
Abstract
Differences in diet appear to contribute substantially to the burden of disease in populations, and therefore changes in diet could lead to major improvements in public health. This is predicated on the reliable identification of causal effects of nutrition on health, and unfortunately nutritional epidemiology has deficiencies in terms of identifying these. This is reflected in the many cases where observational studies have suggested that a nutritional factor is protective against disease, and randomized controlled trials have failed to verify this. The use of genetic variants as proxy measures of nutritional exposure—an application of the Mendelian randomization principle—can contribute to strengthening causal inference in this field. Genetic variants are not subject to bias due to reverse causation (disease processes influencing exposure, rather than vice versa) or recall bias, and if obvious precautions are applied are not influenced by confounding or attenuation by errors. This is illustrated in the case of epidemiological studies of alcohol intake and various health outcomes, through the use of genetic variants related to alcohol metabolism (in ALDH2 and ADH1B). Examples from other areas of nutritional epidemiology and of the informative nature of gene–environment interactions interpreted within the Mendelian randomization framework are presented, and the potential limitations of the approach addressed.
Similar content being viewed by others
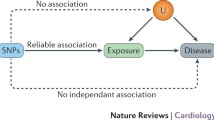

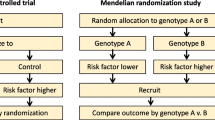
Introduction
A range of classical epidemiological studies—including migration studies and the analysis of secular trends and ecological differences in disease rates—demonstrate that for most common complex diseases environmentally modifiable risk factors account for much of the burden of disease. Twin studies—that by definition exclude time trends and geographical differences in disease risk and thus provide lower (and often substantially lower) estimates of the modifiable aspects of disease risk than apply in practice—support this contention [1]. Identifying modifiable causes of disease, which can be then manipulated to improve individual and public health, is thus a key task for epidemiology. In this paper, I will argue that, paradoxically, incorporating germline genetic variants—which are essentially fixed—into epidemiological studies can strengthen evidence regarding the undeniably major role of modifiable risk processes in determining population health.
There are, however, important limitations to the ability of observational studies to reliably identify causes of disease, which have been particularly evident in the nutrition field. Consider the following two examples, from many that could be presented. Several observational studies suggested that the use of vitamin E supplements was associated with a reduced risk of coronary heart disease, two of the most influential coming from the Health Professionals Follow-Up Study [2] and the Nurses’ Health Study [3], both published in the New England Journal of Medicine in 1993. Findings from one of these studies are presented in Fig. 1, where it can be seen that even short-term use of vitamin E supplements was associated with reduced coronary heart disease risk (CHD), which persisted after adjustment for confounding factors. Nearly half of US adults are taking either vitamin E supplements or multivitamin/multimineral supplements that generally contain vitamin E [4], and data from the three available time points suggest there has been a particular increase in vitamin E use following 1993 [5], possibly consequent upon the publication of the two observational studies mentioned above, which have received over 3,000 citations between them since publication. The apparently strong observational evidence with respect to vitamin E and reduced CHD risk, which may have influenced the very high current use of vitamin E supplements in developed countries, was unfortunately not realised in randomized controlled trials (Fig. 2), in which no benefit from vitamin E supplementation use is seen. In this example, it is important to note that the observational studies and the randomized controlled trials were testing precisely the same exposure—short-term vitamin E supplement use—and yet yielded very different findings with respect to the apparent influence on risk.

Observed effect of duration of vitamin E use compared to no use on CHD events in the Health Professional Follow-up Study [2]

A similar scenario has been played out in regard to vitamin C. In 2001, the Lancet published an observational study demonstrating an inverse association between circulating vitamin C levels and incident coronary heart disease [6]. The left-hand side of Fig. 3 summarises these data, presenting the relative risk for 15.7 μmol/l higher plasma vitamin C level, assuming a log-linear association. As can be seen, adjustment for confounders had little impact on this association. However, a large-scale randomized controlled trial, the Heart Protection Study, examined the effect of a supplement that increased average plasma vitamin C levels by 15.7 μmol/l. In this study, randomization to the supplement was associated with no decrement in coronary heart disease risk [7].

Estimates of the effects of an increase of 15.7 μmol/l plasma vitamin C on CHD 5-year mortality estimated from the observational epidemiological EPIC [6] and the randomised controlled Heart Protection Study [7] (EPIC m men, age-adjusted; EPIC m* men, adjusted for systolic blood pressure, cholesterol, BMI, smoking, diabetes and vitamin supplement use; EPIC f women, age-adjusted; EPIC f* women, adjusted for systolic blood pressure, cholesterol, BMI, smoking, diabetes and vitamin supplement use)
What underlies the discrepancy between these findings? One possibility is that there is considerable confounding between vitamin C levels and other exposures that could increase the risk of coronary heart disease. In the British Women’s Heart and Health study (BWHHS), for example, women with higher plasma vitamin C levels were less likely to be in a manual social class, have no car access, be a smoker or be obese and more likely to exercise, be on a low-fat diet, have a daily alcoholic drink, and be tall [8]. Furthermore for these women in their 60s and 70s those with higher plasma vitamin C levels were less likely to have come from a home many decades ago in which the head of household was in a manual job, or had no bathroom or hot water, or within which they had to share a bedroom. They were also less likely to have limited educational attainment. In short, a substantial amount of confounding by factors from across the life course that predict elevated risk of coronary heart disease was seen.
In the BWHHS, 15.7 mmol/l higher plasma vitamin C level was associated with a relative risk of incident coronary heart disease of 0.88 (95% CI 0.80–0.97), in the same direction as the estimates seen in the observational study summarized in Fig. 3. When adjusted for the same confounders as were adjusted for in the observational study reported in Fig. 3, the estimate changed very little—to 0.90 (95% CI 0.82–0.99). When additional adjustment for confounders acting across the life course was made, considerable attenuation was seen, with a residual relative risk of 0.95 (95% CI 0.85–1.05) [9]. It is obvious that given inevitable amounts of measurement imprecision in the confounders, or a limited number of missing unmeasured confounders, the residual association is essentially null and close to the finding of the randomized controlled trial. Most studies have more limited information on potential confounders than is available in the BWHHS, and in other fields we may know less about the confounding factors we should measure. In these cases, inferences drawn from observational epidemiological studies may be seriously misleading. As the major and compelling rationale for doing these observational studies is to underpin public health prevention strategies, their repeated failures are a major concern for public health policy makers, researchers and funders. Whilst sophisticated methods of taking measurement error into account, including measurement error in confounders, have been introduced into nutritional epidemiology [10–12], they cannot guarantee that observational study effects are reliable estimates of underlying causal effects [13, 14].
Other processes in addition to confounding can generate robust, but non-causal, associations in observational studies. Reverse causation—where the disease influences the apparent exposure, rather than vice versa, may generate strong and replicable associations. For example, many studies have found that people with low circulating cholesterol levels are at increased risk of several cancers, including colon cancer. If causal, this is an important association as it might mean that efforts to lower cholesterol levels would increase the risk of cancer. However, it is possible that the early stages of cancer may, many years before diagnosis or death, lead to a lowering in cholesterol levels, rather than low cholesterol levels increasing the risk of cancer. Reverse causation can also occur through behavioural processes—for example, people with early stages and symptoms of cardiovascular disease may reduce their consumption of alcohol, which would generate a situation in which alcohol intake appears to protect against cardiovascular disease. A form of reverse causation can also occur through reporting bias, with the presence of disease influencing reporting disposition. In retrospective case–control studies, people with the disease under investigation may report on their prior exposure history in a different way than do controls—perhaps because the former will think harder about potential reasons to account for why they have developed the disease.
The problems of confounding and bias discussed above relate to the production of associations in observational studies that are not reliable indicators of the true direction of causal associations. A separate issue is that the strength of associations between causal risk factors and disease in observational studies will generally be underestimated due to random measurement imprecision in indexing the exposure. A century ago Charles Spearman demonstrated mathematically how such measurement imprecision would lead to what he termed the ‘attenuation by errors’ of associations [15, 16]. This has more latterly been renamed ‘regression dilution bias’.
Observational studies in the nutritional epidemiology field can and do produce findings that either spuriously enhance or downgrade estimates of causal associations between modifiable exposures and disease. This has serious consequences for the appropriateness of interventions that aim to reduce disease risk in populations. It is for these reasons that alternative approaches—including those within the Mendelian randomization framework—need to be applied.
Background to Mendelian randomization
The basic principle utilized in the Mendelian randomization approach is that if genetic variants either alter the level of, or mirror the biological effects of, a modifiable environmental exposure that itself alters disease risk, then these genetic variants should be related to disease risk to the extent predicted by their influence on exposure to the risk factor. Common genetic polymorphisms that have a well-characterized biological function (or are markers for such variants) can therefore be utilized to study the effect of a suspected environmental exposure on disease risk [17–21]. The variants should not have an association with the disease outcome except through their link with the modifiable risk process of interest.
It may seem counter intuitive to study genetic variants as proxies for environmental exposures rather than measure the exposures themselves. However, there are several crucial advantages of utilizing functional genetic variants (or their markers) in this manner, which relate to the problems with observational studies outlined above. First, unlike environmental exposures, genetic variants are not generally associated with the wide range of behavioural, social and physiological factors that can confound associations. This means that if a genetic variant is used as a proxy for an environmentally modifiable exposure, it is unlikely to be confounded in the way that direct measures of the exposure will be. Further, aside from the effects of population structure [22], such variants will not be associated with other genetic variants, except through linkage disequilibrium (the association of alleles located close together on a chromosome).
Second, inferences drawn from observational studies may be subject to bias due to reverse causation. Disease processes may influence exposure levels such as alcohol intake, or measures of intermediate phenotypes, such as cholesterol levels and C-reactive protein. However, germline genetic variants associated with average alcohol intake or circulating levels of intermediate phenotypes will not be influenced by the onset of disease. This will also be true with respect to reporting bias generated by knowledge of disease status in case–control studies, or of differential reporting bias in any study design.
Finally, a genetic variant will indicate long-term levels of exposure, and, if the variant is considered to be a proxy for such exposure, it will not suffer from the measurement error inherent in phenotypes that have high levels of variability. For example, differences between groups defined by cholesterol level–related genotype will, over a long period, reflect the cumulative differences in absolute cholesterol levels between the groups. For individuals, blood cholesterol is variable over time, and the use of single measures of cholesterol will underestimate the true strength of association between cholesterol and, for instance, coronary heart disease. Indeed, use of the Mendelian randomization approach predicts a strength of association that is in line with randomized controlled trial findings of effects of cholesterol lowering, when the increasing benefits seen over the relatively short trial period are projected to the expectation for differences over a lifetime [18]. A particular strength of Mendelian randomization approaches is that genetic variants generally proxy for long-term differences in exposure levels. For intermediate phenotypes (circulating cholesterol or C-reactive protein levels), genetic variants tend to be associated with differences of a similar order of magnitude throughout life. For some behavioural factors, such as alcohol intake, associations will only emerge at the stage of life when the behaviour is instigated.
In the Mendelian randomization framework, the associations of genotype with outcomes are of interest because of the strengthened inference they allow about the action of the environmental modifiable risk factors that the genotypes proxy for, rather than what they say about genetic mechanisms per se. Mendelian randomization studies are aimed at informing strategies to reduce disease risk through influencing the non-genetic component of modifiable risk processes.
The principle of Mendelian randomization relies on the basic (but approximate) laws of Mendelian genetics. If the probability that a postmeiotic germ cell that has received any particular allele at segregation contributes to a viable concepts is independent of environment (following from Mendel’s first law), and if genetic variants sort independently (following from Mendel’s second law), then at a population level these variants will not be associated with the confounding factors that generally distort conventional observational studies. Empirical evidence that there is lack of confounding of genetic variants with factors that confound exposures in conventional observational epidemiological studies comes from several sources. For example, consider the virtually identical allele frequencies in the British 1958 birth cohort and British blood donors [23]. Blood donors are clearly a very selected sample of the population, whereas the 1958 birth cohort comprised all births in 1 week in Britain with minimal selection bias. Blood donors and the general population sample would differ considerably with respect to the behavioural, socio-economic and physiological risk factors that are often the confounding factors in observational epidemiological studies. However, they hardly differ in terms of allele frequencies. Similarly, we have demonstrated the lack of association between a range of SNPs of known phenotypic effects and nearly 100 socio-cultural, behavioural and biological risk factors for disease [24].
Mendelian randomization and nutrition-related exposures
The principle of using genetic variation to proxy for a modifiable exposure was explicitly applied in observational studies from the 1960s, with a series of studies that utilized genetically–determined lactase persistence as an indicator of milk intake, and used this marker to inform evidence regarding the effect of consuming milk on several health-related outcomes [25–27]. The approach was hypothetically proposed for investigating whether low circulating cholesterol levels causally influenced cancer risk by Martijn Katan in 1986 [28]. The term Mendelian randomization was introduced by Richard Gray and Keith Wheatley in 1991 [29], in the context of an innovative genetically informed observational approach to assess the effects of bone marrow transplantation in the treatment of childhood acute myeloid leukaemia. More recently, the term has been widely used in discussions of observational epidemiological studies [17, 30–33]. Further discussion of the origins of this approach is given elsewhere [34], and recent reviews have dealt explicitly with the application of Mendelian randomization within nutritional epidemiology [35, 36].
There are several categories of inference that can be drawn from studies utilizing the Mendelian randomization approach. In the most direct forms, genetic variants can be related to the probability or level of exposure (“exposure propensity”) or to intermediate phenotypes believed to influence disease risk. Less direct evidence can come from genetic variant-disease associations that indicate that a particular biological pathway may be of importance, perhaps because the variants modify the effects of environmental exposures [17, 18, 21, 37, 38]. I illustrate some of these categories within investigations of the effects of alcohol on various health outcomes.
Alcohol intake and blood pressure
The consequences of alcohol drinking for health range from the well established (effects on liver cirrhosis and accidents) to the uncertain (coronary heart disease, depression and dementia). For example, the possible protective effect of moderate alcohol consumption on coronary heart disease (CHD) risk remains highly controversial [39–41]. Non-drinkers may be at a higher risk of CHD because health problems (perhaps induced by previous alcohol abuse) dissuade them from drinking [42]. In addition to this form of reverse causation, confounding could play a role, with non-drinkers being more likely to display an adverse profile of socioeconomic or other behavioural risk factors for CHD. Alternatively, alcohol may have a direct biological effect that lessens the risk of CHD—for example by increasing the levels of protective high-density lipoprotein (HDL) cholesterol [43]. It is, however, unlikely that an RCT of differential levels of alcohol intake, adequate to test whether there is a protective effect of alcohol on CHD events, will ever be carried out.
Alcohol is oxidized to acetaldehyde, which in turn is oxidized by aldehyde dehydrogenases (ALDHs) to acetate. Half of Japanese people are heterozygotes or homozygotes for a null variant of ALDH2, and peak blood acetaldehyde concentrations post alcohol challenge are 18 times and 5 times higher, respectively, among homozygous null variant and heterozygous individuals compared with homozygous wild-type individuals [44]. This renders the consumption of alcohol unpleasant through inducing facial flushing, palpitations, drowsiness and other symptoms, and there are very considerable differences in alcohol consumption according to genotype. The principles of Mendelian randomization are seen to apply—two factors that would be expected to be associated with alcohol consumption, age and cigarette smoking, which would confound conventional observational associations between alcohol and disease, are not related to genotype despite the strong association of genotype with alcohol consumption [45].
It would be expected that ALDH2 genotype influences diseases known to be related to alcohol consumption and as proof of principle it has been shown that ALDH2 null variant homozygosity—associated with low alcohol consumption—is indeed related to a lower risk of liver cirrhosis [46]. Considerable evidence, including data from short-term randomized controlled trials, suggests that alcohol increases HDL cholesterol levels [47, 48] (which should protect against CHD). In line with this, ALDH2 genotype is strongly associated with HDL cholesterol in the expected direction [45]. With respect to blood pressure, observational evidence suggests that long-term alcohol intake produces an increased risk of hypertension and higher prevailing blood pressure levels. However the results from different studies vary and there is clearly a very large degree of potential confounding between alcohol and other exposures that would influence blood pressure. As in the case of vitamin E intake and coronary heart disease discussed earlier, we could be looking at a confounded rather than a causal association. Indeed evidence of controversy in this area is reflected by newspaper coverage of a recent study suggesting that moderate alcohol consumption has beneficial effects, even for hypertensive men [49], with headlines like “Moderate drinking may help men with high blood pressure”.
Evidence regarding the causal nature of the association of alcohol drinking with blood pressure can come from studies of ALDH2 genotype and blood pressure. A meta-analysis of such studies suggests there is indeed a substantial positive effect of alcohol on blood pressure [50]. As shown in Fig. 4, alcohol consumption is strongly related to genotype among men, and despite higher levels of overall alcohol consumption in some studies compared with others the shape of the association remains similar. Among women, however, who drink very little compared to men, there is no evidence of association between drinking and genotype. Figure 5 demonstrates that men who are homozygous for the wild type have nearly two and half times the risk of hypertension than men who are homozygous for the null variant. Heterozygous men who drink an intermediate amount of alcohol have a more modest elevated risk of hypertension compared to men who are homozygous for the null variant. Thus, a dose–response association of hypertension and genotype is seen, in line with the dose–response association between genotype and alcohol intake. Among men homozygous for the null variant, who drink considerably less alcohol than those homozygous for the wild type, systolic and diastolic blood pressures are considerably lower. By contrast, among women, for whom genotype is unrelated to alcohol intake, there is no association between genotype and blood pressure. The differential genotype—blood pressure associations in men and women suggest that there is no other mechanism linking genotype and blood pressure than that relating to alcohol intake. If alternative pathways existed, both men and women would be expected to have the same genotype–blood pressure association.

ALDH2 genotype by alcohol consumption, g/day: 5 studies, n = 6,815 [50]

Forest plot of studies of ALDH2 genotype and hypertension [50]
In this example, the interaction is between a genetic variant and gender. Gender indicates substantial differences in alcohol consumption, which lead to the genotype being strongly associated with alcohol consumption in one group (males), but not associated in the other group (females), because of very low levels of alcohol consumption, irrespective of genotype, among the latter group. The power of this interaction is that it indicates that it is the association with alcohol intake and not any other aspects of the function of the genotype that is influencing blood pressure. If it were due to a pleiotropic effect of the genetic variation then the association between genotype and blood pressure would be seen for women as well as men.
Alcohol and illegal substance use: testing the “gateway hypothesis”
In many contexts, people who drink alcohol manifest higher rates of illegal substance use. This could reflect common social and environmental factors that increase uptake of several behaviours, or underlying genetic vulnerability factors. An alternative is the “gateway hypothesis” that postulates that alcohol use itself increases liability to initiate and maintain non-alcohol substance use [51–53]. The Mendelian randomization approach has been applied in a study of East Asian Americans, all born in Korea but living in the United States from infancy, among whom ALDH2 status was associated with alcohol use and alcohol use was associated with tobacco, marijuana, and other illegal drug use. ALDH2 variation was not robustly associated with non-alcohol substance use, however, which was taken to provide evidence against the “gateway hypothesis” [51].
Maternal drinking, the intrauterine environment and offspring outcomes
The influence of high levels of alcohol intake by pregnant women on the health and development of their offspring is well recognized for very high levels of intake, in the form of foetal alcohol syndrome [54]. However, the influence outside of this extreme situation is less easy to assess, particularly as higher levels of alcohol intake will be related to a wide array of potential socio-cultural, behavioural and environmental confounding factors. Furthermore, there may be systematic bias in how mothers report alcohol intake during pregnancy, which could distort associations with health outcomes. Therefore, outside of the case of very high alcohol intake by mothers, it is difficult to establish a causal link between maternal alcohol intake and offspring developmental characteristics. Some studies have approached this in ways that can be interpreted within the Mendelian randomization framework by investigating alcohol-metabolizing genotypes in mothers and offspring outcomes.
Studies have generally utilized a variant in the alcohol dehydrogenase gene (ADH1B*3 allele). Alcohol dehydrogenase metabolises alcohol to acetaldehyde and the ADH1B variant influences the rate of such metabolism. The ADH1B*3 variant has a reasonable prevalence among African Americans and is related to faster alcohol metabolism. This can be associated with a lower level of drinking, possibly because the faster metabolism leads to a more rapid spike in acetaldehyde, with its aversive effects. At a given level of drinking, faster metabolism will clear blood alcohol more rapidly, so less high levels will be reached and these will more quickly return to low levels. Both of these processes, if occurring in the mother, would protect the foetus from the effects of alcohol. Some studies have selected mothers who have a universally high level of alcohol consumption and among these mothers the alcohol-metabolizing genotypes will relate to alcohol levels that could have a toxic effect on the developing foetus, but not to their drinking, which is universally high. In this circumstance, the genotypic differences will mimic the differences in level of alcohol intake with regard to the foetal exposure to maternal circulating alcohol. Although sample sizes have been low and the analysis strategies not optimal, studies applying this approach provide some evidence to support the influence of maternal genotype, and thus of alcohol, on offspring outcomes [54–56]. Studies that have been able to analyse both maternal genotype and foetal genotype find that it is the maternal genotype that is related to offspring outcomes, as anticipated if the crucial exposure related to maternal alcohol intake and alcohol levels.
As in other examples of Mendelian randomization, these studies are of relevance because they provide evidence of the influence of maternal alcohol levels on offspring development, rather than because they highlight a particular maternal genotype that is of importance. In the absence of alcohol drinking, the maternal genotype would presumably have no influence on offspring outcomes. Studies utilizing maternal genotype as a proxy for environmentally modifiable influences on the intrauterine environment can be analysed in a variety of ways. First, the mothers of offspring with a particular outcome can be compared to a control group of mothers who have offspring without the outcome, in a conventional case–control design, but with the mother as the exposed individual (or control) rather than the offspring with the particular health outcome (or the control offspring). Fathers could serve as a control group when autosomal genetic variants are being studied. If the exposure is mediated by the mother, maternal genotype, rather than offspring genotype, will be the appropriate exposure indicator. Clearly, maternal and offspring genotype are associated, but conditional on each other, it should be the maternal genotype that shows the association with the health outcome among the offspring. Indeed, in theory it would be possible to simply compare genotype distributions of mother and offspring, with a higher prevalence among mothers providing evidence that maternal genotype, through an intrauterine pathway, is of importance. However, the statistical power of such an approach is low, and an external control group, whether fathers or women who have offspring without the health outcome, is generally preferable.
Other examples of Mendelian randomization in relation to nutritional exposures
With respect to exposure propensity Mendelian randomization can be applied to milk consumption (through use of genetic variants related to lactase persistence), although given the low strength of association of such genetic variation and milk consumption sample sizes need to be large [57]. Molecular genetic variation in taste receptors relates to different patterns of dietary intake, in particular with respect to bitter taste perception and cruciferous vegetable intake [58]; however, differences in taste are likely to be related to a range of dietary differences and therefore do not serve as specific proxies for any particular component of diet.
There is considerably greater potential for the application of Mendelian randomization in testing the causal nature of the associations observed between nutritionally influenced intermediate phenotypes and disease outcomes. This can provide good evidence on the influence of nutritional factors on disease. For example, many studies demonstrate robust effects of differences in dietary fat intake on circulating cholesterol levels, and Mendelian randomization studies demonstrate that genetic variants associated with higher cholesterol levels are associated with higher risk of coronary heart disease [38]. This proof-of-principle example confirms what has been demonstrated in randomized controlled trials of cholesterol lowering through the use of statins, that cholesterol levels are causally related to coronary heart disease risk. The implication is that various methods of modifying cholesterol levels, such as dietary changes, are likely to influence coronary heart disease risk, although of course there could be other influences of the dietary changes that counterbalance such an effect.
As indicated earlier, there is considerable interest in the possibility that circulating antioxidants may protect against various disease states, and therefore molecular genetic variants associated with different levels of circulating antioxidants can be utilized to determine if these associations are causal. For example a variant in the SLC23A1 gene, which codes for the Sodium Dependent Vitamin C Transporter protein 1 (SVCT1), is associated with a reasonably large difference in circulating vitamin C levels [59]. This can be utilized to test whether the apparent protective effects of higher circulating vitamin C levels against a variety of adverse health outcomes are causal. It would be expected that higher dietary intake of vitamin C—that results in higher circulating levels—would reduce the risk of these adverse health outcomes to the extent predicted by any causal associations identified using the Mendelian randomization approach. Similarly, molecular genetic variation related to circulating α-tocopherol [60] and carotenoids [61] can be utilized to elucidate the causal effects of these factors.
Another example of a nutritionally influenced intermediate phenotype is seen in studies of the association of high body mass index (BMI) and a variety of cardiovascular risk factors. A variant in the FTO gene is robustly associated with differences in BMI, and as shown in Fig. 6 FTO, variation predicts risk factor levels to the degree expected, given its effect on BMI and a causal association between BMI and these risk factors [62]. With considerably greater statistical power, the causal association of BMI on blood pressure level and hypertension has been demonstrated [63] and been shown to persist into old age, whilst the observational associations (perhaps due to a greater degree of confounding and disease-related weight loss) attenuate with age. A causal nature for the positive association between body mass index and bone mineral density—possibly responsible for the protective effect of greater body mass index on fracture risk—has also been suggested utilizing this approach [64].

The observed effects of FTO variation on metabolic traits are as predicted by the associations of body mass index with the same metabolic traits [62]
Mendelian randomization and randomized controlled trials
RCTs are clearly the definitive means of obtaining evidence on the effects of modifying disease risk processes. There are similarities in the logical structure of RCTs and Mendelian randomization, relating to the unconfounded nature of exposures for which genetic variants serve as proxies (analogous to the unconfounded nature of a randomized intervention), the impossibility of reverse causation as an influence on exposure-outcome associations in both Mendelian randomization and RCT settings, and the importance of intention to treat analyses—i.e., analysis by group defined by genetic variant, irrespective of associations between the genetic variant and the exposure for which this is a proxy for any particular individual.
The analogy with RCTs is also useful with respect to one objection that has been raised in conjunction with Mendelian randomization studies. This is that the environmentally modifiable exposure for which genetic variants serve as proxies (such as alcohol intake) is influenced by many other factors in addition to the genetic variants [65]. This is of course true. However, consider an RCT of blood pressure–lowering medication. Blood pressure is mainly influenced by factors other than taking blood pressure lowering medication—obesity, alcohol intake, salt consumption and other dietary factors, smoking, exercise, physical fitness, genetic factors and early-life developmental influences are all of importance. However, the randomization that occurs in trials ensures that these factors are balanced between the groups that receive the blood pressure lowering medication and those that do not. Thus, the fact that many other factors are related to the modifiable exposure does not compromise the power of RCTs; neither does it diminish the strength of Mendelian randomization designs. A related objection is that the genetic variants often explain only a trivial proportion of the variance in the environmentally modifiable risk factor for which the genetic variants are surrogate variables [66]. Again, consider an RCT of blood pressure-lowering medication, where 50% of participants receive the medication and 50% received a placebo. If the antihypertensive therapy reduced blood pressure by a quarter of a standard deviation (i.e., a 5 mmHg reduction in systolic blood pressure, given systolic blood pressure has a standard deviation of 20 mmHg in the population) then within the whole study group, treatment assignment (i.e., antihypertensive use vs. placebo) will explain 5/202 = 1.25% of the variance. In the example of ALDH2 variation and alcohol, the genetic variant explains about 2% of the variance in alcohol intake in the largest study available on this issue [45]. As can be seen, the quantitative association of genetic variants as instruments can be similar to that of randomized treatments with respect to biological processes that such treatments modify. Genetic variants are often as strong—if not stronger—predictors of unconfounded differences in exposures as are the randomized treatments in RCTs. The use of haplotypes or multiple independent genetic variants at different loci related to the exposure of interest can also be used to increase statistical power.
Mendelian randomization and instrumental variable approaches
In addition to the analogy with RCTs, Mendelian randomization can also be likened to instrumental variable approaches that have been heavily utilized in econometrics and social science, although rather less so in epidemiology. In an instrumental variable approach, the instrument is a variable that is only related to the outcome through its association with the modifiable exposure of interest. The instrument is not related to confounding factors nor is its assessment biased in a manner that would generate a spurious association with the outcome. Furthermore, the instrument will not be influenced by the development of the outcome (i.e., there will be no reverse causation). The development of instrumental variable methods within econometrics, in particular, has led to a sophisticated suite of statistical methods for estimating causal effects, and these have now been applied within Mendelian randomization studies [20, 63, 67]. The parallels between Mendelian randomization and instrumental variable approaches are discussed in more detail elsewhere [20, 68]. The instrumental variable method allows for the estimation of the causal effect size of the modifiable environmental exposure of interest and the outcome, together with estimates of the precision of the effect. Thus, in the example of alcohol intake (indexed by ALDH2 genotype) and blood pressure, it is possible to utilize the joint associations of ALDH2 genotype and alcohol intake and ALDH2 genotype and blood pressure to estimate the causal influence of alcohol intake on blood pressure. There are convenient rules of thumb, such as the rule that the first stage F test should be over 10 for an instrument to be of adequate strength, which can be adopted from the econometrics field, in which instrumental variables methods have been well developed [69], and applied to the Mendelian randomization setting.
Gene-by-environment interactions interpreted within a Mendelian randomization framework
Mendelian randomization is one way in which genetic epidemiology can inform understanding about environmental determinants of disease. A more conventional approach to the joint study of genes and environment has been to study interactions between environmental exposures and genotype [70–72]. From epidemiological and Mendelian randomization perspectives, several issues arise with gene–environment interactions.
The most reliable findings in genetic association studies relate to the main effects of polymorphisms on disease risk [32]. The power to detect meaningful gene–environment interaction is low [73], with the result being that there are a large number of reports of spurious gene–environment interactions in the medical literature [74]. The presence or absence of statistical interactions depends upon the scale (e.g., linear or logarithmic with respect to the exposure-disease outcome association) and the meaning of observed deviation from either an additive or multiplicative model is not clear. Furthermore, the biological implications of interactions (however defined) are generally uncertain [75]. Mendelian randomization is most powerful when studying modifiable exposures that are difficult to measure and/or considerably confounded, such as dietary factors. Given measurement error—particularly if this is differential with respect to other factors influencing disease risk—interactions are both difficult to detect and often misleading when, apparently, they are found [32].
Given these caveats, gene-by-environment interactions can be informative with respect to both cause and mechanism of disease. This can be demonstrated with respect to the investigation of alcohol as a potential cause of head and neck and oesophageal cancer. For these cancers, alcohol intake appears to increase the risk, although some have questioned the importance of its role [76].
A rare variant in the alcohol dehydrogenase 1B gene has been shown to be associated with lower levels of alcohol intake [77], and this same variant provides substantial protection against the risk of head and neck cancer [78]. If this association was due to the influence of alcohol consumption, it would be expected that no genotypic effect would be seen within never drinkers, and this is indeed what is seen (Fig. 7, top panel). Thus, this qualitative gene-by-environment interaction—of an effect of genotype in alcohol consumers and no effect in never drinkers—supports the role of alcohol consumption in increasing the risk of head and neck cancer.

Risk of upper aerodigestive cancer by ADH1B genetic variation, stratified by drinking intensity and smoking status. Odds ratio (OR) of upper aerodigestive cancer by re1229984 (ADH1B). Rare allele (dominant model) carriers versus common allele homozygous genotype. ORs are standardised by age, sex, center, cumulative alcohol consumption and, when relevant, smoking. ORs and 95% CI are derived from fixed effects models. Source: Hashibe et al. [78]
In relation to ALDH2 genotype, a meta-analysis of studies of its association with oesophageal cancer risk [79] found that people who are homozygous for the null variant, who therefore consume considerably less alcohol, have a greatly reduced risk of oesophageal cancer. The reduction in risk is close to that predicted from the size of effect of genotype on alcohol consumption and the dose–response association of alcohol and oesophageal cancer risk [80]. A similar picture is seen when head and neck cancer is the outcome [81].
Thus, with respect to the homozygous null variant versus homozygous wild type, the situation is similar to that of our blood pressure example—the genotypic association provides evidence of the effect of alcohol consumption, through allowing comparison of a group of low drinkers to a group who drink considerable amounts of alcohol, with no confounding factors differing between these groups. With respect to both oesophageal and head and neck cancer, acetaldehyde (the metabolite that is increased in people carrying the null variant who do drink alcohol) is considered to be carcinogenic [82]. Thus, drinkers who carry the null variant have higher levels of acetaldehyde than those who do not carry the variant. As shown above, people who are homozygous for the null variant drink very little alcohol, but heterozygous individuals do drink. When the heterozygotes are compared with wild type homozygotes, an interesting picture emerges—the risk of oesophageal cancer is higher in the heterozygotes, although they drink less alcohol than the homozygotes. If alcohol itself acted directly as the immediate causal factor, cancer risk would be intermediate in the heterozygotes compared with the other two groups. Acetaldehyde is the more likely causal factor, as heterozygotes as a group drink some alcohol but metabolize it inefficiently, leading to accumulation of higher levels of acetaldehyde than would occur in homozygotes for the common variant, who metabolize alcohol efficiently, and homozygotes for the null variant, who drink insufficient alcohol to produce raised acetaldehyde levels. Examination of the difference in oesophageal cancer risk between ALDH2 heterozygotes and those homozygous for the wild type, stratified by drinking status, reveals that in non-drinkers there is no robust evidence of any association between genotype and oesophageal cancer outcomes, as would be expected if the underlying environmentally modifiable causal factor were alcohol intake and the mechanism was through acetaldehyde levels. In further support of the hypothesis, amongst people who were drinking alcohol there was increased risk amongst heterozygotes, who have higher acetaldehyde levels, and this was especially marked in heavy drinkers, who would have the greatest difference in acetaldehyde levels according to genotype (Fig. 8). A similar analysis has been performed for head and neck cancer and again demonstrates no association of genotype and cancer risk in never drinkers and a graded association according to level of alcohol intake among alcohol drinkers [81].

Risk of oesophageal cancer in individuals with the ALDH2*1*2 versus *1*1 genotype [75]. The “other” category are alcohol drinkers who fall outside of the heavy drinking categories
Identifying the causal element within complex dietary mixtures
Particular dietary intakes tend to correlate with each other, such that individuals with high fruit and vegetable consumption would be more likely to have low saturated fat intake, for example. Furthermore, different micronutrient intakes will show correlations, such that high vitamin C intake would be associated with higher on-average beta-carotene and vitamin E intake, for example. Separating out which specific aspects of the diet are causally related to disease is problematic in this context. For example, studies of neural tube defects (NTDs) demonstrate that mothers of offspring with NTDs were different with respect to many aspects of their dietary intake from control mothers [83, 84]. The mothers of cases have lower intakes of many vitamins, for example. In this situation, a test of folic acid metabolism—the FIGLU test [85]—pointed to folate as the crucial element [86]. With molecular genetic approaches, demonstration of gene-by-environment interactions can help identify which particular dietary factor is related to disease risk. However, as demonstrated in Fig. 7 with regard to alcohol and cigarette smoking, the correlated nature of exposures will lead to interactions with relevant genotypes being seen both for the causative factor (which the genotype may well modify absorption or metabolism of) and the non-causal factor, but the interaction will be stronger for the casual factor. In this situation, identifying the strongest gene-by-environment interactions—in particular when the genotype is known to modify absorption or metabolism of one of the dietary factors under study—can help isolate the specific nutritional factor having a causal influence on the disease outcome.
Problems and limitations of Mendelian randomization
The Mendelian randomization approach provides useful evidence on the influence of modifiable exposures on health outcomes. However, there are several limitations to this approach, in particular relating to the need for large sample sizes and adequate statistical power. These have been discussed at considerable length elsewhere [17, 21, 35] and therefore the focus here is on implications of these for interpretation of gene-by-environment interaction.
Confounding of genotype-environmentally modifiable risk factor-disease associations
The power of Mendelian randomization lies in its ability to avoid the often substantial confounding seen in conventional observational epidemiology. However, confounding can be reintroduced into Mendelian randomization studies and when interpreting the results, this possibility needs to be considered. First, it is possible that the locus under study is in linkage disequilibrium—i.e., is associated—with another polymorphic locus, with the former being confounded by the latter. It may seem unlikely, given the relatively short distances over which linkage disequilibrium is seen in the human genome, that a polymorphism influencing, for instance, CHD risk, would be associated with another polymorphism influencing CHD risk (and thus producing confounding). There are, nevertheless, examples of different genes influencing the same metabolic pathway being in physical proximity. For example, different polymorphisms influencing alcohol metabolism appear to be in linkage disequilibrium [87].
Second, Mendelian randomization is most useful when it can be used to relate a single intermediate phenotype to a disease outcome. However, polymorphisms may (and probably often will) influence more than one intermediate phenotype, and this may mean they proxy for more than one environmentally modifiable risk factor. This pleiotropy can be generated through multiple effects mediated by their RNA expression or protein coding, through alternative splicing, where one polymorphic region contributes to alternative forms of more than one protein [88], or through other mechanisms. The most robust interpretations will be possible when the functional polymorphism appears to directly influence the level of the intermediate phenotype of interest, but such examples are probably going to be less common in Mendelian randomization than in cases where the polymorphism could in principle influence several systems, with different potential interpretations of how the effect on outcome is generated.
Linkage disequilibrium and pleiotropy can reintroduce confounding and thus reduce the potential value of the Mendelian randomization approach. Genomic knowledge may help in estimating the degree to which these are likely to be problems in any particular Mendelian randomization study, through, for instance, explication of genetic variants that may be in linkage disequilibrium with the variant under study, or the function of a particular variant and its known pleiotropic effects. Furthermore, genetic variation can be analyzed in relation to measures of potential confounding factors in each study and the magnitude of such confounding estimated. Empirical studies to date suggest that common genetic variants are largely unrelated to the behavioural and socioeconomic factors considered to be important confounders in conventional observational studies [24]. However, relying on measurement of confounders does, of course, remove the central purpose of Mendelian randomization, which is to balance unmeasured as well as measured confounders.
In some circumstances, the genetic variant will be related to the environmentally modifiable exposure of interest in some population subgroups but not in others. The alcohol, ALDH2 genotype and blood pressure association affecting men but not women, discussed earlier, is an example of this. If ALDH2 genetic variation influenced blood pressure for reasons other than its influence on alcohol intake, for example, if it was in linkage disequilibrium with another genetic variant that influenced blood pressure through another pathway or if there was a direct pleiotropic effect of the genetic variant on blood pressure, the same genotype-blood pressure association should be seen among both men and women. If the genetic variant only influences blood pressure through its effect on alcohol intake, an effect should only be seen in men, which is what is observed. This further strengthens the evidence that the genotype–blood pressure association depends upon the genotype influencing alcohol intake and that the associations do indeed provide causal evidence of an influence of alcohol intake on blood pressure.
In some cases, it may be possible to identify two separate genetic variants, which are not in linkage disequilibrium with each other, but which both serve as proxies for the environmentally modifiable risk factor of interest. If both variants are related to the outcome of interest and point to the same underlying association, then it becomes much less plausible that reintroduced confounding explains the association, since it would have to be acting in the same way for these two unlinked variants. This can be likened to RCTs of different blood pressure–lowering agents, which work through different mechanisms and have different potential side effects. If the different agents produce the same reductions in cardiovascular disease risk, then it is unlikely that this is through agent-specific (pleiotropic) effects of the drugs; rather, it points to blood pressure lowering as being key. The latter is indeed what is in general observed [89]. In another context, two distinct genetic variants acting as instruments for higher body fat content have been used to demonstrate that greater adiposity is related to higher bone mineral density [63]. With the large number of genetic variants that are being identified in genome wide association studies in relation to particular phenotypes—e.g., >50 independent variants that are related to height; >90 that are related to total cholesterol and >20 related to fasting glucose—it is possible to generate many independent combinations of such variants and from these many independent instrumental variable estimates of the causal associations between an environmentally modifiable risk factor and a disease outcome. The independent estimates will not be plausibly influenced by any common pleiotropy or LD-induced confounding, and therefore if they display consistency this provides strong evidence against the notion that reintroduced confounding is generating the associations.
Special issues with confounding in studies of gene-by-environment interactions
It must be recognized that gene-by-environment interactions interpreted within the Mendelian randomization framework as evidence regarding the causal nature of environmentally modifiable exposures are not protected from confounding to the same extent as main genetic effects. In the ADH1B/alcohol/head and neck cancer example, any factor related to alcohol consumption—such as smoking—will tend to show greater association with head and neck cancer within the more rapid alcohol metabolizers, because smokers are more likely to drink alcohol and alcohol drinking interacts with ADH1B genotype in determining head and neck cancer risk. Because there is not a 1-to-1 association of smoking with alcohol consumption, this will not produce the quantitative interaction of essentially no effect of the genotype amongst never drinkers and an effect in the other drinking stratum, but rather a qualitative interaction of a greater effect in the smoking groups amongst whom alcohol consumption is more prevalent and a smaller, but still evident, effect in the non-smoking group amongst whom alcohol consumption tends to be less prevalent. This is indeed what is seen (Fig. 7). Situations in which both the biological basis of an expected interaction is well understood and in which a qualitative (effect vs. no effect) interaction may be postulated are the ones that are most amenable to interpretations with respect to the causal nature of the environmentally modifiable risk factor.
Non-linear associations
Mendelian randomization is most powerful when examining linear exposure-disease associations, such as those between circulating cholesterol levels and coronary heart disease. For possible non-linear associations—such as have been suggested for alcohol intake and CHD—the situation may be more complex. First, the observed non-linear associations (U-shaped in the case of alcohol and coronary heart disease in many studies) may reflect confounding and bias, as discussed above. The suggested linear effects from a Mendelian randomization study may be the correct one. Second, it is possible to use single genetic variants or combinations of variants to define the proportion of individuals in a range of alcohol intake groups (from none to high) and investigate non-linear associations in this way. For example, a very large proportion of individuals homozygous for the ALDH2 null variant are non-drinkers, and if there was truly an elevated risk of coronary heart disease among non drinkers compared to moderate alcohol consumers this group would be expected to be at higher risk than heterozygotes.
Canalization and developmental stability
Perhaps a greater potential problem for Mendelian randomization than reintroduced confounding arises from the developmental compensation that may occur through a polymorphic genotype being expressed during foetal or early post-natal development and thus influencing development in such a way as to buffer against the effect of the polymorphism. Such compensatory processes have been discussed since Waddington introduced the notion of canalization in the 1940s [90]. Canalization refers to the buffering of the effects of either environmental or genetic forces attempting to perturb development and Waddington’s ideas have been well developed both empirically and theoretically [91–97]. Such buffering can be achieved either through genetic redundancy (more than one gene having the same or similar function) or through alternative metabolic routes, where the complexity of metabolic pathways allows recruitment of different pathways to reach the same phenotypic endpoint. In effect, a functional polymorphism expressed during foetal development or post-natal growth may influence the expression of a wide range of other genes, leading to changes that may compensate for the influence of the polymorphism. Put crudely, if a person has developed and grown from the intrauterine period onwards within an environment in which one factor is perturbed (e.g., there is elevated cholesterol levels due to genotype) then they may be rendered resistant to the influence of life-long elevated circulating cholesterol, through permanent changes in tissue structure and function that counterbalance its effects. In intervention studies—for example, RCTs of cholesterol-lowering drugs—the intervention is generally randomized to participants during their middle age; similarly, in observational studies of this issue, cholesterol levels are ascertained during adulthood. In Mendelian randomization, on the other hand, randomization occurs before birth. This leads to important caveats when attempting to relate the findings of conventional observational epidemiological studies to the findings of studies carried out within the Mendelian randomization paradigm.
In some Mendelian randomization designs, developmental compensation is not an issue. For example, when maternal genotype is utilized as an indicator of the intrauterine environment (e.g., maternal ADH variation discussed above), then the response of the foetus will not differ whether the effect is induced by maternal genotype or by environmental perturbation and the effect on the foetus can be taken to indicate the effect of environmental influences during the intrauterine period. Also in cases where a variant influences an adulthood environmental exposure—e.g., ALDH2 variation and alcohol intake—developmental compensation to genotype will not be an issue. In many cases of gene-by-environment interaction interpreted with respect to causality of the environmental factor, the same applies, since development will not have occurred in the presence of the modifiable risk factor of interest and thus developmental compensation will not have occurred.
Lack of suitable genetic variants to proxy for exposure of interest
An obvious limitation of Mendelian randomization is that it can only examine areas for which there are functional polymorphisms (or genetic markers linked to such functional polymorphisms) that are relevant to the modifiable exposure of interest. In the context of genetic association studies, it has been pointed out more generally that in many cases, even if a locus is involved in a disease-related metabolic process, there may be no suitable marker or functional polymorphism to allow study of this process [98]. In an earlier paper on Mendelian randomization [17], we discussed the example of vitamin C, since observational epidemiology appeared to have got the wrong answer regarding associations between vitamin C levels and disease. We considered whether the association between vitamin C and CHD could have been studied utilizing the principles of Mendelian randomization. We stated that polymorphisms existed that had been related to lower circulating vitamin C levels—for example, in the haptoglobin gene [99]—but in this case the effect on vitamin C was not direct and these other phenotypic differences could have an influence on CHD risk that would distort examination of the influence of vitamin C levels through relating genotype to disease. SLC23A1—a gene encoding for the vitamin C transporter SVCT1, which is involved in vitamin C transport by intestinal cells—was an attractive candidate for Mendelian randomization studies. However, by 2003 (the date of the earlier paper), a search for variants had failed to find any common SNP that could be used in such a way [100]. We therefore used this as an example of a situation where suitable polymorphisms for studying the modifiable risk factor of interest could not be located. However, since the earlier paper was written, functional variation in SLC23A1 has been identified that is related to circulating vitamin C levels [59]. This example is used not to suggest that the obstacle of locating relevant genetic variation for particular problems in observational epidemiology will always be overcome, but to point out that rapidly developing knowledge of human genomics will identify more variants that can serve as instruments for Mendelian randomization studies.
Conclusions
Mendelian randomization is not predicated on the assumption that genetic variants are major determinants of health and disease within or between populations. There are many cogent critiques of genetic reductionism and the over-selling of “discoveries” in genetics that reiterate obvious truths so clearly (albeit somewhat repetitively) that there is no need to repeat them here [101–104]. Mendelian randomization does not depend upon there being “genes for” particular traits, and certainly not in the strict sense of a gene “for” a trait being one that is maintained by selection because of its causal association with that trait [105]. The association of genotype and the environmentally modifiable factor that it proxies for will be like most genotype–phenotype associations, one that is contingent and cannot be reduced to individual level prediction, but within environmental limits will pertain at a group level [106]. This is analogous to an RCT of antihypertensive agents, where at a collective level the group randomized to active medication will have lower mean blood pressure than the group randomized to placebo, but at an individual level many participants randomized to active treatment will have higher blood pressure than many individuals randomized to placebo. It is group level differences that create the analogy between Mendelian randomization and RCTs.
Finally, the associations that Mendelian randomization depend upon do need to pertain to a definable group at a particular time, but do not need to be immutable. Thus, ALDH2 variation will not be related to alcohol consumption in a society where alcohol is not consumed; the association will vary by gender, by cultural group and may change over time [107, 108]. Within the setting of a study of a well-defined group, however, the genotype will be associated with group-level differences in alcohol consumption and group assignment will not be associated with confounding variables.
Nutrition contributes importantly to population health, but the tools of nutritional epidemiology have proved fallible and led to misleading findings. Mendelian randomization offers one way in which the exciting developments in molecular genetics can help improve our understanding of nutritional determinants of population health. This approach is clearly distinct from the usual nutrigenomics approaches that promise personalized interventions tailored to individual genomes, but perhaps it offers at least as much in terms of ultimately identifying ways in which health can be improved. Use must be made of the optimal observational data for understanding the potential effects of interventions. Mendelian randomization approaches can help identify the most promising nutritional candidates for formal evaluation within randomized controlled trials of dietary manipulation, which must be carried out before such findings are considered ready for implementation. In this way, genetic epidemiology can be linked with conventional epidemiology, and in turn with intervention research, in a truly translational fashion.
References
Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, Pukkala E, Skytthe A, Hemminki K (2000) Environmental and heritable factors in the causation of cancer—analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med 343:78–85
Rimm EB, Stampfer MJ, Ascherio A, Giovannucci E, Colditz GA, Willett WC (1993) Vitamin E consumption and the risk of coronary heart disease in men. New Engl J Med 328:1450–1456
Stampfer MJ, Hennekens CH, Manson JE, Colditz GA, Rosner B, Willett WC (1993) Vitamin E consumption and the risk of coronary disease in women. New Engl J Med 328:1444–1449
Radimer K, Bindewald B, Hughes J, Ervin B, Swanson C, Picciano MF (2004) Dietary supplement use by US adults: data from the National Health and Nutrition Examination Survey 1999–2000. Am J Epidemiol 160:339–349
Millen AE, Dodd KW, Subar AF (2004) Use of vitamin, mineral, nonvitamin, and nonmineral supplements in the United States: the 1987, 1992, and 2000 National Health Interview Survey results. J Am Diet Assoc 104:942–950
Khaw K-T, Bingham S, Welch A et al (2001) Relation between plasma ascorbic acid and mortality in men and women in EPIC-Norfolk prospective study: a prospective population study. Lancet 357:657–663
Heart Protection Study Collaborative Group (2002) MRC/BHF Heart Protection Study of antioxidant vitamin supplementation in 20536 high-risk individuals: a randomised placebo-controlled trial. Lancet 360:23–33
Lawlor DA, Davey Smith G, Bruckdorfer KR, Kundu D, Ebrahim S (2004) Those confounded vitamins: what can we learn from the differences between observational versus randomised trial evidence? Lancet 363:1724–1727
Lawlor DA, Ebrahim S, Kundu D, Bruckdorfer KR, Whincup PH, Davey Smith G (2005) Vitamin C is not associated with coronary heart disease risk once life course socioeconomic position is taken into account: prospective findings from the British Women’s Heart and Health Study. Heart 91:1086–1087
Rosner B, Michels KB, Chen Y-H, Day NE (2008) Measurement error correction for nutritional exposures with correlated measurement error: use of the method of triads in a longitudinal setting. Stat Med 27:3466–3489
Ferraril P, Carroll RJ, Gustafson P (2008) A Bayesian multilevel model for estimating the diet/disease relationship in a multicenter study with exposures measured with error: the EPIC study. Stat Med 27:6037–6054
Kaaks R, Ferrari P (2006) Dietary intake assessments in epidemiology: can we know what we are measuring? Ann Epidemiol 16:377–380
Phillips A, Davey Smith G (1991) How independent are “independent” effects? Relative risk estimation when correlated exposures are measured imprecisely. J Clin Epidemiol 44:1223–1231
Phillips AN, Davey Smith G (1992) Bias in relative odds estimation owing to imprecise measurement of correlated exposures. Stat Med 11:953–961
Spearman C (1904) The proof and measurement of association between two things. Am J Psychol 15:72–101
Davey Smith G, Phillips AN (1996) Inflation in epidemiology: ‘the proof and measurement of association between two things’ revisited. Br Med J 312:1659–1661
Davey Smith G, Ebrahim S (2003) ‘Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol 32:1–22
Davey Smith G, Ebrahim S (2004) Mendelian randomization: prospects, potentials, and limitations. Int J Epidemiol 33:30–42
Davey Smith G, Ebrahim S (2005) What can Mendelian randomization tell us about modifiable behavioural and environmental exposures. BMJ 330:1076–1079
Lawlor DA, Harbord RM, Sterne JAC, Timpson NJ, Davey Smith G (2008) Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med 27:1133–1163
Ebrahim S, Davey Smith G (2008) Mendelian randomization: can genetic epidemiology help redress the failures of observational epidemiology? Hum Genet 123:15–33
Palmer L, Cardon L (2005) Shaking the tree: mapping complex disease genes with linkage disequilibrium. Lancet 366:1223–1234
Wellcome Trust Case Control Consortium (2007) Genome-wide association study of 14, 000 cases of seven common diseases and 3, 000 shared controls. Nature 447:661–678
Davey Smith G, Lawlor D, Harbord R, Timpson N, Day I, Ebrahim S (2008) Clustered environments and randomized genes: a fundamental distinction between conventional and genetic epidemiology. PLoS Med 4:1985–1992
Birge SJ, Keutmann HT, Cuatrecasas P, Wheedon GD (1967) Osteoporosis, intestinal lactase deficiency and low dietary calcium intake. N Eng J Med 276:445–448
Newcomer AD, Hodgson SF, Douglas MD, Thomas PJ (1978) Lactase deficiency: prevalence in osteoporosis. Ann Int Med 89:218–220
Honkanen R, Pulkkinen P, Järvinen R, Kröger H, Lindstedt K, Tuppurainen M et al (1996) Does lactose intolerance predispose to low bone density? A population-based study of perimenopausal Finnish women. Bone 19:23–28
Katan MB (1986) Apolipoprotein E isoforms, serum cholesterol, and cancer. Lancet i:507–508
Wheatley K, Gray R (2004) Commentary: Mendelian randomization—an update on its use to evaluate allogeneic stem cell transplantation in leukaemia. Int J Epidemiol 33:15–17
Youngman LD, Keavney BD, Palmer A et al (2000) Plasma fibrinogen and fibrinogen genotypes in 4685 cases of myocardial infarction and in 6002 controls: test of causality by ‘Mendelian randomization’. Circulation 102(Suppl II):31–32
Fallon UB, Ben-Shlomo Y, Elwood P, Ubbink JB, Davey Smith G (2001) Homocysteine and coronary heart disease—Author’s reply. Heart, published online 14 Mar (http://heart.bmjjournals.com/cgi/eletters/85/2/153)
Clayton D, McKeigue PM (2001) Epidemiological methods for studying genes and environmental factors in complex diseases. Lancet 358:1356–1360
Keavney B (2002) Genetic epidemiological studies of coronary heart disease. Int J Epidemiol 31:730–736
Davey Smith G (2006) Capitalising on Mendelian randomization to assess the effects of treatments. James Lind Library (http://www.jameslindlibrary.org)
Schatzkin A, Abnet CC, Cross AJ, Gunter M, Pfeiffer R, Gail M, Lim U, Davey Smith G (2009) Mendelian randomization: how it can-and cannot-help confirm causal relations between nutrition and cancer. Cancer Prev Res 2:104–113
Qi L (2009) Mendelian randomization in nutritional epidemiology. Nutr Rev 67:439–450
Davey Smith G (2006) Cochrane lecture: randomised by (your) god: robust inference from an observational study design. J Epidemiol Community Health 60:382–388
Davey Smith G, Timpson N, Ebrahim S (2008) Strengthening causal inference in cardiovascular epidemiology through Mendelian randomization. Ann Med 12:524–541
Marmot M (2001) Reflections on alcohol and coronary heart disease. Int J Epidemiol 30:729–734
Bovet P, Paccaud F (2001) Alcohol, coronary heart disease and public health: which evidence-based policy? Int J Epidemiol 30:734–737
Klatsky AL (2001) Commentary: could abstinence from alcohol be hazardous to your health? Int J Epidemiol 30:739–742
Shaper AG (1993) Editorial: alcohol, the heart, and health. Am J Public Health 83:799–801
Rimm E (2001) Commentary: alcohol and coronary heart disease—laying the foundation for future work. Int J Epidemiol 30:738–739
Enomoto N, Takase S, Yasuhara M, Takada A (1991) Acetaldehyde metabolism in different aldehyde dehydrogenase-2 genotypes. Alcohol Clin Exp Res 15:141–144
Takagi S, Iwai N, Yamauchi R, Kojima S, Yasuno S, Baba T et al (2002) Aldehyde dehydrogenase 2 gene is a risk factor for myocardial infarction in Japanese men. Hypertens Res 25:677–681
Chao Y-C, Liou S-R, Chung Y-Y, Tang H-S, Hsu C-T, Li T-K et al (1994) Polymorphism of alcohol and aldehyde dehydrogenase genes and alcoholic cirrhosis in Chinese patients. Hepatology 19:360–366
Haskell WL, Camargo C, Williams PT, Vranizan KM, Krauss RM, Lindgren FT et al (1984) The effect of cessation and resumption of moderate alcohol intake on serum high-density-lipoprotein subfractions. N Engl J Med 310:805–810
Burr ML, Fehily AM, Butland BK, Bolton CH, Eastham RD (1986) Alcohol and high-density-lipoprotein cholesterol: a randomized controlled trial. Br J Nutr 56:81–86
Beulens JWJ, Rimm EB, Ascherio A, Spiegelman D, Hendriks HFJ, Mukamal KJ (2007) Alcohol consumption and risk for coronary heart disease among men with hypertension. Ann Intern Med 146:10–19
Chen L, Davey Smith G, Harbord R, Lewis S (2008) Alcohol intake and blood pressure: a systematic review implementing Mendelian randomization approach. PLoS Med 5:461
Irons DE, McGue M, Iacono WG, Oetting WS (2007) Mendelian randomization: a novel test of the gateway hypothesis and models of gene—environment interplay. Dev Psychopathol 19:1181–1195
Kandel DB, Yamaguchi K, Chen K (1992) Stages of progression in drug involvement from adolescence to adulthood: further evidence for the gateway theory. J Stud Alcohol 53:447–457
Kandel D, Yamaguchi K (1993) From beer to crack: developmental patterns of drug involvement. Am J Public Health 83:851–855
Gemma S, Vichi S, Testai E (2007) Metabolic and genetic factors contributing to alcohol induced effects and fetal alcohol syndrome. Neurosci Biobehav Rev 31:221–229
Jacobson SW, Carr LG, Croxford J, Sokol RJ, Li TK, Jacobson JL (2006) Protective effects of the alcohol dehydrogenase-ADH1B allele in children exposed to alcohol during pregnancy. J Pediatr 148:30–37
Warren KR, Li TK (2005) Genetic polymorphisms: impact on the risk of fetal alcohol spectrum disorders. Birth Defects Res Clin Mol Teratol 73:195–203
Timpson NJ, Brennan P, Gaborieau V, Moore L, Zaridza D, Matveev V, Szeszenia-Dabrowska N, Lissowska J, Fabianova E, Mates D, Bencko V, Foretova L, Janout V, Chow W-H, Rothman N, Boffetta P, Harbord R, Davey Smith G (2010) Can lactase persistence genotype be used to reassess the relationship between renal cell carcinoma and milk drinking, potentials and problems in the application of Mendelian Randomization. Cancer Epidemiol Biomarkers Prev 19:1341–1348
Sacerdote C, Guarrera S, Davey Smith G, Grioni S, Krogh V, Masala G, Mattiello A, Palli D (2007) Lactase persistence and bitter taste response: instrumental variables and Mendelian randomization in epidemiologic studies of dietary factors and cancer risk. Am J Epidemiol 166:576–581
Timpson NJ, Forouhi NG, Brion MJ, Harbord RM, Cook DG, Johnson P, McConnachie A, Morris R, Rodriguez S, Luan J, Ebrahim S, Padmanabhan S, Watt G, Bruckdorfer KR, Wareham NJ, Whincup PH, Chanock S, Sattar N, Lawlor DA, Davey Smith G (2010) Genetic variation at the SLC23A1 locus is associated with circulating levels of L-ascorbic acid (vitamin C). Evidence from 5 independent studies with over 15,000 participants. Am J Clin Nutr 92:375–382
Wright ME, Peters U, Gunter J, Moore SC, Lawson KA, Yeager M, Weinstein SJ, Snyder K, Virtamo J, Albanes D (2009) Association of variants in two vitamin E transport genes with circulating vitamin E concentrations and prostate cancer risk. Cancer Res 69:1429–1438
Ferrucci L, Perry JRB, Matteini A, Perola M, Tanaka T, Silander K, Rice N, Melzer D, Murray A, Cluett C, Fried LP, Albanes D, Corsi A-M, Cherubini A, Guralnik J, Bandinelli S, Singleton A, Virtamo J, Walston J, Semba RD, Frayling TM (2009) Common variation in the beta-carotene 15, 15′-monooxygenase 1 gene affects circulating levels of carotenoids: a genome-wide association study. Am J Hum Genet 84:123–133
Freathy RM, Timpson NJ, Lawlor DA, Pouta A, Ben-Shlomo Y, Ruokonen A et al (2008) Common variation in the FTO gene alters diabetes-related metabolic traits to extent expected, given its effect on BMI. Diabetes 57:1419–1426
Timpson N, Harbord R, Davey Smith G, Zacho J, Tybaerg-Hansen A, Nordestgaard BG (2009) Does greater adiposity increase blood pressure and hypertension risk? Mendelian randomization using Fto/Mc4r genotype. Hypertension 54:84–90
Timpson NJ, Sayers A, Davey Smith G, Tobias JH (2009) How does body fat influence bone mass in childhood? A Mendelian randomisation approach. J Bone Miner Res 24:522–533
Jousilahti P, Salomaa V (2004) Fibrinogen, social position, and Mendelian randomisation. J Epidemiol Community Health 58:883
Glynn RK (2006) Commentary: genes as instruments for evaluation of markers and causes. Int J Epidemiol 35:932–934
Davey Smith G, Harbord R, Milton J, Ebrahim S, Sterne JAC (2005) Does elevated plasma fibrinogen increase the risk of coronary heart disease? Evidence from a meta-analysis of genetic association studies. Arterioscler Thromb Vasc Biol 25:2228–2233
Thomas DC, Conti DV (2004) Commentary on the concept of “Mendelian Randomization”. Int J Epidemiol 33:17–21
Angrist JD, Pischke JS (2008) Mostly harmless econometrics: an empiricist’s companion. Princeton University Press, Princeton
Perera FP (1997) Environment and cancer: who are susceptible? Science 278:1068–1073
Mucci LA, Wedren S, Tamimi RM, Trichopoulos D, Adami HO (2001) The role of gene-environment interaction in the aetiology of human cancer: examples from cancers of the large bowel, lung and breast. J Intern Med 249:477–493
Thomas D (2010) Gene—environment-wide association studies: emerging approaches. Nat Rev Genet 11:259–272
Wright AF, Carothers AD, Campbell H (2002) Gene-environment interactions—the BioBank UK study. Pharmacogenomics J 2:75–82
Colhoun H, KcKeigue PM, Davey Smith G (2003) Problems of reporting genetic associations with complex outcomes. Lancet 361:865–872
Thompson WD (1991) Effect modification and the limits of biological inference from epidemiological data. J Clin Epidemiol 44:221–232
Memik F (2003) Alcohol and esophageal cancer, is there an exaggerated accusation? Hepatogastroenterology 54:1953–1955
Zuccolo L, Fitz-Simon N, Gray R, Ring SM, Sayal K, Davey Smith G, Lewis SJ (2009) A non-synonymous variant in ADH1B is strongly associated with prenatal alcohol use in a European sample of pregnant women. Hum Mol Genet 18:4457–4466
Hashibe M, McKay JD, Curado MP, Oliveira JC, Koifman S, Koifman R et al (2008) Multiple ADH genes are associated with upper aerodigestive cancers. Nat Genet 40:707–709
Lewis S, Davey Smith G (2005) Alcohol, ALDH2 and esophageal cancer: a meta-analysis which illustrates the potentials and limitations of a Mendelian randomization approach. Cancer Epidemiol Biomarkers Prev 14:1967–1971
Gutjahr E, Gmel G, Rehm J (2001) Relation between average alcohol consumption and disease: an overview. Eur Addict Res 7:117–127
Boccia S, Hashibe M, Gallı P, De Feo E, Asakage T, Hashimoto T et al (2009) Aldehyde dehydrogenase 2 and head and neck cancer: a meta-analysis implementing a Mendelian randomization approach. Cancer Epidemiol Biomarkers Prev 18:248–254
Seitz HK, Stickel F (2007) Molecular mechanisms of alcoholmediated carcinogenesis. Nat Rev Cancer 7:599–612
Bower C, Stanley F (1992) The role of nutritional factors in the aetiology of neural tube defects. J Paediatr Child Health 28:12–16
Yates JRW, Ferguson-Smith MA, Shenkin A, Guzman-Rodriguez R, White M, Clark BJ (1987) Is disordered folate metabolism the basis for the genetic predisposition to neural tube defects? Clin Genet 31:279–287
Kohn J, Mollin DL, Rosenbach LM (1961) Conventional voltage electrophoresis for formiminoglutamic-acid determination in folic acid deficiency. J Clin Pathol 14:345–350
Hibbard ED, Smithells RW (1965) Folic acid metabolism and human embryopathy. Lancet 285:1254
Osier MV, Pakstis AJ, Soodyall H, Comas D, Goldman D, Odunsi A et al (2002) A global perspective on genetic variation at the ADH genes reveals unusual patterns of linkage disequilibrium and diversity. Am J Hum Genet 71:84–99
Glebart WM (1998) Databases in genomic research. Science 282:659–661
Law MR, Morris JK, Wald NJ (2009) Use of blood pressure lowering drugs in the prevention of cardiovascular disease: meta-analysis of 147 randomised trials in the context of expectations from prospective epidemiological studies. BMJ 338:b1665
Waddington CH (1942) Canalization of development and the inheritance of acquired characteristics. Nature 150:563–565
Wilkins AS (1997) Canalization: a molecular genetic perspective. BioEssays 19:257–262
Rutherford SL (2000) From genotype to phenotype: buffering mechanisms and the storage of genetic information. BioEssays 22:1095–1105
Gibson G, Wagner G (2000) Canalization in evolutionary genetics: a stabilizing theory? BioEssays 22:372–380
Hartman JL, Garvik B, Hartwell L (2001) Principles for the buffering of genetic variation. Science 291:1001–1004
Debat V, David P (2001) Mapping phenotypes: canalization, plasticity and developmental stability. Trends Ecol Evol 16:555–561
Kitami T, Nadeau JH (2002) Biochemical networking contributes more to genetic buffering in human and mouse metabolic pathways than does gene duplication. Nat Genet 32:191–194
Hornstein E, Shomron N (2006) Canalization of development by microRNAs. Nat Genet 38:S20–S24
Weiss K, Terwilliger J (2000) How many diseases does it take to map a gene with SNPs? Nat Genet 26:151–157
Langlois MR, Delanghe JR, De Buyzere ML, Bernard DR, Ouyang J (1997) Effect of haptoglobin on the metabolism of vitamin C. Am J Clin Nutr 66:606–610
Erichsen HC, Eck P, Levine M, Chanock S (2001) Characterization of the genomic structure of the human vitamin C transporter SVCT1 (SLC23A2). J Nutr 131:2623–2627
Berkowitz A (1996) Our genes, ourselves? Bioscience 46:42–51
Baird P (2000) Genetic technologies and achieving health for populations. Int J Health Serv 30:407–424
Holtzman NA (2001) Putting the search for genes in perspective. Int J Health Serv 31:445
Strohman RC (1993) Ancient genomes, wise bodies, unhealthy people: the limits of a genetic paradigm in biology and medicine. Perspect Biol Med 37:112–145
Kaplan JM, Pigliucci M (2001) Genes “for” phenotypes: a modern history view. Biol Philos 16:189–213
Wolf U (1995) The genetic contribution to the phenotype. Hum Genet 95:127–148
Higuchi S, Matsuushitam S, Imazeki H, Kinoshita T, Takagi S, Kono H (1994) Aldehyde dehydrogenase genotypes in Japanese alcoholics. Lancet 343:741–742
Hasin D, Aharonovich E, Liu X, Mamman Z, Matseoane K, Carr L et al (2002) Alcohol and ADH2 in Israel: Ashkenazis, Sephardics, and recent Russian immigrants. Am J Psychiatry 159:1432–1434
Eidelman RS, Hollar D, Hebert PR, Lamas GA, Hennekens CH (2004) Randomized trials of vitamin E in the treatment and prevention of cardiovascular disease. Arch Intern Med 164:1552–1556
Ackowledgments
George Davey Smith works in a centre that is supported by the MRC (G0600705) and the University of Bristol. Thanks to Tom Palmer for the calculation of variance in alcohol consumption accounted for by ALDH2.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Davey Smith, G. Use of genetic markers and gene-diet interactions for interrogating population-level causal influences of diet on health. Genes Nutr 6, 27–43 (2011). https://doi.org/10.1007/s12263-010-0181-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12263-010-0181-y