that must bind to cyclins, their regulatory subunits, whose expression levels vary throughout the cell cycle phases, to form active catalytic heterodimers [50, 51]. Each Cdk is able to associate with different cyclins, which will in turn determine the proteins to be phosphorylated by a specific Cdk–cyclin complex (Fig. 1).
Several studies support the view that most cell cycle regulators are functionally redundant [52] and the need for a particular molecule is dependent on the cell type and on the niche [53]. This holds also true for the hippocampal neurogenic niche. As an example, studies on the role of Cdk4 and Cdk6 in the adult hippocampus unraveled a crucial role for Cdk6, but not Cdk4, in controlling the expansion of neuronal committed progenitors and thus the rate of neuronal production [54]. In fact, the absence of Cdk6 was shown to induce the lengthening of the G1 phase causing premature cell cycle exit and differentiation [54]. These findings lead to the “cell cycle length hypothesis” [55], which states that proliferative divisions exhibit a short G1 phase whereas neurogenic divisions are characterized by longer G1 phases. In a molecular perspective, it is proposed that the ability of a cell fate determinant to induce any cellular response is related with the time it has to act during G1 [55, 56]. Additionally, overexpression of the Cdk4–cyclin D1 complex in the adult mouse hippocampal niche was shown to increase the expansion of neural progenitor cells (NPCs), in a specific and cell–autonomous manner, while inhibiting neurogenesis [57]. Indeed, stopping the overexpression of the Cdk4–cyclin D1 complex was accompanied by an overproduction of new neurons, corroborating its effect on the expansion of the neural progenitors at the expense of neuronal differentiation. Moreover, no effects were observed in the survival and maturation patterns of DCX + immature neurons [57]. Altogether, results suggest that the Cdk4–cyclin D1 complex is able to decrease the cell cycle length of cells that characteristically present longer cell cycles, whereas it is not able to change the short-length cycles [57]. In line, it is proposed [55, 58] that beyond the cell cycle length itself, it is the length relative variation that may be underlying the changes in the fate of a given cell [57]; however, more studies are needed to clarify this subject.
Cyclins, another important class of cell cycle regulators, are also implicated in the adult neurogenic phenomenon. Indeed, three types of cyclins D (D1, D2 and D3), which control Cdk4/6 activity, were already identified in mammals [59]. Although most cells express more than one cyclin D, several studies have demonstrated cell type-dependent roles for each of them [60–62]. One paradigmatic example of such specificity in the adult brain is the demonstration that cyclin D2, but not cyclin D1, knock-out (KO) mice, have a marked reduction of cell proliferation in the DG, measured by BrdU incorporation [59]. Other studies have corroborated this inability of cyclin D1 to promote neurogenesis in the hippocampus in the absence of cyclin D2 [63, 64].
Cell cycle progression is also negatively regulated, at a post-translational level, by two families of cyclin-dependent kinase inhibitors: the Inhibitor of kinase 4/Alternative reading frame (Ink4/ARF) family and the CDK inhibitory protein/Kinase inhibitory protein (Cip/Kip) family. These intracellular proteins are responsible for slowing or arresting the progression through the cell cycle, by blocking imperative events. The expression pattern and function of some of these Cdk inhibitors in the context of adult neurogenesis have also been characterized. P27kip1 (p27) is an important Cdk inhibitor, that induces cell cycle exit in proliferative cells [65]. In accordance, p27 expression decreases when cells are exposed to mitogenic signals, allowing their entrance in the S phase [66]. In the context of adult hippocampal neurogenesis, p27 protein is expressed in the SGZ of mice [67] and rats [64]; interestingly, many of the p27 cells co-express DCX in this region [67]. In vitro assays, using cultured NPCs, showed rapid increases in p27 expression following differentiation by growth factor withdrawal, thus confirming its role in cell cycle arrest and NPCs differentiation [67]. Additionally, deletion of p27 promoted an increase in the proliferative pool of NPCs [67], in accordance with previously published results in the SEZ [68]. These data were corroborated by in vivo assays showing an increased number of BrdU positive cells in the SGZ and in the SEZ of p27 KO mice. These results, together with the increased levels of proliferating cell nuclear antigen (PCNA) expression in the KO animals, further suggest that the absence of p27 promotes NPCs proliferation in both adult neurogenic niches [67]. Adding to the p27 studies, Pechnick et al. [69, 70] carried out two separate studies exploring the function of p21cip1 (p21) in the mouse hippocampus. Using a BrdU incorporation paradigm (one injection every 2h in a total of three injections, and sacrifice 24 h after the first injection), the authors showed, both in vivo and in vitro, that the absence of p21 lead to an increased proliferation of hippocampal neurons. Ultimately, the work suggests that p21 is responsible for blocking cell cycle progression in the adult hippocampal SGZ [69, 70]. Interestingly, p21 expression is restricted to neuronal committed progenitors [70], unlike p27 that reveals no distinction on lineage preference [67]. Strikingly, the results are not in agreement with a previous report in which p21 deletion showed no impact on the proliferation of neural progenitors in basal conditions [71]. However, it is worth mentioning that a different BrdU incorporation paradigm was used in this latter study. Indeed, the use of BrdU incorporation approaches has limitations as a direct measure of proliferation because does not always discriminates among effects that may underlie an increased S phase labeling; for example, G1/G2 phase shortening, increase in the growth fraction and lengthening of S phase [56]. The appropriate controls, the use of additional thymidine analogs and endogenous markers of proliferation should improve the analyses and may give a broader picture on the cell cycle regulatory mechanisms.
A final word to mention E2F1, an element that is part of a broad family of transcription factors involved in the regulation of cell cycle progression [42], but also with important roles in the adult neurogenic process. Contrary to what is described for most members of this family, E2F1 has been reported to induce cell death, by forcing postmitotic cells to re-entry cell cycle [42, 72]. In the context of adult neurogenesis, E2F1 was shown to be important for cell proliferation and differentiation. Using a single BrdU injection paradigm, 2h before sacrifice, Cooper-Kuhn et al. [72] showed that E2F1-deficient mice have decreased cell proliferation and diminished neurogenesis, both in the hippocampal DG and the SVZ. These authors also described a decrease of about 60-70 % in apoptotic cells, in the hippocampal neurogenic niche of E2F1-deficient mice compared to wild-type (WT), further corroborating the role of this gene in regulating cell death in the context of adult neurogenesis [72]. Figure 1 depicts the cell cycle regulators described in the context of adult hippocampal neurogenesis.
Signaling Pathways
Cell cycle entry promotion and initial progression through G1 phase is induced by mitogens or growth factors present in the extracellular environment [64, 73, 74]. The interplay between cell cycle regulation and cell fate determination is also a topic of great relevance [56]. In particular, the G1 phase length is crucial for the switch from proliferation to differentiation and is modulated by cell cycle regulators and cell fate determinants [56]. Thus, signaling from the niche is suggested to be responsible for key processes in the regulation of adult neurogenesis homeostasis, including: the balance between quiescence versus proliferation, the mode of cell division, and the prevention of stem cell depletion [75, 76]. In this section we will briefly describe some of the signaling pathways activated in the adult hippocampal neurogenic niche.
The role of Notch signaling in NPCs in the adult hippocampus was investigated in vivo through inducible gain- and loss-of-function experiments. Activated Notch1 overexpression induced proliferation of endogenous progenitors, whereas inhibition or ablation of Notch1 signaling promoted cell cycle exit, inducing the transition from neural stem or progenitor cells to transit-amplifying cells or neurons [77]. On the other hand, in maturing neurons, Notch1 proved to be relevant for survival and structural plasticity modulation [77].
Bone morphogenetic proteins (BMPs) are other key regulatory components of the adult hippocampal neurogenic niche, restricting the proliferation of the stem cell pool, through BMP receptor-IA (BMPR-IA) activation, and thus maintaining the equilibrium between stem cell proliferation and quiescence [75]. Downregulation of endogenous BMP signaling promoted an increased proliferation of SOX2+ cells by recruiting quiescent radial cells into the cell cycle. Moreover, the canonical BMP signaling pathway is reactivated shortly after neuronal fate commitment, possibly to promote cell cycle exit of newly born neurons [75].
Sonic hedgehog (Shh) is an evolutionarily conserved secreted protein that plays an important role in many aspects of developmental control [78], as well as in adult hippocampal neurogenesis [79]. Shh signaling pathway was shown to induce a dose-dependent proliferative response in progenitors in vitro, whereas inhibition of Shh signaling reduced proliferation in vivo. These studies confirmed Shh signaling pathway as an important regulator of adult hippocampal neural progenitors [79], suggesting also its involvement in cell cycle regulation.
Like Shh, Wnt proteins are also well-known key regulators of NSC behavior during embryonic development [80]. Wnt signaling has been reported as a regulator of adult hippocampal neurogenesis [81], through the activation of the proneural gene NeuroD1 [82]. Activation of NeuroD1 is important for the generation of granule cells in the hippocampus and cerebellum [83], possibly by promoting cell cycle exit.
Altogether, these data highlight the complex orchestration of the cell cycle process in the context of adult hippocampal neurogenesis, as well as the interplay between cell cycle regulators and upstream molecular signaling pathways. Interestingly, there is a certain level of functional redundancy among cell cycle regulatory components, possibly as an evolutionary mechanism to prevent severe damage upon deficiency of one of these molecules. On the other hand, most of these studies point to tissue and cell specificity as a hallmark of these systems, proving that these regulators may operate at different levels of the cell cycle and implying the need for their fine tuning in the homeostatic control of adult hippocampal neurogenesis.
Cell Cycle Regulation in Neuropathological Scenarios
The molecular mechanisms and pathways regulating adult hippocampal neurogenesis in response to deleterious stimuli, and the contrasting actions of pro-neurogenic drugs, are still largely undisclosed. It is legitimate to consider that these alterations in adult neurogenesis may be attributable to direct or indirect changes in cell cycle regulatory mechanisms. As such, the cell cycle machinery is possibly a convergent pathway through which intrinsic and extrinsic factors, such as stress and toxins, manifest their effects. Indeed, cell cycle deregulation in the context of adult neurogenesis has been associated with the pathogenesis of neurodegenerative disorders, such as Alzheimer’s disease [84] and Parkinson’s disease [85], neuropsychiatric diseases, as is the case of schizophrenia [86] and major depression [87], and injury, namely stroke [88, 89]. These changes in cell cycle dynamics, as observed in several disease states [90–92], further reinforce the need for additional studies examining the role of core cell cycle players as targets for disruption. Next, we will briefly explore the case of depression as a paradigmatic example of how cell cycle deregulation can lead to the development of pathological traits.
Major Depression
Major depression is a chronic and debilitating disease, and one of the most common psychiatric disorders in modern society. It is estimated that about 16 % of the population will be affected by this disease once or several times during lifetime [93]. Like other psychiatric disorders, depression is a complex and heterogeneous clinical entity [94], dependent on the interaction between genetic susceptibility [95, 96] and environmental factors [97]. Depressive patients present symptoms of depressed_mood, learned helplessness, anhedonia and impaired cognition, and present a high co-morbidity with anxiety disorders [94].
Strikingly, depression is characterized by several pathophysiological alterations in the brain such as differences in size of specific brain regions, changes in neuronal morphology, neurochemical and signaling alterations, and changes in genetic and epigenetic regulation [98, 99]. Knowledge of the etiopathogenesis of depression has progressed substantially in the last years [100], in part due to studies employing animal models. Animal models of depression use known etiological factors (etiological validity) to induce behavioral and neurobiological symptoms in animals similar to those of the human disease (face validity). Moreover, a valid animal model for the formulation of hypotheses and for the development of novel therapeutic strategies should respond to clinically effective treatments (predictive validity) [94, 101]. There are several animal models of depression: chronic mild stress (CMS), social stress, early life stress, fear conditioning and olfactory bulbectomy [97]. Although none of them can fully recapitulate the complexity and heterogeneity of the human disease, they are considered robust approaches to study the depression in humans. For example, the CMS animal model presents alterations in the three behavioral domains known to be affected in depressive patients, i.e., mood, anxiety and cognition [94]. Despite this large contribution of data from studies in animal models of depression, and from post-mortem studies of human brains, the neurobiological basis of this disorder is still poorly defined. Importantly, the fact that approximately half of the patients presenting clinical depression show incomplete remission or relapse after treatment with the currently available antidepressants [97] further reinforces the need for finding new molecular targets and more efficient treatments.
There are currently several leading hypotheses that attempt to elucidate the neural and molecular mechanisms of depression. The monoamine hypothesis of depression [102] has been the most prevalent. The main support of this hypothesis is the fact that most classic antidepressants induce an increase of the serotonin and noradrenaline levels [103]. More recently, additional studies have shown that other mechanisms are implicated in the neurobiology of this disorder; this is mostly based on the observation that other factors are altered in depressed individuals [87, 104, 105] and on the efficacy of new antidepressants, in which the mechanisms of action do not rely on the monoamine transmission systems [106–109]. Thus, several other hypotheses on the etiology of depression have been put forward, including: the neurotrophin hypothesis, the cytokine hypothesis, the hypothalamic pituitary adrenal (HPA) axis modulation hypothesis and the neurogenic hypothesis. Although none of these are mutually exclusive, in this discussion we focus mostly on the role of adult hippocampal neurogenesis and on the molecular processes that can regulate it at the cell cycle level.
The Neurogenic Hypothesis of Depression and Cell Cycle (De)regulation
Studies showing reduced hippocampal neurogenesis in several animal models of depression [110–112] constitute the basis for the neurogenic hypothesis of depression. Importantly, all major classes of antidepressants [87, 113], and most of the environmental factors that confer antidepressant-like behavioral effects, such as environmental enrichment [114, 115], physical activity [115] and learning [7], are also known to promote hippocampal neurogenesis. These facts have lead to the proposal that neurogenesis may have a role in the etiopathogenesis of depression; however, the currently available data strongly reinforces the need for restructuring this possibly oversimplified view. Indeed, the functional implications of decreased neurogenesis for the precipitation and maintenance of the depressive state are yet to be completely established, as the experimental approaches and time frames of analysis diverge. Some studies have implicated neurogenesis in the emergence of behavioral deficits observed in animal models of depression and in the actions of antidepressants [116–118]. While, other studies showed that at least the short-term mood-improving actions of antidepressants depend on neuronal remodeling in the hippocampus and prefrontal cortex (PFC), rather than on neurogenesis [110]. Moreover, recently published data from our lab showed that the appropriate incorporation of new cells in the adult rat hippocampus is a key factor for the long-term spontaneous recovery from depressive-like behavior as well as for the action of antidepressants [119]. Using a longer experimental time frame, to allow the full differentiation and integration of newborn cells in the pre-existing neuro-glial circuitry, it was possible to fate-map the new cells generated during antidepressants treatment and understand their impact in distinct behavioral dimensions [119]. These findings further reinforce the need for an integrated time-dependent overview of the neurogenic phenomenon with great emphasis on the functional role of newly generated cells in the adult hippocampus. Importantly, most of the stimuli affecting adult neurogenesis, are also responsible for inducing changes at the cell cycle level in the progenitor cells of the hippocampal niche. Some of the most relevant reports on cell cycle regulation in the context of adult hippocampal neurogenesis and stress-related disorders have disclosed a major role for Cdk inhibitors [64, 69, 70]. Heine et al. [64] evaluated the role of p27 in the regulation of the cell cycle in the DG of rats following exposure to stress. After 3 weeks of chronic exposure to unpredictable stress, rats presented significantly decreased numbers of proliferating cells, measured by ki67 immunostaining, and increased numbers of p27 positive cells in the SGZ. Notably, this effect was not observed upon acute stress exposure. Moreover, the proliferation levels returned to normal after a 3-week recovery period from chronic stress, suggesting a transient p27-dependent G1 arrest in the SGZ cells of chronically stressed animals [64]. Somehow unexpectedly, neither cyclin-E nor cyclin-D1 protein levels were significantly altered in these animals when compared to controls [64].
Other animal studies have focused their attention on the role of Cdk inhibitors regarding the pro-neurogenic action of antidepressants [69, 70]. Pechnick et al. [69]showed that naïve mice chronically treated with imipramine, a tricyclic antidepressant, not only show increased neurogenesis in the DG but also decreased the expression of p21 Cdk inhibitor in the SGZ, when comparing to saline-treated controls. In a more recent study, the same group analyzed the effects of chronic administration of other classes of antidepressants on SGZ p21 expression and neurogenesis; all antidepressants tested (fluoxetine, imipramine and desipramine) were able to specifically inhibit p21 expression in the mice DG and this effect was linked to increased neurogenesis [70]. Unexpectedly, no change was noted in p27 expression following antidepressants administration [70], possibly suggesting specific roles for each of these Cdk inhibitors following different stimuli. It is worth mentioning that Pechnick et al. did not include an animal model of depression in their studies, possibly accounting for these results. Together, these findings support the involvement of cell cycle molecules in the mechanistic association between stress and the action of antidepressants, in the context of neurogenesis regulation.
A recent study has implicated the cyclin dependent kinase 5 (Cdk5)/p35 complex in the development of depressive-like behavior and in the action of antidepressants [120]. Cdk5 still has no recognizable function in the progression of the cell cycle [37, 47], although structurally similar to other Cdks. In fact, Cdk5 expression and activity occur almost exclusively in postmitotic neurons, both in the developing and in the adult brain [37]. This kinase works as a cell cycle inhibitor in postmitotic neurons, repressing aberrant cell cycle reentry, a phenomenon linked to the development of several neurodegenerative disorders [37, 121]. Cdk5 regulation requires activators that are specifically expressed in postmitotic neurons. One of these activators is p35, a regulatory subunit that translocates from the cytosol to the membrane to induce Cdk5 activity [120, 121]. In a recent study, it was reported an increased Cdk5 kinase activity together with the translocation of p35 to the cell membrane, in the DG of rats exposed to CMS. They also observed that inhibition of Cdk5 specifically in the DG, but not in the CA1 or CA3 of the hippocampus, prevented the CMS-induced behavioral impairments, further suggesting the involvement of the Cdk5/p35 complex in the etiology of depressive-like behavior. Remarkably, p35 overexpression blocked the antidepressant behavioral effects of venlafaxine, a selective serotonin reuptake inhibitor (SSRI) antidepressant [120]. These data suggest an association between Cdk5 activity and the development of stress-related disorders [120], similar to what has been previously described for some neurodegenerative disorders [121]. Moreover, the studies may also suggest that the effect of Cdk5 activation is attributable to the impairments typically observed in hippocampal neurogenesis induced by CMS exposure [120]. Complementary studies with analyses of cell proliferation and neurogenesis would help to better define Cdk5 function in the adult hippocampus. Figure. 2b and c show the schematic representations of the adult hippocampal neurogenesis changes observed in animal models of depression and after antidepressant treatment, and the corresponding cell cycle alterations.
Changes in the signaling pathways known to be involved in the modulation of adult hippocampal neurogenesis have also been indirectly associated with the development of depressive-like behavior in animal models. Indeed, Wnt knockdown-mediated neurogenesis ablation was shown to impair several hippocampal-dependent cognitive functions, such as long-term retention of spatial memory and object recognition memory [122]. Importantly, these cognitive behavioral deficits were linked with depression onset and maintenance [94, 123]. Wnt signaling was further implicated in the actions of fluoxetine, an SSRI antidepressant; chronic treatment with this antidepressant was able to stimulate the expression of Wnt3a protein in the hippocampal DG. However, Wnt activity appears to be preferentially implicated in fluoxetine’s reported induction of neural plasticity and not in its pro-neurogenic actions [124].
Notch and BMP signaling have also been shown to be mediators of the pro-neurogenic actions of physical exercise. Physical exercise is a stimulus with recognized antidepressant effects [125, 126]. Moreover, it has been consistently reported to robustly induce adult hippocampal neurogenesis, by promoting the proliferation of progenitors and the survival and maturation of newborn neurons [127-130]. More recently, some studies investigated the molecular signaling correlates of these cellular events [131-133]. Using thymidine analogs incorporation paradigms, Brandt et al. showed that voluntary exercise (i.e., mice that had access to a running wheel) preferentially promotes the proliferation of DCX+ type_3 precursor cells and Notch1-dependent cell cycle exit. Since Notch1 is known to induce proliferation and inhibit differentiation in earlier NPCs (type_1 and 2a cells) [77], it is interesting to notice these contrasting pro-neurogenic functions in more committed progenitors [132]. Altogether the findings support the use of experimental designs that specifically address the role of molecular determinants in each hippocampal cell type. Table 1 summarizes the most relevant studies regarding the cell cycle and signaling alterations implicated in adult hippocampal neurogenesis imbalances in the context of depressive-like behavior.
Conclusions and Perspectives
Sixty years after the first report of ongoing neurogenesis in the adult brain, we are now at the point of evaluating the physiological relevance of the incorporation of new neurons in pre-existing neuronal networks. The integrated studies on adult neurogenesis in its various stages—progenitor cells proliferation, cell cycle exit, migration and differentiation—have brought new players into the complex network of factors and molecular mediators that directly or indirectly participate in the process. Nonetheless, we were not yet able to establish the precise molecular cascades that regulate the homeostasis in adult neurogenic niches. Therefore, the future of this field of research needs to build up an integrated view of the molecular processes, by specifically targeting candidate molecules using conditional approaches to overcome the limitations of full_KO models. This approach will allow the exclusion of possible compensatory mechanisms promoted during embryonic development, a strategy that seems to be of particular importance in the case of cell cycle regulators. Additionally, most of the literature on the regulation of adult neurogenesis relies on the use of thymidine analogs incorporation, such as BrdU. The use of these strategies to study cell cycle regulation in the context of adult hippocampal neurogenesis requires careful interpretation of the data. In this way, the appropriate controls and additional strategies should be considered to ensure that the results definitely reflect the generation of new neural cells. Moreover, caution is needed when comparing different studies, as distinct experimental paradigms may draw contrasting conclusions.
More than a physiological phenomenon, adult hippocampal neurogenesis is a process by which the etiology of many neurodegenerative and neuropsychiatric disorders may be unraveled. More importantly, the neurogenic process is a substrate from which new molecular targets for treating these disorders may arise. The diverse ways of approaching the topic provide unique perspectives on how neurogenesis may be implicated in homeostatic responses and in the development of pathological states. The data reviewed here strongly supports that both direct and indirect cell cycle regulatory events may constitute relevant pieces to elucidate the complex mechanisms underlying the response to anti- and pro-neurogenic stimuli, in both basal conditions and in disease. These reports further emphasize the pertinence of modulating cell cycle regulators as targets for the development of new therapeutic approaches for disorders associated with neuroplastic imbalances.
Particularly in the case of major depression, new theories beyond monoamines have created a broader picture on how etiological factors are translated into disease and the action of antidepressants into the alleviation of the most common symptoms. In this context, much of the mechanisms are now being explored, including those interfering with adult hippocampal neurogenesis. However, the study of depression still represents a challenge for research since it involves the interplay between an individual’s genetic predisposition and molecular responses to environment. Certainly, the discovery of new molecular mediators will give us important clues on susceptibility or predisposition targets, promoting the establishment of novel disease models, in a feedback loop that would nourish the field with new perspectives.
References
Lemaire V, Tronel S, Montaron MF, Fabre A, Dugast E, Abrous DN (2012) Long-lasting plasticity of hippocampal adult-born neurons. J Neurosci 32(9):3101–3108. doi:10.1523/JNEUROSCI.4731-11.2012
Aimone JB, Deng W, Gage FH (2010) Adult neurogenesis: integrating theories and separating functions. Trends Cogn Sci 14(7):325–337. doi:10.1016/j.tics.2010.04.003
Abrous DN, Koehl M, Le Moal M (2005) Adult neurogenesis: From precursors to network and physiology. Physiol Rev 85(2):523–569. doi:10.1152/physrev.00055.2003
Deng W, Aimone JB, Gage FH (2010) New neurons and new memories: how does adult hippocampal neurogenesis affect learning and memory? Nat Rev Neurosci 11(5):339–350. doi:sj.npp.1300091/nrn2822
Shors TJ, Miesegaes G, Beylin A, Zhao M, Rydel T, Gould E (2001) Neurogenesis in the adult is involved in the formation of trace memories. Nature 410(6826):372–376. doi:sj.npp.1300091/35066584
Shors TJ, Anderson ML, Curlik DM 2nd, Nokia MS (2012) Use it or lose it: how neurogenesis keeps the brain fit for learning. Behav Brain Res 227(2):450–458. doi:10.1016/j.bbr.2011.04.023
Zhao C, Deng W, Gage FH (2008) Mechanisms and functional implications of adult neurogenesis. Cell 132(4):645–660. doi:10.1016/j.cell.2008.01.033
Seri B, Garcia-Verdugo JM, McEwen BS, Alvarez-Buylla A (2001) Astrocytes give rise to new neurons in the adult mammalian hippocampus. J Neurosci 21(18):7153–7160. doi:21/18/7153
Pinto L (2008) Molecular mechanisms regulating neurogenesis in the developing mouse cerebral cortex. Dissertation, Faculty of Biology Ludwig-Maximilian-University Munich,
Ehninger D, Kempermann G (2003) Regional effects of wheel running and environmental enrichment on cell genesis and microglia proliferation in the adult murine neocortex. Cereb Cortex 13(8):845–851. doi:10.3410/f.1016143.200028
Benraiss A, Chmielnicki E, Lerner K, Roh D, Goldman SA (2001) Adenoviral brain-derived neurotrophic factor induces both neostriatal and olfactory neuronal recruitment from endogenous progenitor cells in the adult forebrain. J Neurosci 21(17):6718–6731. doi:21/17/6718
Kodama M, Fujioka T, Duman RS (2004) Chronic olanzapine or fluoxetine administration increases cell proliferation in hippocampus and prefrontal cortex of adult rat. Biol Psychiatry 56(8):570–580. doi:10.1016/j.biopsych.2004.07.008
Gould E, Reeves AJ, Graziano MS, Gross CG (1999) Neurogenesis in the neocortex of adult primates. Science 286(5439):548–552. doi:10.1126/science.286.5439.548
Goncalves L, Silva R, Pinto-Ribeiro F, Pego JM, Bessa JM, Pertovaara A, Sousa N, Almeida A (2008) Neuropathic pain is associated with depressive behaviour and induces neuroplasticity in the amygdala of the rat. Exp Neurol 213(1):48–56. doi:10.1016/j.expneurol.2008.04.043
Bernier PJ, Bedard A, Vinet J, Levesque M, Parent A (2002) Newly generated neurons in the amygdala and adjoining cortex of adult primates. Proc Natl Acad Sci U S A 99(17):11464–11469. doi:10.1073/pnas.172403999
Fowler CD, Johnson F, Wang Z (2005) Estrogen regulation of cell proliferation and distribution of estrogen receptor-alpha in the brains of adult female prairie and meadow voles. J Comp Neurol 489(2):166–179. doi:10.1002/cne.20638
Kokoeva MV, Yin H, Flier JS (2005) Neurogenesis in the hypothalamus of adult mice: potential role in energy balance. Science 310(5748):679–683. doi:10.1126/science.1115360
Fowler CD, Liu Y, Ouimet C, Wang Z (2002) The effects of social environment on adult neurogenesis in the female prairie vole. J Neurobiol 51(2):115–128. doi:10.1002/neu.10042
Dayer AG, Cleaver KM, Abouantoun T, Cameron HA (2005) New GABAergic interneurons in the adult neocortex and striatum are generated from different precursors. J Cell Biol 168(3):415–427. doi:10.1083/jcb.200407053
Bedard A, Gravel C, Parent A (2006) Chemical characterization of newly generated neurons in the striatum of adult primates. Exp Brain Res 170(4):501–512. doi:10.1007/s00221-005-0233-5
Zhao M, Momma S, Delfani K, Carlen M, Cassidy RM, Johansson CB, Brismar H, Shupliakov O, Frisen J, Janson AM (2003) Evidence for neurogenesis in the adult mammalian substantia nigra. Proc Natl Acad Sci U S A 100(13):7925–7930. doi:10.1073/pnas.1131955100
Yoshimi K, Ren YR, Seki T, Yamada M, Ooizumi H, Onodera M, Saito Y, Murayama S, Okano H, Mizuno Y, Mochizuki H (2005) Possibility for neurogenesis in substantia nigra of parkinsonian brain. Ann Neurol 58(1):31–40. doi:10.1002/ana.20506
von Bohlen und Halbach O (2011) Immunohistological markers for proliferative events, gliogenesis, and neurogenesis within the adult hippocampus. Cell Tissue Res 345(1):1–19. doi:10.1007/s00441-011-1196-4
Lugert S, Basak O, Knuckles P, Haussler U, Fabel K, Gotz M, Haas CA, Kempermann G, Taylor V, Giachino C (2010) Quiescent and active hippocampal neural stem cells with distinct morphologies respond selectively to physiological and pathological stimuli and aging. Cell Stem Cell 6(5):445–456. doi:10.1016/j.stem.2010.03.017
Hodge RD, Kahoud RJ, Hevner RF (2012) Transcriptional control of glutamatergic differentiation during adult neurogenesis. Cell Mol Life Sci. doi:10.1007/s00018-011-0916-y
Balu DT, Lucki I (2009) Adult hippocampal neurogenesis: regulation, functional implications, and contribution to disease pathology. Neurosci Biobehav Rev 33(3):232–252. doi:10.1016/j.neubiorev.2008.08.007
Carlen M, Cassidy RM, Brismar H, Smith GA, Enquist LW, Frisen J (2002) Functional integration of adult-born neurons. Curr Biol 12(7):606–608. doi:S0960982202007716
Snyder JS, Choe JS, Clifford MA, Jeurling SI, Hurley P, Brown A, Kamhi JF, Cameron HA (2009) Adult-born hippocampal neurons are more numerous, faster maturing, and more involved in behavior in rats than in mice. J Neurosci 29(46):14484–14495. doi:10.1523/JNEUROSCI.1768-09.2009
Gu Y, Arruda-Carvalho M, Wang J, Janoschka SR, Josselyn SA, Frankland PW, Ge S (2012) Optical controlling reveals time-dependent roles for adult-born dentate granule cells. Nat Neurosci 15(12):1700–1706. doi:sj.npp.1300091/nn.3260
Kempermann G, Gast D, Kronenberg G, Yamaguchi M, Gage FH (2003) Early determination and long-term persistence of adult-generated new neurons in the hippocampus of mice. Development 130(2):391–399. doi:10.1242/dev.00203
Nokia MS, Sisti HM, Choksi MR, Shors TJ (2012) Learning to learn: theta oscillations predict new learning, which enhances related learning and neurogenesis. PLoS One 7(2):e31375. doi:10.1371/journal.pone.0031375
Biebl M, Cooper CM, Winkler J, Kuhn HG (2000) Analysis of neurogenesis and programmed cell death reveals a self-renewing capacity in the adult rat brain. Neurosci Lett 291(1):17–20. doi:10.1016/S0304-3940(00)01368-9
Lu Z, Elliott MR, Chen Y, Walsh JT, Klibanov AL, Ravichandran KS, Kipnis J (2011) Phagocytic activity of neuronal progenitors regulates adult neurogenesis. Nat Cell Biol 13(9):1076–1083. doi:sj.npp.1300091/ncb2299
Chesnokova V, Pechnick RN (2008) Antidepressants and Cdk inhibitors: releasing the brake on neurogenesis? Cell Cycle 7(15):2321–2326. doi:6446
Liu DZ, Ander BP, Sharp FR (2010) Cell cycle inhibition without disruption of neurogenesis is a strategy for treatment of central nervous system diseases. Neurobiol Dis 37(3):549–557. doi:10.1016/j.nbd.2009.11.013
Schmetsdorf S, Gartner U, Arendt T (2005) Expression of cell cycle-related proteins in developing and adult mouse hippocampus. Int J Dev Neurosci 23(1):101–112. doi:10.1016/j.ijdevneu.2004.07.019
Jessberger S, Gage FH, Eisch AJ, Lagace DC (2009) Making a neuron: Cdk5 in embryonic and adult neurogenesis. Trends Neurosci 32(11):575–582. doi:10.1016/j.tins.2009.07.002
Borghese L, Dolezalova D, Opitz T, Haupt S, Leinhaas A, Steinfarz B, Koch P, Edenhofer F, Hampl A, Brustle O (2010) Inhibition of notch signaling in human embryonic stem cell-derived neural stem cells delays G1/S phase transition and accelerates neuronal differentiation in vitro and in vivo. Stem Cells 28(5):955–964. doi:10.1002/stem.408
Ye W, Mairet-Coello G, Pasoreck E, Dicicco-Bloom E (2009) Patterns of p57Kip2 expression in embryonic rat brain suggest roles in progenitor cell cycle exit and neuronal differentiation. Dev Neurobiol 69(1):1–21. doi:10.1002/dneu.20680
Legrier ME, Ducray A, Propper A, Chao M, Kastner A (2001) Cell cycle regulation during mouse olfactory neurogenesis. Cell Growth Differ 12(12):591–601
Legrier ME, Ducray A, Propper A, Kastner A (2001) Region-specific expression of cell cycle inhibitors in the adult brain. Neuroreport 12(14):3127–3131. doi:10.1097/00001756-200110080-00029
Yoshikawa K (2000) Cell cycle regulators in neural stem cells and postmitotic neurons. Neurosci Res 37(1):1–14. doi:10.1016/S0168-0102(00)00101-2
Herrup K, Neve R, Ackerman SL, Copani A (2004) Divide and die: cell cycle events as triggers of nerve cell death. J Neurosci 24(42):9232–9239. doi:10.1523/JNEUROSCI.3347-04.2004
Khurana V, Lu Y, Steinhilb ML, Oldham S, Shulman JM, Feany MB (2006) TOR-mediated cell-cycle activation causes neurodegeneration in a Drosophila tauopathy model. Curr Biol 16(3):230–241. doi:10.1016/j.cub.2005.12.042
Di Giovanni S, Movsesyan V, Ahmed F, Cernak I, Schinelli S, Stoica B, Faden AI (2005) Cell cycle inhibition provides neuroprotection and reduces glial proliferation and scar formation after traumatic brain injury. Proc Natl Acad Sci U S A 102(23):8333–8338. doi:10.1073/pnas.0500989102
Kuan CY, Schloemer AJ, Lu A, Burns KA, Weng WL, Williams MT, Strauss KI, Vorhees CV, Flavell RA, Davis RJ, Sharp FR, Rakic P (2004) Hypoxia-ischemia induces DNA synthesis without cell proliferation in dying neurons in adult rodent brain. J Neurosci 24(47):10763–10772. doi:10.1523/JNEUROSCI.3883-04.2004
Herrup K, Yang Y (2007) Cell cycle regulation in the postmitotic neuron: oxymoron or new biology? Nat Rev Neurosci 8(5):368–378. doi:sj.npp.1300091/nrn2124
Frank CL, Tsai LH (2009) Alternative functions of core cell cycle regulators in neuronal migration, neuronal maturation, and synaptic plasticity. Neuron 62(3):312–326. doi:10.1016/j.neuron.2009.03.029
Kim AH, Puram SV, Bilimoria PM, Ikeuchi Y, Keough S, Wong M, Rowitch D, Bonni A (2009) A centrosomal Cdc20-APC pathway controls dendrite morphogenesis in postmitotic neurons. Cell 136(2):322–336. doi:10.1016/j.cell.2008.11.050
Soriano S, Currais A, Hortobagyi T (2009) The neuronal cell cycle as a mechanism of pathogenesis in Alzheimer's disease. Aging-Us 1(4):363–371
Galderisi U, Jori FP, Giordano A (2003) Cell cycle regulation and neural differentiation. Oncogene 22(33):5208–5219. doi:sj.npp.1300091/sj.onc.1206558
Vandenbosch R, Borgs L, Beukelaers P, Foidart A, Nguyen L, Moonen G, Berthet C, Kaldis P, Gallo V, Belachew S, Malgrange B (2007) CDK2 is dispensable for adult hippocampal neurogenesis. Cell Cycle 6(24):3065–3069. doi:10.4161/cc.6.24.5048
Malumbres M, Sotillo R, Santamaria D, Galan J, Cerezo A, Ortega S, Dubus P, Barbacid M (2004) Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell 118(4):493–504. doi:10.1016/j.cell.2004.08.002
Beukelaers P, Vandenbosch R, Caron N, Nguyen L, Belachew S, Moonen G, Kiyokawa H, Barbacid M, Santamaria D, Malgrange B (2011) Cdk6-dependent regulation of G(1) length controls adult neurogenesis. Stem Cells 29(4):713–724. doi:10.1002/stem.616
Calegari F, Huttner WB (2003) An inhibition of cyclin-dependent kinases that lengthens, but does not arrest, neuroepithelial cell cycle induces premature neurogenesis. J Cell Sci 116(Pt 24):4947–4955. doi:10.1242/jcs.00825
Salomoni P, Calegari F (2010) Cell cycle control of mammalian neural stem cells: putting a speed limit on G1. Trends Cell Biol 20(5):233–243. doi:10.1016/j.tcb.2010.01.006
Artegiani B, Lindemann D, Calegari F (2011) Overexpression of cdk4 and cyclinD1 triggers greater expansion of neural stem cells in the adult mouse brain. J Exp Med 208(5):937–948. doi:10.1084/jem.20102167
Calegari F, Haubensak W, Haffner C, Huttner WB (2005) Selective lengthening of the cell cycle in the neurogenic subpopulation of neural progenitor cells during mouse brain development. J Neurosci 25(28):6533–6538. doi:10.1523/JNEUROSCI.0778-05.2005
Kowalczyk A, Filipkowski RK, Rylski M, Wilczynski GM, Konopacki FA, Jaworski J, Ciemerych MA, Sicinski P, Kaczmarek L (2004) The critical role of cyclin D2 in adult neurogenesis. J Cell Biol 167(2):209–213. doi:10.1083/jcb.200404181
Sicinski P, Donaher JL, Geng Y, Parker SB, Gardner H, Park MY, Robker RL, Richards JS, McGinnis LK, Biggers JD, Eppig JJ, Bronson RT, Elledge SJ, Weinberg RA (1996) Cyclin D2 is an FSH-responsive gene involved in gonadal cell proliferation and oncogenesis. Nature 384(6608):470–474. doi:sj.npp.1300091/384470a0
Sicinska E, Aifantis I, Le Cam L, Swat W, Borowski C, Yu Q, Ferrando AA, Levin SD, Geng Y, von Boehmer H, Sicinski P (2003) Requirement for cyclin D3 in lymphocyte development and T cell leukemias. Cancer Cell 4(6):451–461. doi:10.1016/S1535-6108(03)00301-5
Ma C, Papermaster D, Cepko CL (1998) A unique pattern of photoreceptor degeneration in cyclin D1 mutant mice. Proc Natl Acad Sci U S A 95(17):9938–9943. doi:10.1073/pnas.95.17.9938
Glickstein SB, Alexander S, Ross ME (2007) Differences in cyclin D2 and D1 protein expression distinguish forebrain progenitor subsets. Cereb Cortex 17(3):632–642. doi:10.1093/cercor/bhk008
Heine VM, Maslam S, Joels M, Lucassen PJ (2004) Increased P27KIP1 protein expression in the dentate gyrus of chronically stressed rats indicates G1 arrest involvement. Neuroscience 129(3):593–601. doi:10.1016/j.neuroscience.2004.07.048
Cobrinik D (2005) Pocket proteins and cell cycle control. Oncogene 24(17):2796–2809. doi:sj.npp.1300091/sj.onc.1208619
Vidal A, Koff A (2000) Cell-cycle inhibitors: three families united by a common cause. Gene 247(1–2):1–15. doi:10.1016/S0378-1119(00)00092-5
Qiu J, Takagi Y, Harada J, Topalkara K, Wang Y, Sims JR, Zheng G, Huang P, Ling Y, Scadden DT, Moskowitz MA, Cheng T (2009) p27Kip1 constrains proliferation of neural progenitor cells in adult brain under homeostatic and ischemic conditions. Stem Cells 27(4):920–927. doi:10.1002/stem.1
Doetsch F, Verdugo JM, Caille I, Alvarez-Buylla A, Chao MV, Casaccia-Bonnefil P (2002) Lack of the cell-cycle inhibitor p27Kip1 results in selective increase of transit-amplifying cells for adult neurogenesis. J Neurosci 22(6):2255–2264. doi:22/6/2255
Pechnick RN, Zonis S, Wawrowsky K, Pourmorady J, Chesnokova V (2008) p21Cip1 restricts neuronal proliferation in the subgranular zone of the dentate gyrus of the hippocampus. Proc Natl Acad Sci U S A 105(4):1358–1363. doi:10.1073/pnas.0711030105
Pechnick RN, Zonis S, Wawrowsky K, Cosgayon R, Farrokhi C, Lacayo L, Chesnokova V (2011) Antidepressants Stimulate Hippocampal Neurogenesis by Inhibiting p21 Expression in the Subgranular Zone of the Hipppocampus. PLoS One 6(11):e27290. doi:10.1371/journal.pone.0027290
Qiu J, Takagi Y, Harada J, Rodrigues N, Moskowitz MA, Scadden DT, Cheng T (2004) Regenerative response in ischemic brain restricted by p21cip1/waf1. J Exp Med 199(7):937–945. doi:10.1084/jem.20031385
Cooper-Kuhn CM, Vroemen M, Brown J, Ye H, Thompson MA, Winkler J, Kuhn HG (2002) Impaired adult neurogenesis in mice lacking the transcription factor E2F1. Mol Cell Neurosci 21(2):312–323. doi:10.1006/mcne.2002.1176
Sherr CJ (1993) Mammalian G1 cyclins. Cell 73(6):1059–1065. doi:10.1016/0092-8674(93)90636-5
Sherr CJ, Kato J, Quelle DE, Matsuoka M, Roussel MF (1994) D-type cyclins and their cyclin-dependent kinases: G1 phase integrators of the mitogenic response. Cold Spring Harb Symp Quant Biol 59:11–19. doi:10.1101/SQB.1994.059.01.004
Mira H, Andreu Z, Suh H, Lie DC, Jessberger S, Consiglio A, San Emeterio J, Hortiguela R, Marques-Torrejon MA, Nakashima K, Colak D, Gotz M, Farinas I, Gage FH (2010) Signaling through BMPR-IA regulates quiescence and long-term activity of neural stem cells in the adult hippocampus. Cell Stem Cell 7(1):78–89. doi:10.1016/j.stem.2010.04.016
Morrison SJ, Spradling AC (2008) Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell 132(4):598–611. doi:10.1016/j.cell.2008.01.038
Breunig JJ, Silbereis J, Vaccarino FM, Sestan N, Rakic P (2007) Notch regulates cell fate and dendrite morphology of newborn neurons in the postnatal dentate gyrus. Proc Natl Acad Sci U S A 104(51):20558–20563. doi:10.1073/pnas.0710156104
Suh H, Deng W, Gage FH (2009) Signaling in adult neurogenesis. Annu Rev Cell Dev Biol 25:253–275. doi:10.1146/annurev.cellbio.042308.113256
Lai K, Kaspar BK, Gage FH, Schaffer DV (2003) Sonic hedgehog regulates adult neural progenitor proliferation in vitro and in vivo. Nat Neurosci 6(1):21–27. doi:sj.npp.1300091/nn983
Kleber M, Sommer L (2004) Wnt signaling and the regulation of stem cell function. Curr Opin Cell Biol 16(6):681–687. doi:10.1016/j.ceb.2004.08.006
Lie DC, Colamarino SA, Song HJ, Desire L, Mira H, Consiglio A, Lein ES, Jessberger S, Lansford H, Dearie AR, Gage FH (2005) Wnt signalling regulates adult hippocampal neurogenesis. Nature 437(7063):1370–1375. doi:sj.npp.1300091/nature04108
Kuwabara T, Hsieh J, Muotri A, Yeo G, Warashina M, Lie DC, Moore L, Nakashima K, Asashima M, Gage FH (2009) Wnt-mediated activation of NeuroD1 and retro-elements during adult neurogenesis. Nat Neurosci 12(9):1097–1105. doi:sj.npp.1300091/nn.2360
Miyata T, Maeda T, Lee JE (1999) NeuroD is required for differentiation of the granule cells in the cerebellum and hippocampus. Genes Dev 13(13):1647–1652. doi:10.1101/gad.13.13.1647
Mu Y, Gage FH (2011) Adult hippocampal neurogenesis and its role in Alzheimer's disease. Mol Neurodegener 6:85. doi:10.1186/1750-1326-6-85
Winner B, Kohl Z, Gage FH (2011) Neurodegenerative disease and adult neurogenesis. Eur J Neurosci 33(6):1139–1151. doi:10.1111/j.1460-9568.2011.07613.x
Reif A, Fritzen S, Finger M, Strobel A, Lauer M, Schmitt A, Lesch KP (2006) Neural stem cell proliferation is decreased in schizophrenia, but not in depression. Mol Psychiatry 11(5):514–522. doi:sj.npp.1300091/sj.mp.4001791
Sahay A, Hen R (2007) Adult hippocampal neurogenesis in depression. Nat Neurosci 10(9):1110–1115. doi:sj.npp.1300091/nn1969
Zhang RL, Zhang ZG, Lu M, Wang Y, Yang JJ, Chopp M (2006) Reduction of the cell cycle length by decreasing G1 phase and cell cycle reentry expand neuronal progenitor cells in the subventricular zone of adult rat after stroke. J Cereb Blood Flow Metab 26(6):857–863. doi:sj.npp.1300091/sj.jcbfm.9600237
Zhang RL, Zhang ZG, Roberts C, LeTourneau Y, Lu M, Zhang L, Wang Y, Chopp M (2008) Lengthening the G(1) phase of neural progenitor cells is concurrent with an increase of symmetric neuron generating division after stroke. J Cereb Blood Flow Metab 28(3):602–611. doi:sj.npp.1300091/sj.jcbfm.9600556
Hoglinger GU, Breunig JJ, Depboylu C, Rouaux C, Michel PP, Alvarez-Fischer D, Boutillier AL, Degregori J, Oertel WH, Rakic P, Hirsch EC, Hunot S (2007) The pRb/E2F cell-cycle pathway mediates cell death in Parkinson's disease. Proc Natl Acad Sci U S A 104(9):3585–3590. doi:10.1073/pnas.0611671104
Fan Y, Abrahamsen G, McGrath JJ, Mackay-Sim A (2012) Altered cell cycle dynamics in schizophrenia. Biol Psychiatry 71(2):129–135. doi:10.1016/j.biopsych.2011.10.004
Cunningham KA, Watson CS (2008) Cell cycle regulation, neurogenesis, and depression. Proc Natl Acad Sci U S A 105(7):2259–2260. doi:10.1073/pnas.0800029105
Lee S, Jeong J, Kwak Y, Park SK (2010) Depression research: where are we now? Mol Brain 3:8. doi:10.1186/1756-6606-3-8
Bessa JM, Mesquita AR, Oliveira M, Pego JM, Cerqueira JJ, Palha JA, Almeida OF, Sousa N (2009) A trans-dimensional approach to the behavioral aspects of depression. Front Behav Neurosci 3:1. doi:10.3389/neuro.08.001.2009
Lesch KP, Bengel D, Heils A, Sabol SZ, Greenberg BD, Petri S, Benjamin J, Muller CR, Hamer DH, Murphy DL (1996) Association of anxiety-related traits with a polymorphism in the serotonin transporter gene regulatory region. Science 274(5292):1527–1531. doi:10.1126/science.274.5292.1527
Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H, McClay J, Mill J, Martin J, Braithwaite A, Poulton R (2003) Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science 301(5631):386–389. doi:10.1126/science.1083968
Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM (2002) Neurobiology of depression. Neuron 34(1):13–25. doi:10.1016/S0896-6273(02)00653-0
Tsankova N, Renthal W, Kumar A, Nestler EJ (2007) Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci 8(5):355–367. doi:sj.npp.1300091/nrn2132
Mateus-Pinheiro A, Pinto L, Sousa N (2011) Epigenetic (de)regulation of adult hippocampal neurogenesis: implications for depression. Clinical Epigenetics 3(1):5. doi:10.1186/1868-7083-3-5
Krishnan V, Nestler EJ (2008) The molecular neurobiology of depression. Nature 455(7215):894–902. doi:sj.npp.1300091/nature07455
Berton O, Hahn CG, Thase ME (2012) Are we getting closer to valid translational models for major depression? Science 338(6103):75–79. doi:10.1126/science.1222940
Heninger GR, Delgado PL, Charney DS (1996) The revised monoamine theory of depression: a modulatory role for monoamines, based on new findings from monoamine depletion experiments in humans. Pharmacopsychiatry 29(1):2–11. doi:10.1055/s-2007-979535
Wong ML, Licinio J (2001) Research and treatment approaches to depression. Nat Rev Neurosci 2(5):343–351. doi:sj.npp.1300091/35072566
Pariante CM, Lightman SL (2008) The HPA axis in major depression: classical theories and new developments. Trends Neurosci 31(9):464–468. doi:10.1016/j.tins.2008.06.006
Berton O, Nestler EJ (2006) New approaches to antidepressant drug discovery: beyond monoamines. Nat Rev Neurosci 7(2):137–151. doi:sj.npp.1300091/nrn1846
Hanoun N, Mocaer E, Boyer PA, Hamon M, Lanfumey L (2004) Differential effects of the novel antidepressant agomelatine (S 20098) versus fluoxetine on 5-HT1A receptors in the rat brain. Neuropharmacology 47(4):515–526. doi:10.1016/j.neuropharm.2004.06.003
Zoladz PR, Muñoz C, Diamond DM (2010) Beneficial Effects of Tianeptine on Hippocampus-Dependent Long-Term Memory and Stress-Induced Alterations of Brain Structure and Function. Pharmaceuticals 3(10):3143–3166. doi:10.3390/ph3103143
Papp M, Gruca P, Boyer PA, Mocaer E (2003) Effect of agomelatine in the chronic mild stress model of depression in the rat. Neuropsychopharmacology 28(4):694–703. doi:sj.npp.1300091/sj.npp.1300091
Kasper S, McEwen BS (2008) Neurobiological and clinical effects of the antidepressant tianeptine. CNS Drugs 22(1):15–26. doi:10.2165/00023210-200822010-00002
Bessa JM, Ferreira D, Melo I, Marques F, Cerqueira JJ, Palha JA, Almeida OF, Sousa N (2009) The mood-improving actions of antidepressants do not depend on neurogenesis but are associated with neuronal remodeling. Mol Psychiatry 14 (8):764–773, 739. doi:sj.npp.1300091/mp.2008.119
Pham K, Nacher J, Hof PR, McEwen BS (2003) Repeated restraint stress suppresses neurogenesis and induces biphasic PSA-NCAM expression in the adult rat dentate gyrus. Eur J Neurosci 17(4):879–886. doi:10.1046/j.1460-9568.2003.02513.x
Vollmayr B, Simonis C, Weber S, Gass P, Henn F (2003) Reduced cell proliferation in the dentate gyrus is not correlated with the development of learned helplessness. Biol Psychiatry 54(10):1035–1040. doi:S0006-3223(03)00527-4
Malberg JE, Eisch AJ, Nestler EJ, Duman RS (2000) Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J Neurosci 20(24):9104–9110. doi:sj.npp.1300091/mp.2010.34
Schloesser RJ, Lehmann M, Martinowich K, Manji HK, Herkenham M (2010) Environmental enrichment requires adult neurogenesis to facilitate the recovery from psychosocial stress. Mol Psychiatry 15(12):1152–1163. doi:sj.npp.1300091/mp.2010.34
Brown J, Cooper-Kuhn CM, Kempermann G, Van Praag H, Winkler J, Gage FH, Kuhn HG (2003) Enriched environment and physical activity stimulate hippocampal but not olfactory bulb neurogenesis. Eur J Neurosci 17(10):2042–2046. doi:10.1046/j.1460-9568.2003.02647.x
Santarelli L, Saxe M, Gross C, Surget A, Battaglia F, Dulawa S, Weisstaub N, Lee J, Duman R, Arancio O, Belzung C, Hen R (2003) Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 301(5634):805–809. doi:10.1126/science.1083328
Surget A, Saxe M, Leman S, Ibarguen-Vargas Y, Chalon S, Griebel G, Hen R, Belzung C (2008) Drug-dependent requirement of hippocampal neurogenesis in a model of depression and of antidepressant reversal. Biol Psychiatry 64(4):293–301. doi:10.1016/j.biopsych.2008.02.022
David DJ, Samuels BA, Rainer Q, Wang JW, Marsteller D, Mendez I, Drew M, Craig DA, Guiard BP, Guilloux JP, Artymyshyn RP, Gardier AM, Gerald C, Antonijevic IA, Leonardo ED, Hen R (2009) Neurogenesis-dependent and -independent effects of fluoxetine in an animal model of anxiety/depression. Neuron 62(4):479–493. doi:10.1016/j.neuron.2009.04.017
Mateus-Pinheiro A, Pinto L, Bessa JM, Morais M, Alves ND, Monteiro S, Patricio P, Almeida OF, Sousa N (2013) Sustained remission from depressive-like behavior depends on hippocampal neurogenesis. Transl Psychiatry 3:e210. doi:sj.npp.1300091/tp.2012.141
Zhu WL, Shi HS, Wang SJ, Xu CM, Jiang WG, Wang X, Wu P, Li QQ, Ding ZB, Lu L (2011) Increased Cdk5/p35 activity in the dentate gyrus mediates depressive-like behaviour in rats. Int J Neuropsychopharmaco 1:1–15. doi:10.1017/S1461145711000915
Lopes JP, Agostinho P (2011) Cdk5: multitasking between physiological and pathological conditions. Prog Neurobiol 94(1):49–63. doi:10.1016/j.pneurobio.2011.03.006
Jessberger S, Clark RE, Broadbent NJ, Clemenson GD Jr, Consiglio A, Lie DC, Squire LR, Gage FH (2009) Dentate gyrus-specific knockdown of adult neurogenesis impairs spatial and object recognition memory in adult rats. Learn Mem 16(2):147–154. doi:10.1101/lm.1172609
Disner SG, Beevers CG, Haigh EA, Beck AT (2011) Neural mechanisms of the cognitive model of depression. Nat Rev Neurosci 12(8):467–477. doi:sj.npp.1300091/nrn3027
Pinnock SB, Blake AM, Platt NJ, Herbert J (2010) The roles of BDNF, pCREB and Wnt3a in the latent period preceding activation of progenitor cell mitosis in the adult dentate gyrus by fluoxetine. PLoS One 5(10):e13652. doi:10.1371/journal.pone.0013652
Ernst C, Olson AK, Pinel JP, Lam RW, Christie BR (2006) Antidepressant effects of exercise: evidence for an adult-neurogenesis hypothesis? J Psychiatry Neurosci 31(2):84–92. doi:20060300s00002p84
Lucassen PJ, Meerlo P, Naylor AS, van Dam AM, Dayer AG, Fuchs E, Oomen CA, Czeh B (2010) Regulation of adult neurogenesis by stress, sleep disruption, exercise and inflammation: Implications for depression and antidepressant action. Eur Neuropsychopharmacol 20(1):1–17. doi:10.1016/j.euroneuro.2009.08.003
Clark PJ, Bhattacharya TK, Miller DS, Kohman RA, DeYoung EK, Rhodes JS (2012) New neurons generated from running are broadly recruited into neuronal activation associated with three different hippocampus-involved tasks. Hippocampus 22(9):1860–1867. doi:10.1002/hipo.22020
van Praag H, Kempermann G, Gage FH (1999) Running increases cell proliferation and neurogenesis in the adult mouse dentate gyrus. Nat Neurosci 2(3):266–270. doi:sj.npp.1300091/6368
Kronenberg G, Reuter K, Steiner B, Brandt MD, Jessberger S, Yamaguchi M, Kempermann G (2003) Subpopulations of proliferating cells of the adult hippocampus respond differently to physiologic neurogenic stimuli. J Comp Neurol 467(4):455–463. doi:10.1002/cne.10945
Snyder JS, Glover LR, Sanzone KM, Kamhi JF, Cameron HA (2009) The effects of exercise and stress on the survival and maturation of adult-generated granule cells. Hippocampus 19(10):898–906. doi:10.1002/hipo.20552
Gobeske KT, Das S, Bonaguidi MA, Weiss C, Radulovic J, Disterhoft JF, Kessler JA (2009) BMP signaling mediates effects of exercise on hippocampal neurogenesis and cognition in mice. PLoS One 4(10):e7506. doi:10.1371/journal.pone.0007506
Brandt MD, Maass A, Kempermann G, Storch A (2010) Physical exercise increases Notch activity, proliferation and cell cycle exit of type-3 progenitor cells in adult hippocampal neurogenesis. Eur J Neurosci 32(8):1256–1264. doi:10.1111/j.1460-9568.2010.07410.x
Bruel-Jungerman E, Veyrac A, Dufour F, Horwood J, Laroche S, Davis S (2009) Inhibition of PI3K-Akt signaling blocks exercise-mediated enhancement of adult neurogenesis and synaptic plasticity in the dentate gyrus. PLoS One 4(11):e7901. doi:10.1371/journal.pone.0007901
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Patrício, P., Mateus-Pinheiro, A., Sousa, N. et al. Re-cycling Paradigms: Cell Cycle Regulation in Adult Hippocampal Neurogenesis and Implications for Depression. Mol Neurobiol 48, 84–96 (2013). https://doi.org/10.1007/s12035-013-8422-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-013-8422-x