
Abstract
Cancer cells are distinguished by several distinct characteristics, such as self-sufficiency in growth signal, resistance to growth inhibition, limitless replicative potential, evasion of apoptosis, sustained angiogenesis, and tissue invasion and metastasis. Tumor cells acquire these properties due to the dysregulation of multiple genes and associated cell signaling pathways, most of which are linked to inflammation. For that reason, rationally designed drugs that target a single gene product are unlikely to be of use in preventing or treating cancer. Moreover, targeted drugs can cause serious and even life-threatening side effects. Therefore, there is an urgent need for safe and effective promiscuous (multitargeted) drugs. “Mother Nature” produces numerous such compounds that regulate multiple cell signaling pathways, are cost effective, exhibit low toxicity, and are readily available. One among these is tocotrienol, a member of the vitamin E family, which has exhibited anticancer properties. This review summarizes data from in vitro and in vivo studies of the effects of tocotrienol on nuclear factor-κB, signal transducer and activator of transcription (STAT) 3, death receptors, apoptosis, nuclear factor (erythroid-derived 2)-like 2 (Nrf2), hypoxia-inducible factor (HIF) 1, growth factor receptor kinases, and angiogenic pathways.
Similar content being viewed by others


Introduction
Vitamin E, which exhibits antioxidant properties, is a generic term that represents derivatives of tocopherol and tocotrienol (Kamal-Eldin and Appelqvist 1996). Antioxidants are generally believed to inhibit the development of cardiovascular disease and cancer by neutralizing free radical damage (Meydani 1995). Therefore, consumption of vitamin E–enriched foods may reduce the incidence of these two diseases. Vitamin E is present in most edible oils extracted from wheat, rice bran, barley, oat, coconut, and palm. The richest sources of tocotrienol specifically are the rice bran, palm, and annatto oils, for which the ratio of tocopherol to tocotrienol is 50:50; 25:75, and 0.1:99.9, respectively (Tan, spacedoc.net). Other natural sources of tocotrienol are rye, amaranth, walnut, hazelnut, poppy, safflower, maize, and the seeds of flax, grape, and pumpkin.
Tocotrienol and tocopherol, which are fat soluble, are structurally similar in that they have a common chromanol ring and a side chain at the C-2 position. But whereas tocopherol has a saturated phytyl tail, tocotrienol possesses an unsaturated isoprenoid side chain (Fig. 1). Depending on the number and position of methyl substitutions on the chromanol ring, tocotrienol, and tocopherol are further classified into α, β, γ, and δ isoforms (Fig. 1).

Chemical structure and natural sources of tocotrienols. TP, tocopherol
Tocotrienol derivatives did not attract much attention from researchers until the late 1980s, when their cholesterol-lowering potential (Qureshi et al. 1986) and anticancer effects were described (Kato et al. 1985; Sundram et al. 1989). Evidence now suggests that tocotrienols have potentially greater physiologic functions than tocopherols do (Ahmad et al. 2005; Ong et al. 1993; Yu et al. 1999; Kannappan et al. 2010a; Kannappan et al. 2010b). For instance, the ability to inhibit the inflammatory transcription factors nuclear factor (NF)-κB and signal transducer and activator of transcription (STAT) 3, 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase, mammalian DNA polymerases, and certain protein tyrosine kinases is unique to tocotrienols.
In the last few years, much emphasis has been placed on designing drugs that hit a single target (“smart drugs” or “magic bullets”). Well-known targeted therapies include celecoxib (which inhibits cyclooxygenase (COX)-2), cetixumab (epidermal growth factor receptor (EGFR)), bortezomib (NF-κB), etanercep (tumor necrosis factor (TNF)), trastuzumab (human epidermal growth factor receptor (HER)-2), imatinib mesylate (breakpoint cluster region (Bcr)-abl), bevacizumab (vascular endothelial growth factor (VEGF)), paclitaxel (tubulin), and camptothecin (topoisomerase). Yet several of these expensive drugs have been found to be ineffective and unsafe. Most diseases, especially cancer, are a result of dysregulation of as many as 500 gene products (Gupta et al. 2010b; Vogelstein and Kinzler 2004), and inhibition of a single target or signaling pathway is unlikely to prevent or control the disease. Therefore, drugs that modulate multiple targets (multitargeted therapy), formally referred to as “dirty drugs,” are needed (Kannappan et al. 2011). In this review, we examine in detail how tocotrienol modulates various pathways (Table 1) and contribute to cancer.
Tocotrienol and signaling pathways
NF-κB and tocotrienol
First discovered in 1986, the transcription factor NF-κB is closely connected to the process of tumorogenesis (Sen and Baltimore 1986). NF-κB is activated in response to tobacco, stress, dietary agents, obesity, alcohol, infectious agents, irradiation, and other environmental stimuli that account for as much as 95% of all cancers (Gupta et al. 2010a). It has been linked with the transformation of normal cells and is constitutively active in most tumor cells. Furthermore, NF-κB-regulated gene products have been implicated in cell transformation, proliferation, survival, invasion, angiogenesis, metastasis, and chemoresistance.
We studied the effect of γ-tocotrienol on the NF-κB pathway and found that it completely abolished NF-κB activation induced by TNF, phorbol myristate acetate, okadaic acid, lipopolysaccharide, cigarette smoke, interleukin (IL)-1β, and epidermal growth factor (Ahn et al. 2007). Constitutive NF-κB activation expressed by certain tumor cells was also abrogated by γ-tocotrienol. When we investigated the mechanism, we found that tocotrienol blocked TNF-induced phosphorylation and degradation of IκBα by inhibiting IκBα kinase activation, thus leading to the suppression of the phosphorylation and nuclear translocation of p65 (Ahn et al. 2007). Yap et al. (2008) investigated the antiproliferative effect of tocotrienol-rich fraction (TRF) on prostate cancer cells and showed that the γ-tocotrienol-induced apoptosis was associated with the activation of pro-caspases and suppression of NF-κB, EGFR, and Id family proteins. The group also found that the c-jun N-terminal kinase-specific inhibitor SP600125 partially reversed the effect of γ-tocotrienol. Similar results were observed by Wu et al. (2008), who investigated the effects of TRF on lipopolysaccharide-induced inflammatory response. TRF dose dependently protected human monocytic (THP-1) cells against lipopolysaccharide-induced cell death through the inhibition of nitric oxide, prostaglandin E2, and proinflammatory cytokines. TRF also blocked lipopolysaccharide induction of inducible nitric oxide synthase, COX-2, and NF-κB expression (Wu et al. 2008).
Recently, we showed that tocotrienol potentiates the effect of gemcitabine, a standard part of clinical treatment against pancreatic cancer, against human pancreatic cancer cells both in vitro and in vivo. Pancreatic cancers generally respond poorly to chemotherapy, and agents that could sensitize these tumors to treatment need to be identified. In our study, tocotrienol inhibited NF-κB activation and suppressed key cellular regulators, including cyclin D1, c-myc, COX-2, B-cell lymphoma protein 2 (Bcl-2), cellular inhibitor of apoptosis protein, survivin, VEGF, intracellular adhesion molecule-1, and chemokine (C-X-C motif) receptor 4. Furthermore, in an orthotopic nude mouse model of human pancreatic cancer, oral administration of tocotrienol inhibited tumor growth and enhanced the antitumor properties of gemcitabine by inhibiting NF-κB (Kunnumakkara et al. 2010).
STAT3 and tocotrienol
STAT3, which was discovered almost 15 years ago as an acute-phase response factor, belongs to one of six members of a family of transcription factors that are linked with inflammation, cellular transformation, survival, proliferation, invasion, angiogenesis, and metastasis of cancer. Like NF-κB, STAT3 is activated by various types of carcinogens, radiation, viruses, growth factors, oncogenes, and inflammatory cytokines and is constitutively active in most tumor cells. STAT3 regulates the expression of genes that mediate proliferation (e.g., c-fos, c-myc, and cyclin D1), suppress apoptosis (e.g., bcl-xL and survivin), invasion (e.g., matrix metalloproteinase-2), and promote angiogenesis (e.g., the VEGF gene). Activation of STAT3 has also been associated with chemoresistance and radioresistance. STAT3 mediates these effects through its collaboration with other transcription factors, including NF-κB, hypoxia-inducible factor (HIF)-1, and peroxisome proliferator-activated receptor (PPAR)-γ. Thus, inhibitors of STAT3 activation might be used for cancer prevention and therapy. Small peptides, oligonucleotides, and small molecules have been identified as potential STAT-3 inhibitors, including synthetic molecules (e.g., AG 490, decoy peptides, and oligonucleotides) and plant polyphenols (e.g., curcumin, resveratrol, flavopiridol, indirubin, magnolol, piceatannol, parthenolide, epigallocatechin gallate, and cucurbitacin).
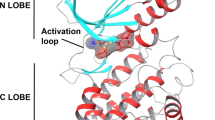
In our laboratory, we investigated whether γ-tocotrienol can modulate the STAT3 cell signaling pathway and discovered that γ-tocotrienol, but not γ-tocopherol, inhibited constitutive activation of STAT3 in multiple myeloma cells (Kannappan et al. 2010b) (Fig. 2). Tocotrienol inhibited the activation of Src kinase, Janus kinase (JAK) 1, and JAK2, which are believed to be the regulators of STAT3. We further found that γ-tocotrienol-induced expression of the tyrosine phosphatase SHP-1 and that gene silencing of SHP-1 with small interfering RNA reversed the ability of tocotrienol to inhibit STAT3 activation; this result suggested that SHP-1 plays a vital role in the action of γ-tocotrienol. Tocotrienol down-modulated the activation of STAT3 and induced SHP-1 in vivo as well. Eventually, γ-tocotrienol down-regulated the expression of STAT3-regulated antiapoptotic, proliferative, and angiogenic gene products (Kannappan et al. 2010b).

Tocotrienol targets multiple cell signaling pathways. Modern mono-targeted therapies include celecoxib, avastin, erbitux, enbrel, herceptin, gleevec, and avastin (inhibits COX2, EGFR, TNF, HER2, bcr-abl and VEGF, respectively) and also inhibits other targets including transcription factors, enzymes, growth factors and its receptors, kinases, and anti-apoptotic proteins. Note: Celecoxib, a non-steroidal anti-inflammatory drug that selectively inhibit COX-2; avastin (bevacizumab), monoclonal antibody against VEGF; Herceptin (Trastuzumab), humanized monoclonal antibody against HER-2; Gleevac (imatinib), a multiple tyrosine kinase inhibitor. AGP, human alpha1-acid glycoprotein; AP-1, activating protein-1; Bcl-2, B-cell lymphoma protein 2; COX-2, cyclooxygenase; DNA poly, DNA polymerase; DR, death receptor; EGFR, EGF-receptor; ER, estrogen receptor; ERK, extracellular receptor kinase; GST, glutathione-S-transferase; HER-2, human epidermal growth factor receptor-2; HIF-1α, hypoxia-inducible factor-1 alpha; IAP, inhibitory apoptosis protein; ICAM-1, intracellular adhesion molecule-1; IL, interleukin; iNOS, inducible nitric oxide synthase; JAK, janus kinase; NF-κB, nuclear factor kappa B; MMP, Matrix metalloproteinase; PKC, protein kinase C, STAT, signal transducers, and activators of transcription; VCAM-1, vascular cell adhesion molecule-1; VEGF, vascular endothelial growth factor; VEGFR, VEGF receptor; IFN-γ, interferon gamma; PF-4, Platelet factor 4; 12-LOX, 12-lipoxygenase; eNOS, endothelial nitric oxide synthase; GGT, gamma-glutamyl transpeptidase; GSTP-1, glutathione-S-transferase pi; HMGCR, 3-hydroxy-3-methylglutaryl-CoA reductase; hTERT, human telomerase reverse transcriptase; NQO1, NAD(P)H dehydrogenase, quinone1; PLA-2, phospholipase A2; TRAF-1, TNF receptor-associated factor-1; XIAP, X-linked inhibitor of apoptosis protein; c-FLIP, cellular FLICE inhibitory protein, CDK, cyclin-dependent kinase; IKK, IκB kinase; Pdk-1, phosphoinositide-dependent kinase-1; PI3K, phosphoinositide 3-kinase; C/EBP-α, CCAAT/enhancer-binding protein alpha; PPAR-γ, peroxisome proliferator-activated receptor gamma; PGE-2, prostaglandin E2; TX-B2, thromboxane B2
Bachawal et al. (2010) investigated the effect of combined treatment of γ-tocotrienol and a tyrosine kinase inhibitor (erlotinib or gefitinib) on STAT and Akt signaling in murine mammary tumor cells. In their study 3 μM γ-tocotrienol with 0.25 μM erlotinib or 0.5 μM gefitinib significantly inhibited anchorage-independent cell growth and reduced levels of cyclin D1 and phosphorylated (i.e., active) Pdk-1, Akt, Stat3, and Stat5. Those researchers concluded that treatment with γ-tocotrienol plus erlotinib or gefitinib prevents ErbB receptor heterodimer cooperation and inhibits epidermal growth factor–dependent mitogenic signaling in anchorage-independent murine mammary tumor cells (Bachawal et al. 2010).
Death receptors and tocotrienol
Recent innovative strategy in cancer therapy is the induction of apoptosis in tumor cells through agents that induces death receptors (DRs). The best characterized DRs are CD95 (Fas/Apo-1), TNF receptor, and TNF-related apoptosis-inducing ligand (TRAIL) receptors. TRAIL is emerging as the most promising agent for cancer therapy because it induces apoptosis in a variety of tumor and transformed cells without being toxic to normal cells. In 1995, the ligand itself was discovered on the basis of its sequence homology to TNF and Fas ligand. TRAIL interacts with at least six proteins, of which DR4 and DR5 recruit the adaptor molecule FADD and either the FLICE caspase or caspase-8. Activation of caspase-8 leads directly to the activation of caspase-3 and subsequently apoptosis; this is the extrinsic apoptosis pathway. Cross-talk exists between this extrinsic pathway and the mitochondria-dependent intrinsic apoptosis pathway. Activated caspase-8 cleaves Bid, which then translocates to the mitochondria to induce cytochrome c release, which in turn leads to sequential activation of caspase-9 and caspase-3. Although TRAIL can induce apoptosis in most cancer cells, the emergence of resistance to TRAIL limits its utility as a therapeutic agent. Thus, agents that up-regulate DRs have the potential to sensitize tumors to TRAIL.
We investigated whether tocotrienol can promote TRAIL-induced apoptosis in colon cancer cells and found that it can sensitize human colon cancer cells to TRAIL (Kannappan et al. 2010a). We also discovered that tocotrienol induced DR expression in a non-cell-type-specific fashion that is mediated through reactive oxygen species/extracellular signal-regulated kinase/p53. Similarly, Park et al. (2010) observed that tocotrienol induces apoptosis in breast cancer cell lines via an endoplasmic reticulum stress-dependent increase in extrinsic death receptor signaling. However, they found that DR5 induction by tocotrienol was transcriptionally regulated by C/EBP homologous protein. These researchers concluded that up-regulation of DR5 by γ-tocotrienol treatment is dependent on the activation of c-jun N-terminal kinase and p38 mitogen-activated protein kinase and that the activation is mediated by endoplasmic reticulum stress (Park et al. 2010).
Apoptosis and tocotrienol
Every cell has a finite life span. Cell death is due to passive necrotic processes or to the active process of apoptosis. During apoptosis, the most common mechanism by which the body eliminates damaged or unneeded cells, the cells die without local inflammation from leakage of cell contents (Raff 1998). Morphologic changes, such as cell shrinkage, condensation, fragmentation of the nucleus and bubbling of the plasma membrane (“blebbing”), and chromatin condensation and nucleosomal fragmentation are characteristics of cells undergoing apoptosis (Heermeier et al. 1996; Hockenbery et al. 1990; Nass et al. 1996). Cancer cells can be eliminated from the body through the apoptosis mechanism (Kerr et al. 1972).
Sundram et al. (1989) were the first to demonstrate that palm oil possesses anticancer properties in female Sprague–Dawley rats. Yu et al. (1999) reported that all tocotrienol derivatives and RRR-γ-tocopherol induce apoptosis in MDA-MB-435 human breast cancer cell lines. However, accumulating evidence suggests that tocopherols have little or no apoptotic activity (Kimmick et al. 1997; Kline et al. 2004; Aggarwal et al. 2010; Neuzil et al. 2002; Schneider 2005). How tocotrienol triggers this natural process of cell death has been investigated extensively. Studies of the intracellular mechanisms mediating tocotrienol‐induced apoptosis in neoplastic mammary epithelial cells revealed that γ‐tocotrienol induced caspases‐8 and ‐3 but not caspase‐9 activation (Shah et al. 2003; Shah and Sylvester 2004). Caspase‐8 processing and activation are associated with DR‐mediated apoptotic signaling, whereas caspase‐9 activation and processing are associated with mitochondrial stress‐mediated apoptotic signaling. Takahashi and Loo (2004) observed disrupted mitochondria in γ-tocotrienol-treated human breast cancer cells, collapsed mitochondrial membrane potential, and cytochrome c release from mitochondria, but the level of Bcl‐2 protein and the induction of caspase‐9 activation were unchanged in these cells. In a study of human colon carcinoma cells, tocotrienol altered the ratio of Bax to Bcl‐2, which induced mitochondrial disruption, cytochrome c release, and caspase‐9 activation (Agarwal et al. 2004). Taken together, these results demonstrate that tocotrienol mediates its apoptotic effect through the activation of different intracellular signaling mechanisms in different types of cancer cells. Many other investigators have also reported that tocotrienol exhibits anticancer properties by inducing apoptosis (Shah et al. 2003; Shah and Sylvester 2004; Takahashi and Loo 2004; Agarwal et al. 2004; McIntyre et al. 2000a, 2000b; Osakada et al. 2004; Sakai et al. 2004; Shun et al. 2004).
Nrf2 and tocotrienol
Nuclear factor (erythroid derived 2)-like 2 (Nrf2, also known as NFE2L2) plays a critical role in protecting cells against oxidative stress. Nrf2 is sequestered in cytoplasm by the cytoskeletal protein Keapl. Under stress conditions, when electrophiles or reactive oxygen species (or both) cause dissociation of Nrf2 from Keap1, the former is translocated to the nucleus, leading to activation of the phase I and II enzymes responsible for the inactivation and removal of mutagenic and carcinogenic factors. Induction of phase II enzymes confers protection against insult by reactive metabolites of carcinogens or by reactive oxygen species. Nrf is a potential target for cancer chemoprevention because it can induce these enzymes, and agents that induce Nrf2 are under investigation. Hsieh et al. (2010) found that tocotrienol treatment induced Nrf2 expression, as indicated by a corresponding decrease in Keap1 levels in estrogen receptor-negative MDA-MB-231 cells but not in estrogen receptor-positive MCF-7 cells.
HIF-1 and tocotrienol
Hypoxia-inducible factors (HIFs) are principal regulators of hypoxic adaptation. They regulate gene expression involved in glycolysis, erythropoiesis, angiogenesis, proliferation, and stem cell function under low-oxygen conditions. Evidence accumulated in recent years suggests an additional important regulatory role for HIFs in inflammation: HIF-1α promotes survival under oxygen-deprived conditions and mediates blood vessel extravasation by modulating β2-integrin expression. The method by which hypoxia and HIFs control properties of tumor-associated macrophages and their relationship to tumor formation and progression have been established.
HIF-1α, its downstream target VEGF, and other angiogenic factors such as IL-8 and COX-2 play critical roles in neovascularization. Shibata et al. (2008a) investigated whether the inhibitory effect of tocotrienol on tumor angiogenesis is via regulation of these angiogenic factors. They found that δ-tocotrienol suppressed hypoxia-induced VEGF and IL-8 expression in DLD-1 cells at both the mRNA and the protein levels and that δ-tocotrienol reduced HIF-1α protein expression or increased HIF-1α degradation. Similarly, Bi et al. (2010) showed that γ-tocotrienol decreased the expression of HIF-1α protein and the paracrine secretion of VEGF under both normoxic and hypoxic conditions in the human gastric adenocarcinoma SGC-7901 cell line.
Growth factor receptor kinases and tocotrienol
A variety of growth factor receptor kinases have been discovered and many of them are linked to tumorigenesis. For example, Src tyrosine kinases are involved in different signal transduction pathways in cells. The corresponding genes participate in such vital processes as growth, differentiation, adhesion, and transcription. Specific structural changes confer oncogenic properties to the Src protein (Guarino 2010). We and other research groups have found that tocotrienol inhibits Src kinase activity and induces apoptosis in various cell types (Kannappan et al. 2010b; Sen et al. 2000; Kashiwagi et al. 2009).
JAK plays an essential and non-redundant role in promoting biologic responses to multiple major cytokine receptor families (Rodig et al. 1998), and it has been shown to mediate IL-6-induced STAT3 activation (Ueda et al. 2002). Thus, inhibition of JAK activation by IL-6 should lead to down-regulation of STAT3 activation. We investigated whether tocotrienol can modulate the effect of IL-6-induced STAT3 and found that tocotrienol inhibited STAT3 activation through inhibition of JAK activation in U266 multiple myeloma cells and that it also inhibited IL-6-induced STAT3 activation in MM.1 s cells (Kannappan et al. 2010b).
Phosphatidylinositol 3-kinase (PI3K) is frequently mutated in cancer cells, and many of these mutations lead to aggressive activation of the kinase. Dysregulation of PI3K activity contributes significantly to cell growth, survival, motility, and metabolism (Ikenoue et al. 2005; Mizoguchi et al. 2004; Samuels and Velculescu 2004). A number of PI3K pathway inhibitors have been developed and are being evaluated in preclinical studies and early clinical trials (Courtney et al. 2010). Tocotrienol has been shown to inhibit PI3K/Pl3K-dependent kinase 1 (Pdk-1)/Akt signaling in a phosphatase and tensin homologue deleted from chromosome 10 (PTEN) and in protein phosphatase type 2A independent manner in neoplastic mammary epithelial cells (Sylvester et al. 2005; Shah and Sylvester 2005a). Samant and Sylvester (2006) found that the effect of tocotrienol on PI3K/Pdk-1/Akt signaling is mediated through the suppression of ErbB3-receptor tyrosine phosphorylation in neoplastic mammary epithelial cells. Finally, Shibata et al. (2008b) investigated the effect of tocotrienol on DLD-1-CM-induced-PI3K/Pdk-1/Akt activation in human umbilical vein endothelial cells (HUVEC). They found that tocotrienol reversed the activation of PDK, Akt, and PTEN as well as their respective downstream gene products, endothelial-inducible nitric oxide synthase, glycogen synthase kinase 3 α/β, and extracellular receptor kinase (ERK) 1/2.
Angiogenic pathways and tocotrienol
Angiogenesis is a process of forming new blood vessels from an existing vascular bed. It normally involves a series of steps, including endothelial cell activation, breakdown of the basement membrane, migration, proliferation, and tube formation of the endothelial cell (Folkman and Shing 1992). Modulation of angiogenesis is now a recognized strategy for preventing various angiogenesis-mediated disorders, including solid tumors. Miyazawa and colleagues investigated whether tocotrienol can modulate VEGF-stimulated tube formation in HUVEC. They found that tocotrienol inhibited tube formation and concluded that it might be useful as a therapeutic dietary supplement to minimize tumor angiogenesis (Inokuchi et al. 2003; Miyazawa et al. 2004a; Miyazawa et al. 2004b). Later, this research group reported that tocotrienol also inhibited angiogenesis in in vivo animal models, a dorsal air sac model, and a chick chorioallantoic membrane model (Miyazawa et al. 2009). They found that in the dorsal air sac model, increased neovascularization in mice that had been implanted with human colon carcinoma DLD-1 cells was suppressed by daily dietary supplementation of 10 mg of tocotrienol-rich oil (equivalent to 4.4 mg of pure tocotrienol). In the chorioallantoic membrane model, they found that tocotrienol treatment (500–1000 μg/egg) inhibited the angiogenic response (Miyazawa et al. 2009).
Because VEGF receptor-2 is the key protein in growth factor signal transduction in endothelial cells (Ferrara et al. 2003), regulation of VEGFR-2 activation is a potential molecular target of antiangiogenic compounds (Schlaeppi and Wood 1999). Several researchers have investigated whether the antiangiogenic activity of tocotrienol works via the modulation of VEGF-induced VEGFR-2 phosphorylation. In HUVEC, tocotrienol inhibited the VEGF-induced VEGFR-2 phosphorylation, whereas tocopherol had no effect (Miyazawa et al. 2009; Shibata et al. 2009). Weng-Yew et al. (2009) investigated the molecular mechanism involved in the inhibition of VEGF-stimulated tube formation by tocotrienol and observed that treatment of BALB/c mice with TRF significantly reduced the serum VEGF level (Weng-Yew et al. 2009).
Conclusion
Whereas more than 25,000 studies on tocopherol have been carried out, relatively little is known about tocotrienol. This review suggests that tocotrienol affects numerous pathways linked with tumorigenesis and thus has potential in both the prevention and the treatment of cancer. Additional studies are required to determine the specific intracellular sites of action that tocotrienol and derivatives target in order to fully understand the mechanisms of action mediating their anticancer and apoptotic effects. In addition, the potential value of these compounds as chemotherapeutic agents in the prevention and treatment of various cancers needs to be clarified.
References
Agarwal MK, Agarwal ML, Athar M, Gupta S (2004) Tocotrienol-rich fraction of palm oil activates p53, modulates Bax/Bcl2 ratio and induces apoptosis independent of cell cycle association. Cell Cycle 3:205–211
Aggarwal BB, Sundaram C, Prasad S, Kannappan R (2010) Tocotrienols, the vitamin E of the 21st century: its potential against cancer and other chronic diseases. Biochem Pharmacol 80:1613–1631
Ahmad NS, Khalid BA, Luke DA, Ima Nirwana S (2005) Tocotrienol offers better protection than tocopherol from free radical-induced damage of rat bone. Clin Exp Pharmacol Physiol 32:761–770
Ahn KS, Sethi G, Krishnan K, Aggarwal BB (2007) Gamma-tocotrienol inhibits nuclear factor-kappaB signaling pathway through inhibition of receptor-interacting protein and TAK1 leading to suppression of antiapoptotic gene products and potentiation of apoptosis. J Biol Chem 282:809–820
Bachawal SV, Wali VB, Sylvester PW (2010) Combined gamma-tocotrienol and erlotinib/gefitinib treatment suppresses Stat and Akt signaling in murine mammary tumor cells. Anticancer Res 30:429–437
Bi S, Liu JR, Li Y et al (2010) gamma-Tocotrienol modulates the paracrine secretion of VEGF induced by cobalt(II) chloride via ERK signaling pathway in gastric adenocarcinoma SGC-7901 cell line. Toxicology 274:27–33
Birringer M, Pfluger P, Kluth D, Landes N, Brigelius-Flohe R (2002) Identities and differences in the metabolism of tocotrienols and tocopherols in HepG2 cells. J Nutr 132:3113–3118
Chang PN, Yap WN, Lee DT et al (2009) Evidence of gamma-tocotrienol as an apoptosis-inducing, invasion-suppressing, and chemotherapy drug-sensitizing agent in human melanoma cells. Nutr Cancer 61:357–366
Comitato R, Nesaretnam K, Leoni G et al (2009) A novel mechanism of natural vitamin E tocotrienol activity: involvement of ERbeta signal transduction. Am J Physiol Endocrinol Metab 297:E427–E437
Constantinou C, Hyatt JA, Vraka PS et al (2009) Induction of caspase-independent programmed cell death by vitamin E natural homologs and synthetic derivatives. Nutr Cancer 61:864–874
Courtney KD, Corcoran RB, Engelman JA (2010) The PI3K pathway as drug target in human cancer. J Clin Oncol 28:1075–1083
Ferrara N, Gerber HP, LeCouter J (2003) The biology of VEGF and its receptors. Nat Med 9:669–676
Folkman J, Shing Y (1992) Angiogenesis. J Biol Chem 267:10931–10934
Goh SH, Hew NF, Norhanom AW, Yadav M (1994) Inhibition of tumour promotion by various palm-oil tocotrienols. Int J Cancer 57:529–531
Guarino M (2010) Src signaling in cancer invasion. J Cell Physiol 223:14–26
Gupta SC, Sundaram C et al (2010a) Inhibiting NF-kappaB activation by small molecules as a therapeutic strategy. Biochim Biophys Acta 1799(10–12):775–787
Gupta SC, Kim JH et al (2010b) Regulation of survival, proliferation, invasion, angiogenesis, and metastasis of tumor cells through modulation of inflammatory pathways by nutraceuticals. Cancer Metastasis Rev 29(3):405–434
Guthrie N, Gapor A, Chambers AF, Carroll KK (1997) Inhibition of proliferation of estrogen receptor-negative MDA-MB-435 and -positive MCF-7 human breast cancer cells by palm oil tocotrienols and tamoxifen, alone and in combination. J Nutr 127:544S–548S
Har CH, Keong CK (2005) Effects of tocotrienols on cell viability and apoptosis in normal murine liver cells (BNL CL.2) and liver cancer cells (BNL 1ME A.7R.1), in vitro. Asia Pac J Clin Nutr 14:374–380
Heermeier K, Benedict M, Li M et al (1996) Bax and Bcl-xs are induced at the onset of apoptosis in involuting mammary epithelial cells. Mech Dev 56:197–207
Hockenbery D, Nunez G, Milliman C, Schreiber RD, Korsmeyer SJ (1990) Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature 348:334–336
Hsieh TC, Wu JM (2008) Suppression of cell proliferation and gene expression by combinatorial synergy of EGCG, resveratrol and gamma-tocotrienol in estrogen receptor-positive MCF-7 breast cancer cells. Int J Oncol 33:851–859
Hsieh TC, Elangovan S, Wu JM (2010) Differential suppression of proliferation in MCF-7 and MDA-MB-231 breast cancer cells exposed to alpha-, gamma- and delta-tocotrienols is accompanied by altered expression of oxidative stress modulatory enzymes. Anticancer Res 30:4169–4176
Hussein D, Mo H (2009) d-Dlta-tocotrienol-mediated suppression of the proliferation of human PANC-1, MIA PaCa-2, and BxPC-3 pancreatic carcinoma cells. Pancreas 38:e124–e136
Ikenoue T, Kanai F, Hikiba Y et al (2005) Functional analysis of PIK3CA gene mutations in human colorectal cancer. Cancer Res 65:4562–4567
Inokuchi H, Hirokane H, Tsuzuki T et al (2003) Anti-angiogenic activity of tocotrienol. Biosci Biotechnol Biochem 67:1623–1627
Kamal-Eldin A, Appelqvist LA (1996) The chemistry and antioxidant properties of tocopherols and tocotrienols. Lipids 31:671–701
Kannappan R, Gupta SC et al. (2011) Neuroprotection by Spice-Derived Nutraceuticals: You are What You Eat! Mol Neurobiol (in press)
Kannappan R, Ravindran J, Prasad S et al (2010a) Gamma-tocotrienol promotes TRAIL-induced apoptosis through reactive oxygen species/extracellular signal-regulated kinase/p53-mediated upregulation of death receptors. Mol Cancer Ther 9:2196–2207
Kannappan R, Yadav VR, Aggarwal BB (2010b) {Gamma}-tocotrienol but not {gamma}-tocopherol blocks STAT3 cell signaling pathway through induction of protein-tyrosine phosphatase SHP-1 and sensitizes tumor cells to chemotherapeutic agents. J Biol Chem 285:33520–33528
Kashiwagi K, Harada K, Yano Y et al (2008) A redox-silent analogue of tocotrienol inhibits hypoxic adaptation of lung cancer cells. Biochem Biophys Res Commun 365:875–881
Kashiwagi K, Virgona N, Harada K et al (2009) A redox-silent analogue of tocotrienol acts as a potential cytotoxic agent against human mesothelioma cells. Life Sci 84:650–656
Kato A, Yamaoka M, Tanaka A, Komiyama Ka, Umezawa I (1985) Physiological effect of tocotrienol. J Jpn Oil Chem Soc (Yukugaku) 34:375–376
Kerr JF, Wyllie AH, Currie AR (1972) Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 26:239–257
Kimmick GG, Bell RA, Bostick RM (1997) Vitamin E and breast cancer: a review. Nutr Cancer 27:109–117
Kline K, Yu W, Sanders BG (2004) Vitamin E and breast cancer. J Nutr 134:3458S–3462S
Komiyama K, Iizuka K, Yamaoka M et al (1989) Studies on the biological activity of tocotrienols. Chem Pharm Bull (Tokyo) 37:1369–1371
Kunnumakkara AB, Sung B, Ravindran J et al (2010) {Gamma}-tocotrienol inhibits pancreatic tumors and sensitizes them to gemcitabine treatment by modulating the inflammatory microenvironment. Cancer Res 70:8695–8705
Liu HK, Wang Q, Li Y et al (2010) Inhibitory effects of gamma-tocotrienol on invasion and metastasis of human gastric adenocarcinoma SGC-7901 cells. J Nutr Biochem 21:206–213
Mazlan M, Sue Mian T, Mat Top G, Zurinah Wan Ngah W (2006) Comparative effects of alpha-tocopherol and gamma-tocotrienol against hydrogen peroxide induced apoptosis on primary-cultured astrocytes. J Neurol Sci 243:5–12
McAnally JA, Gupta J, Sodhani S, Bravo L, Mo H (2007) Tocotrienols potentiate lovastatin-mediated growth suppression in vitro and in vivo. Exp Biol Med (Maywood) 232:523–531
McIntyre BS, Briski KP, Gapor A, Sylvester PW (2000a) Antiproliferative and apoptotic effects of tocopherols and tocotrienols on preneoplastic and neoplastic mouse mammary epithelial cells. Proc Soc Exp Biol Med 224:292–301
McIntyre BS, Briski KP, Tirmenstein MA et al (2000b) Antiproliferative and apoptotic effects of tocopherols and tocotrienols on normal mouse mammary epithelial cells. Lipids 35:171–180
Meydani M (1995) Vitamin E. Lancet 345:170–175
Miyazawa T, Inokuchi H, Hirokane H et al (2004a) Anti-angiogenic potential of tocotrienol in vitro. Biochemistry (Mosc) 69:67–69
Miyazawa T, Tsuzuki T, Nakagawa K, Igarashi M (2004b) Antiangiogenic potency of vitamin E. Ann N Y Acad Sci 1031:401–404
Miyazawa T, Shibata A, Sookwong P et al (2009) Antiangiogenic and anticancer potential of unsaturated vitamin E (tocotrienol). J Nutr Biochem 20:79–86
Mizoguchi M, Nutt CL, Mohapatra G, Louis DN (2004) Genetic alterations of phosphoinositide 3-kinase subunit genes in human glioblastomas. Brain Pathol 14:372–377
Nass SJ, Li M, Amundadottir LT, Furth PA, Dickson RB (1996) Role for Bcl-xL in the regulation of apoptosis by EGF and TGF beta 1 in c-myc overexpressing mammary epithelial cells. Biochem Biophys Res Commun 227:248–256
Nesaretnam K, Guthrie N, Chambers AF, Carroll KK (1995) Effect of tocotrienols on the growth of a human breast cancer cell line in culture. Lipids 30:1139–1143
Nesaretnam K, Stephen R, Dils R, Darbre P (1998) Tocotrienols inhibit the growth of human breast cancer cells irrespective of estrogen receptor status. Lipids 33:461–469
Neuzil J, Zhao M, Ostermann G et al (2002) Alpha-tocopheryl succinate, an agent with in vivo anti-tumour activity, induces apoptosis by causing lysosomal instability. Biochem J 362:709–715
Ong FB, Wan Ngah WZ, Shamaan NA et al (1993) Glutathione S-transferase and gamma-glutamyl transpeptidase activities in cultured rat hepatocytes treated with tocotrienol and tocopherol. Comp Biochem Physiol C 106:237–240
Osakada F, Hashino A, Kume T et al (2004) Alpha-tocotrienol provides the most potent neuroprotection among vitamin E analogs on cultured striatal neurons. Neuropharmacology 47:904–915
Park SK, Sanders BG, Kline K (2010) Tocotrienols induce apoptosis in breast cancer cell lines via an endoplasmic reticulum stress-dependent increase in extrinsic death receptor signaling. Breast Cancer Res Treat 124:361–375
Pierpaoli E, Viola V, Pilolli F et al (2010) Gamma- and delta-tocotrienols exert a more potent anticancer effect than alpha-tocopheryl succinate on breast cancer cell lines irrespective of HER-2/neu expression. Life Sci 86:668–675
Qureshi AA, Burger WC, Peterson DM, Elson CE (1986) The structure of an inhibitor of cholesterol biosynthesis isolated from barley. J Biol Chem 261:10544–10550
Raff M (1998) Cell suicide for beginners. Nature 396:119–122
Rickmann M, Vaquero EC, Malagelada JR, Molero X (2007) Tocotrienols induce apoptosis and autophagy in rat pancreatic stellate cells through the mitochondrial death pathway. Gastroenterology 132:2518–2532
Rodig SJ, Meraz MA, White JM et al (1998) Disruption of the Jak1 gene demonstrates obligatory and nonredundant roles of the Jaks in cytokine-induced biologic responses. Cell 93:373–383
Sakai M, Okabe M, Yamasaki M, Tachibana H, Yamada K (2004) Induction of apoptosis by tocotrienol in rat hepatoma dRLh-84 cells. Anticancer Res 24:1683–1688
Sakai M, Okabe M, Tachibana H, Yamada K (2006) Apoptosis induction by gamma-tocotrienol in human hepatoma Hep3B cells. J Nutr Biochem 17:672–676
Samant GV, Sylvester PW (2006) gamma-Tocotrienol inhibits ErbB3-dependent PI3K/Akt mitogenic signalling in neoplastic mammary epithelial cells. Cell Prolif 39:563–574
Samant GV, Wali VB, Sylvester PW (2010) Anti-proliferative effects of gamma-tocotrienol on mammary tumour cells are associated with suppression of cell cycle progression. Cell Prolif 43:77–83
Samuels Y, Velculescu VE (2004) Oncogenic mutations of PIK3CA in human cancers. Cell Cycle 3:1221–1224
Schlaeppi JM, Wood JM (1999) Targeting vascular endothelial growth factor (VEGF) for anti-tumor therapy, by anti-VEGF neutralizing monoclonal antibodies or by VEGF receptor tyrosine-kinase inhibitors. Cancer Metastasis Rev 18:473–481
Schneider C (2005) Chemistry and biology of vitamin E. Mol Nutr Food Res 49:7–30
Sen R, Baltimore D (1986) Inducibility of kappa immunoglobulin enhancer-binding protein Nf-kappa B by a posttranslational mechanism. Cell 47(6):921–928
Sen CK, Khanna S, Roy S, Packer L (2000) Molecular basis of vitamin E action. Tocotrienol potently inhibits glutamate-induced pp60(c-Src) kinase activation and death of HT4 neuronal cells. J Biol Chem 275:13049–13055
Shah S, Sylvester PW (2004) Tocotrienol-induced caspase-8 activation is unrelated to death receptor apoptotic signaling in neoplastic mammary epithelial cells. Exp Biol Med (Maywood) 229:745–755
Shah SJ, Sylvester PW (2005a) Gamma-tocotrienol inhibits neoplastic mammary epithelial cell proliferation by decreasing Akt and nuclear factor kappaB activity. Exp Biol Med (Maywood) 230:235–241
Shah SJ, Sylvester PW (2005b) Tocotrienol-induced cytotoxicity is unrelated to mitochondrial stress apoptotic signaling in neoplastic mammary epithelial cells. Biochem Cell Biol 83:86–95
Shah S, Gapor A, Sylvester PW (2003) Role of caspase-8 activation in mediating vitamin E-induced apoptosis in murine mammary cancer cells. Nutr Cancer 45:236–246
Shibata A, Nakagawa K, Sookwong P et al (2008a) Tocotrienol inhibits secretion of angiogenic factors from human colorectal adenocarcinoma cells by suppressing hypoxia-inducible factor-1alpha. J Nutr 138:2136–2142
Shibata A, Nakagawa K, Sookwong P et al (2008b) Tumor anti-angiogenic effect and mechanism of action of delta-tocotrienol. Biochem Pharmacol 76:330–339
Shibata A, Nakagawa K, Sookwong P et al (2009) delta-Tocotrienol suppresses VEGF induced angiogenesis whereas alpha-tocopherol does not. J Agric Food Chem 57:8696–8704
Shun MC, Yu W, Gapor A et al (2004) Pro-apoptotic mechanisms of action of a novel vitamin E analog (alpha-TEA) and a naturally occurring form of vitamin E (delta-tocotrienol) in MDA-MB-435 human breast cancer cells. Nutr Cancer 48:95–105
Srivastava JK, Gupta S (2006) Tocotrienol-rich fraction of palm oil induces cell cycle arrest and apoptosis selectively in human prostate cancer cells. Biochem Biophys Res Commun 346:447–453
Sun W, Wang Q, Chen B et al (2008) Gamma-tocotrienol-induced apoptosis in human gastric cancer SGC-7901 cells is associated with a suppression in mitogen-activated protein kinase signalling. Br J Nutr 99:1247–1254
Sun W, Xu W, Liu H et al (2009) gamma-Tocotrienol induces mitochondria-mediated apoptosis in human gastric adenocarcinoma SGC-7901 cells. J Nutr Biochem 20:276–284
Sundram K, Khor HT, Ong AS, Pathmanathan R (1989) Effect of dietary palm oils on mammary carcinogenesis in female rats induced by 7, 12-dimethylbenz(a)anthracene. Cancer Res 49:1447–1451
Sylvester PW, Shah S (2005a) Intracellular mechanisms mediating tocotrienol-induced apoptosis in neoplastic mammary epithelial cells. Asia Pac J Clin Nutr 14:366–373
Sylvester PW, Shah SJ (2005b) Mechanisms mediating the antiproliferative and apoptotic effects of vitamin E in mammary cancer cells. Front Biosci 10:699–709
Sylvester PW, Nachnani A, Shah S, Briski KP (2002) Role of GTP-binding proteins in reversing the antiproliferative effects of tocotrienols in preneoplastic mammary epithelial cells. Asia Pac J Clin Nutr 11(Suppl 7):S452–S459
Sylvester PW, Shah SJ, Samant GV (2005) Intracellular signaling mechanisms mediating the antiproliferative and apoptotic effects of gamma-tocotrienol in neoplastic mammary epithelial cells. J Plant Physiol 162:803–810
Takahashi K, Loo G (2004) Disruption of mitochondria during tocotrienol-induced apoptosis in MDA-MB-231 human breast cancer cells. Biochem Pharmacol 67:315–324
Ueda T, Bruchovsky N, Sadar MD (2002) Activation of the androgen receptor N-terminal domain by interleukin-6 via MAPK and STAT3 signal transduction pathways. J Biol Chem 277:7076–7085
Uto-Kondo H, Ohmori R, Kiyose C et al (2009) Tocotrienol suppresses adipocyte differentiation and Akt phosphorylation in 3T3–L1 preadipocytes. J Nutr 139:51–57
Vogelstein B, Kinzler KW (2004) Cancer genes and the pathways they control. Nat Med 10:789–799
Wada S, Satomi Y, Murakoshi M et al (2005) Tumor suppressive effects of tocotrienol in vivo and in vitro. Cancer Lett 229:181–191
Wali VB, Bachawal SV, Sylvester PW (2009a) Suppression in mevalonate synthesis mediates antitumor effects of combined statin and gamma-tocotrienol treatment. Lipids 44:925–934
Wali VB, Bachawal SV, Sylvester PW (2009b) Endoplasmic reticulum stress mediates gamma-tocotrienol-induced apoptosis in mammary tumor cells. Apoptosis 14:1366–1377
Weng-Yew W, Selvaduray KR, Ming CH, Nesaretnam K (2009) Suppression of tumor growth by palm tocotrienols via the attenuation of angiogenesis. Nutr Cancer 61(3):367–373
Wu SJ, Ng LT (2010) Tocotrienols inhibited growth and induced apoptosis in human HeLa cells through the cell cycle signaling pathway. Integr Cancer Ther 9:66–72
Wu SJ, Liu PL, Ng LT (2008) Tocotrienol-rich fraction of palm oil exhibits anti-inflammatory property by suppressing the expression of inflammatory mediators in human monocytic cells. Mol Nutr Food Res 52:921–929
Xu WL, Liu JR, Liu HK et al (2009) Inhibition of proliferation and induction of apoptosis by gamma-tocotrienol in human colon carcinoma HT-29 cells. Nutrition 25:555–566
Yap WN, Zaiden N, Tan YL et al (2010) Id1, inhibitor of differentiation, is a key protein mediating anti-tumor responses of gamma-tocotrienol in breast cancer cells. Cancer Lett 291:187–199
Yang Z, Xiao H, Jin H et al (2010) Synergistic actions of atorvastatin with gamma-tocotrienol and celecoxib against human colon cancer HT29 and HCT116 cells. Int J Cancer 126:852–863
Yano Y, Satoh H, Fukumoto K et al (2005) Induction of cytotoxicity in human lung adenocarcinoma cells by 6-O-carboxypropyl-alpha-tocotrienol, a redox-silent derivative of alpha-tocotrienol. Int J Cancer 115:839–846
Yap WN, Chang PN, Han HY et al (2008) Gamma-tocotrienol suppresses prostate cancer cell proliferation and invasion through multiple-signalling pathways. Br J Cancer 99:1832–1841
Yap WN, Zaiden N, Luk SY et al (2010) In vivo evidence of gamma-tocotrienol as a chemosensitizer in the treatment of hormone-refractory prostate cancer. Pharmacology 85:248–258
Yu W, Simmons-Menchaca M, Gapor A, Sanders BG, Kline K (1999) Induction of apoptosis in human breast cancer cells by tocopherols and tocotrienols. Nutr Cancer 33:26–32
Author information
Authors and Affiliations
Corresponding author
Additional information
This paper is part of a Special Issue entitled “Tocotrienols in health and disease”.
Rights and permissions
About this article
Cite this article
Kannappan, R., Gupta, S.C., Kim, J.H. et al. Tocotrienols fight cancer by targeting multiple cell signaling pathways. Genes Nutr 7, 43–52 (2012). https://doi.org/10.1007/s12263-011-0220-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12263-011-0220-3