
Abstract
The consumption of high-calorie foods combined with less physical exercise has increased the prevalence of obesity. Obesity is also associated with high blood pressure, atherosclerosis, insulin resistance, diabetes, impaired host defense, and the risk of some cancers. Because PPARγ is a central player that participates in various biological responses, including lipid metabolism, inflammation, and cell proliferation, further understanding of the lipid metabolic sensor PPARγ is necessary to reduce the incidence of metabolic diseases and cancer.
Similar content being viewed by others

Obesity lies at the crossroads of various metabolic diseases
Obesity, which is recognized as one of the most significant global health concerns, is a chronic condition of excess body fat (Hill et al. 2003). Because current human living conditions tend to encourage high-calorie food intake with less energy expenditure, the obesity epidemic will show continuous increases in the future (Yach et al. 2006). In addition, obesity contributes to several metabolic diseases, including various cardiovascular diseases, insulin resistance, type 2 diabetes, impaired immunity, and the risk of cancer (Kopelman 2000; Pi-Sunyer 2003). The treatment of obesity has focused on behavioral changes, and education to consume a healthy diet with physical activity can be successful, but it is difficult to maintain a healthy life style. Other strategies such as surgical procedures and medications are limited due to safety, economic reasons, and effectiveness (Fisher and Schauer 2002). During the last few decades, more players in food intake, energy expenditure, and nutrient storage have been elucidated (Turner et al. 2014). With the identification of new molecular targets and better understanding of energy metabolism, new therapies may become available to keep pace with the obesity prevalence in future.
PPARγ plays a central role in obesity, immunity, and cancer
Three distinct peroxisome proliferator-activated receptor types—PPARα (NR1C1), PPARβ/δ (NR1C2), and PPARγ (NR1C3)—have been identified. The most extensively studied PPARγ is highly expressed in adipose tissues and plays a role in glucose and lipid metabolism, whereas PPARα is a major player in the mitochondrial and peroxisomal β-oxidation pathway expressed most abundantly in liver (van Raalte et al. 2004). Ubiquitously expressed PPARβ/δ in hypertension, insulin resistance, and anti-inflammation has been documented (Fredenrich and Grimaldi 2005). Although PPAR family members seem to contain similar functions in inflammation, energy metabolism, and vascular tone, their affinities for lipid metabolites and their expression patterns can be discriminated.
Metabolic PPARγ
At the intersection of obesity, insulin resistance, inflammation, and cardiovascular disease, the nuclear receptor peroxisome proliferator-activated receptor gamma (PPARγ) is a critical metabolic regulator (Tontonoz and Spiegelman 2008) (Fig. 1). PPARγ, a ligand-activated transcription factor, heterodimerizes with RXR (retinoic X receptor) and translocates into the nucleus to regulate downstream target gene expression upon activation by its agonists (Tontonoz and Spiegelman 2008). PPARγ induces the expression of genes involved in lipid synthesis and adipocyte differentiation. These target genes include CD36, fatty acid binding protein 4, adiponectin, and CCAAT/enhancer binding protein α. PPARγ is most famous for its roles in adipogenesis. Defects in PPARγ result in lipodystrophy in mice and humans. Forced expression of PPARγ in fibroblasts can induce adipocyte-like phenotypic changes. PPARγ can incorporate various signaling cascades of WNT, IGF, FGF, TGF-β, and BMP, cell density, and the composition of the extracellular matrix to coordinate adipogenic processes (Hong and Park 2010). PPARγ also orchestrates adipogenesis through its interaction with other transcriptional mediators such as the C/EBP family (α, β, and δ), SREBP, PRDM, PGC-1, ZFP423, REV-ERBα, GATA3, and several miRNAs. Thus, it is believed that PPARγ is the orchestral conductor in adipogenesis through harmonizing the actions of these multiple players (Cristancho and Lazar 2011).

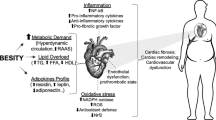
Pleiotropic effects of PPARγ in metabolism, the immune system, and cancer. The lipid metabolic sensor PPARγ conveys the cellular environment to control metabolic gene expression in adipocytes, hepatocytes, the immune systems, and cancer. In these tissues, PPARγ behaves as a key signaling integrator of growth factors, nutrients, and transcriptional cascades to coordinate metabolism, immunity, cell proliferation, and differentiation
Investigations of the molecular actions of insulin sensitizing chemical drugs have revealed that PPARγ can play a significant role in systemic glucose homeostasis. Troglitazone a class of thiazolidinediones (TZDs) drug were first identified in animal models as an insulin sensitizer. Subsequent studies demonstrated that PPARγ in adipose tissues unexpectedly acts as the biological receptor for TZDs that include ciglitazone, englitazone, and pioglitazone (Lehmann et al. 1995). These studies were further verified by the identification of mutations in PPARγ associated with insulin resistance and diabetes in both animal models and humans (Tontonoz and Spiegelman 2008). Although TZDs are being questioned due to their adverse effects (Nissen and Wolski 2007; Graham et al. 2010), they have broadened the understanding of PPAR actions in systemic glucose metabolism beyond adipogenesis.
There are at least two different types of adipocyte tissues: white adipose tissue (WAT), which stores excess calories for energy partitioning into peripheral organs under energy-deprived conditions, and brown adipose tissue (BAT), which dissipates heat in response to thermogenic stimuli (Peirce et al. 2014). Recent studies have shown that multilocular brown adipocytes containing more mitochondria with uncoupling protein-1 expression generate energy expensive heat resulting in increased energy expenditure, body weight decrease, and improved insulin sensitivity. Therefore, BAT is considered as health beneficial fats that counteract obesity and metabolism. Most players in white adipogenesis described above, including PPARγ, similarly act to induce brown adipogenesis (Bartelt and Heeren 2014). However, BAT- or WAT-specific players are also described, suggesting the existence of alternate routes for the generation of the two different fats. For example, PRDM16 and PGC-1α play critical roles only for brown adipogenesis, whereas the players RIP140 and TIF2 induce white adipogenesis selectively (Peirce et al. 2014). Further studies also suggest functional links between the immune system and thermogenic adipocytes (Wernstedt Asterholm et al. 2014). Eosinophil and type 2 cytokine signaling in macrophages can translate cold sensing into thermogenesis in adipocytes, leading to body weight decrease (Qiu et al. 2014). Elucidation of the molecular mechanisms that generate the brown-like phenotype in white adipocytes can provide a new defense against obesity and metabolic diseases. Likewise, further dissection of immune and thermogenic adipocytes are intriguing new avenues for targeting obesity and related diseases.
PPARγ also functions in other metabolic tissues. PPARγ is detected at a low level in skeletal muscles compared with that in fat tissues but indirectly affects skeletal muscle by systemic lipid partitioning and secretion of adipokine expression (Tontonoz and Spiegelman 2008). Similarly, PPARγ is expressed in the liver, but its level compared with the expression in adipose tissues is also significantly lower. However, the expression of PPARγ is induced in hepatic fatty liver, indicating the pathophysiological roles of PPARγ in the liver (Gavrilova et al. 2003). TZD treatments in diabetic mice exacerbate hepatic steatosis in a PPARγ-dependent manner (Yu et al. 2003). Thus, overcoming this obstacle can lead to better insulin sensitization under diabetic conditions.
Global PPARγ deletion caused embryonic lethality due to the defects in placenta (Barak et al. 1999). Subsequently, the placental defects rescued PPARγ deleted embryos demonstrated the roles of PPARγ in lipodystrophy and insulin resistance (Barak et al. 1999; Duan et al. 2007). Mice lacking PPARγ in muscle developed insulin resistance (Hevener et al. 2003; Norris et al. 2003). Hepatocyte specific disruption of PPARγ improved fatty liver but decreased insulin sensitivity in diabetic mice (Matsusue et al. 2003). The effects of PPARγ in whole body metabolism and insulin sensitivity can be also attributed by the adipose PPARγ. Adipocyte specific deletion by the adipocyte protein2 (aP2) promoter driven Cre line on a high fat diet showed the roles of adipose PPARγ in fat mass and fatty liver (He et al. 2003; Jones et al. 2005). The critical role of adipose PPARγ in lipid metabolism and insulin resistance were recently verified by the generation of another fat specific knockout model using adiponectin promoter driven Cre (Wang et al. 2013). This fat specific deletion of PPARγ exhibited lipoatrophy, insulin resistance, increased bone mass, and abnormal marry glands, further indicating that the adipose PPARγ is necessary for fat development, fat related tissues, and whole body metabolisms. Therefore, it is reasonable to consider that the PPARγ in various metabolic tissues crosstalk to maintain whole body insulin sensitivity and lipid metabolism.
Immunogenic PPARγ
Obesity is often considered as a low-grade, chronically inflamed state in addition to involving excessive nutrient stores. In obese and insulin-resistant mice, increased macrophage infiltration has been observed in fat tissues (Weisberg et al. 2003; Xu et al. 2003). Enhanced cytokine production from the infiltrated macrophages contributes to insulin resistance (Ferrante 2013). In addition, PPARγ also play a role in macrophage biology and insulin sensitivity. Macrophages can be activated to classically activated inflammatory (M1) macrophages or to more metabolism-friendly alternatively activated (M2) macrophages (Odegaard and Chawla 2011). Infiltrated macrophages in adipose tissues can also lead to alteration of alternatively activated macrophages to classically activated macrophages. Macrophages defective in PPARγ were shown to fail to differentiate into M2 macrophages and are associated with insulin resistance (Odegaard et al. 2007).
Additionally, PPARγ exerts an anti-inflammatory response in murine and human macrophages. Although the mechanism of repression needs to be further clarified, liganded PPARγ, instead of binding to DNA elements, inhibits the inflammatory activities of activator protein-1 (AP-1), signal transducers and activators of transcription 1 (STAT-1), nuclear factor κB (NF-κB), and nuclear factor of activated T cells (NFAT) transcription factor by physical interaction, referred to as transrepression (Pascual et al. 2005). Furthermore, it has been shown that defects in NF-κB signaling in macrophages improve insulin sensitivity (Shoelson et al. 2003). Thus, anti-inflammatory roles of PPARγ in macrophages can participate, at least in part, in insulin-sensitizing actions.
Macrophage PPARγ also plays important roles in atherosclerosis. 9-Hydroxy octadecadienoic acid (9-HODE) and 13-HODE catabolized from atherogenic oxidized LDL cholesterols activate PPARγ and increase lipid uptake through the induction of CD36, a scavenger receptor for LDL, ultimately converting into atherogenic foam cells (Nagy et al. 1998; Tontonoz et al. 1998). It was also reported that PPARγ ligands could be anti-atherogenic by suppressing cytokine release from macrophages (Ricote et al. 1998). This effect was shown to be mediated by the induction of another nuclear receptor liver X receptor (LXR) followed by the increase of its target gene ATP-binding cassette transporter A1 (ABCA1), a reverse cholesterol transporter with reduction of SR-A, a second LDL scavenger receptor in mouse macrophages (Chawla et al. 2001). Nevertheless, PPARγ acts to regulate cholesterol uptake in macrophages, and activation of PPARγ by agonists reduces atherosclerosis in rodents and human patients.
Regarding its anti-inflammatory actions in macrophages, PPARγ also negatively affects the maturation of dendritic cells (DCs) (Szanto and Nagy 2008; Kiss et al. 2013). Activation of PPARγ in DCs decrease cytokine IL-12, chemokine CXCL10 and CCL5, and costimulatory molecules CD80, CD83, and CD40 with induction of the coinhibitory molecule B7H1 in cultured human DCs (Nencioni et al. 2002; Szatmari et al. 2006). These negative effects on the immunostimulatory capacity in DCs by PPARγ activation are shown to be mediated through inhibition of the signaling cascade for the NF-κB family of transcription factors (Appel et al. 2005). Activated DCs downregulate chemokine receptor CCR7 by PPARγ activation, which in turn responds poorly to CCL19 and CCL21 for homing DCs to secondary lymphoid tissues (Nencioni et al. 2002; Hammad et al. 2004; Appel et al. 2005). Together, these data show the roles of PPARγ in innate immune cells to further shape immune responses.
In addition to the roles in the innate immune system, several studies have demonstrated that PPARγ also tunes adaptive immune responses (da Rocha Junior et al. 2013; Kidani and Bensinger 2012). PPARγ negatively regulates T cell survival and activation by inhibiting IL-2 production. PPARγ is also involved in the balance of Th1/Th2 by increasing Th2 cytokine and IL-4 production but decreasing IFN-γ and Th1 cytokine production, although Th2 preference by PPARγ activation can be dependent on the cell type and selective ligand (Tontonoz and Spiegelman 2008; da Rocha Junior et al. 2013). PPARγ activation also impairs Th17 cell development and affects Th17-dependent autoimmune diseases such as experimental autoimmune encephalomyelitis, inflammatory bowel disease, and arthritis (Straus and Glass 2007; Klotz et al. 2009). Regulatory T cells (Treg) are decreased in T-cell-specific PPARγ-deleted mice, suggesting that both the survival of regulatory T cells and their effects on effector CD4+ T cells can be controlled by PPARγ (Hontecillas and Bassaganya-Riera 2007; Guri et al. 2010). Similar to the effects in T cells, PPARγ activation in B cells is anti-proliferative, but the effects are not consistent with those reported in the literature (da Rocha Junior et al. 2013). Given that various autoimmune diseases are associated with the aberrant activation of adaptive immunity, further understanding of PPARγ effects in the context of immune diseases will be required.
Anti-cancerous PPARγ
PPARγ is expressed in various cancer tissues, as well as in metabolic and immune cells (Campbell et al. 2008). Several reports have shown that PPARγ displays anti-tumorigenic functions (Koeffler 2003). Many studies have identified that the activation of PPARγ represses lung, breast, colon and prostate cancer (Bren-Mattison et al. 2008; Bonofiglio et al. 2006; Cesario et al. 2006; Sikka et al. 2012). Christen A. et al. suggested that depletion of PPARγ increases colon cancer in APCmin/+ mice (McAlpine et al. 2006). Corroborating these observations, the survival rate of colorectal cancer patients was found to be increased more under the positive expression of PPARγ (Ogino et al. 2009). Adipocyte-specific knockout PPARγ mice display more susceptibility toward DMBA-induced breast cancer than wild-type mice (Skelhorne-Gross et al. 2012). In prostate cancer, upregulated NcoR1, a PPARγ negative cofactor, demonstrates negative effects on tumorigenesis (Battaglia et al. 2010).
Additionally, agonists of PPARγ regulate the tumorigenesis of various cancers. For example, TZDs have anti-tumorigenic effects on cancer tissues by inducing cell cycle arrest and apoptosis, as well as by suppressing EMT. One PPARγ agonist, troglitazone, suppresses pancreatic carcinoma cell growth through the increased p27 protein levels (Motomura et al. 2000). The proliferation of the breast cancer cell line MCF-7 was blocked by troglitazone, resulting in the reduction of Rb phosphorylation and leading to decreased CDK2 and -4 activities (Yin et al. 2001). Another TZD, ciglitazone, can also increase p27 protein levels, leading to the suppression of cell proliferation (Chen and Harrison 2005).
Several studies have shown that activation of PPARγ by its ligands could modulate apoptosis via various signaling mechanisms. For example, TZD, upon binding PPARγ, can inhibit post-translational modification such as phosphorylation of β-catenin, ultimately suppressing Wnt pathways and inducing cell death (Sharma et al. 2004; Wei et al. 2007). Activation of PPARγ by rosiglitazone extensively increases PTEN levels, suppressing growth factor-dependent AKT activation (Farrow and Evers 2003). PPARγ can also directly regulate phosphorylation of AKT and PI3K activities, inducing apoptosis (Kim et al. 2006; Yan et al. 2010). Furthermore, PPARγ activation is able to inhibit BCL family function, accelerate the turnover rate of FLIP, and increase the expression of BAX and BAD, resulting in apoptosis (Shiau et al. 2005; Kim et al. 2002; Zander et al. 2002; Bae and Song 2003). Finally, many cancers with a related inflammation mechanism are also regulated by PPARγ activation. PPARγ agonists suppress the expression of pro-inflammatory proteins, IL-6, TNF, and MCP1 (McKinnon et al. 2012; Hwa et al. 2011; Wang et al. 2011; Nguyen et al. 2012). Agonists also contribute to PPARγ sumoylation, which negatively interfere with the activation of NF-κB activity, leading to cell death (Ramkalawan et al. 2012). On the other hand, some of the studies report that patients of diabetes mellitus treated with TZD or rosiglitazone displayed a significant positive correlation with a diagnosis of cancer (Ramos-Nino et al. 2007). TZD treatment also accelerated polyp formation in the colon of APCMin mice (Saez et al. 1998). However, a population-based cohort study in the patients with diabetes mellitus treated with TZD displayed reduction in lung cancer risk but not in colorectal and prostate cancer (Govindarajan et al. 2007). It is not clear at the moment whether the systemic activation of PPARγ using its activators could reduce the risk of cancer development. Thus, the effects of PPARγ agonists on relieving the symptom of diabetes mellitus and controlling the risk of cancer are still to be pursued. In summary, these data suggest that PPARγ activation by its agonist could have repressive roles in tumor formation by inducing cell cycle arrest, accelerating apoptosis and suppressing the production of pro-inflammatory proteins. However, its systemic effects on risk of cancer in patients with diabetes mellitus will require further investigations.
Miscellaneous PPARγ
Bone homeostasis is controlled by bone formation by osteoblasts and resorption by osteoclasts (Sims and Gooi 2008; Manolagas 1998). PPARγ inhibits the generation of bone-forming osteoblasts at the expense of adipocytes from common precursor cells. PPARγ silencing in bone marrow skews toward osteoblasts with significant reduction of osteoclasts and adipocytes (Park et al. 2008a). Conversely, aging and PPARγ activation increase lipid droplet formation and osteoclast activity and reduce osteoblast formation in bone marrow cells (Moerman et al. 2004). A number of signaling events, including the Wnt and hedgehog pathways, control the inverse balance between osteogenic and adipogenic differentiation (Park et al. 2008a). The reciprocal decision of adipogenic or osteogenic fate was further elucidated by a high bone mass resulting from the loss of bone marrow fat in adipose-specific PPARγ knockout mice (Wang et al. 2013). PPARγ is also involved in neuronal death, differentiation, and axon polarity (Quintanilla et al. 2014). Taken together, the data show that PPARγ, a lipid metabolic sensor, exerts its functions through survival, proliferation, and differentiation and is a regulator of metabolic homeostasis in various tissues (Fig. 1).
PPARγ as a therapeutic target
Drugs of the TZD class are PPARγ agonists that have been therapeutically proven to be useful in the treatment of type 2 diabetes (Lehmann et al. 1995). Genetic evidence, including gain-of function and loss-of-function experiments, showed that PPARγ in adipose tissues is the primary target for the glucose-lowering effects of TZDs (Tontonoz and Spiegelman 2008). TZDs activate a set of genes to increase lipid synthesis and lipid uptake in adipocytes. Thus, activation of PPARγ is thought to repartition insulin-resistant lipids from insulin-acting tissues through the induction of lipid flux in adipocytes, which in turn improves insulin sensitivity in the liver and muscles (Waki and Tontonoz 2007). Unfortunately, the use of TZD is questioned by unfavorable side effects such as weight gain, fluid retention, edema, and a higher risk of congestive heart failure (Nesto et al. 2004; Nissen and Wolski 2007). Therefore, the limitation of TZD requires other therapeutic approaches to treat type 2 diabetes.
To circumvent the issue of TZDs, significant efforts are being made for differential PPARγ activation. To separate the beneficial metabolic effects from the unwanted side effects, highly potent and selective PPARα modulators (SPPARMα) and PPARγ modulators (SPPARMγ) are being investigated (Balint and Nagy 2006; DePaoli et al. 2014; Feldman et al. 2008). Dual PPAR-α/γ agonists were also studied and have displayed some promising metabolic effects. Partial agonists, pan agonists, and antagonists are also being developed for the generation of safe and effective drugs. Similarly, significant efforts have led to the identification of SPPARMγ, and partial or dual agonists from herbal natural products (Wang et al. 2014a). These selective compounds have already shown improvement regarding metabolic parameters in diabetic animals with few side effects compared with TZD treatments.
Since the identification of PPARγ as a biological receptor of anti-diabetic TZDs, PPARγ regulation has been actively investigated. Recently, newly identified small-molecule inducers of PPARγ have proposed alternative PPARγ activation for the anti-diabetic effects. Harmine appears to act by inducing PPARγ mRNA expression in preadipocytes. Chronic treatment with harmine in diabetic mice lowered serum glucose levels and improved insulin sensitivity without the detrimental effects of weight gain and liver failure (Waki et al. 2007). Further mechanistic studies showed that harmine induced adipocyte differentiation through acting on the Wnt signaling pathway (Park et al. 2008b). Phenamil was also reported to induce PPARγ expression and adipocyte differentiation (Park et al. 2010). Thus, chemicals that regulate PPARγ expression can represent novel approaches without the side effects as shown in TZD treatments.
PPARγ activation through post-translational modifications of PPARγ can also be viewed as new modulators for PPARγ activity (Hauser et al. 2000; van Beekum et al. 2009). Indeed, recent studies have shown that the phosphorylation of PPARγ by CDK5 lowers glucose levels, and non-TZD drugs post translationally modify PPARγ through acting on CDK5 (Choi et al. 2010; 2011). In addition, PPARγ can be ubiquitinated and sumoylated (Kilroy et al. 2009; Floyd and Stephens 2004). Sumoylation of PPARγ seems to mediate anti-inflammatory activities by repressing inflammatory gene expression in macrophages (Pascual et al. 2005). The ubiquitin ligase Siah2 affects PPARγ protein stability and activity (Kilroy et al. 2012). Similarly, MKRN1 affects PPARγ stability through direct association (Kim et al. 2014). These studies bring new insights into the improvement of insulin sensitivity by managing PPARγ post-translational modification. Furthermore, newly developed PPARγ modulators at the transcriptional and post-translational levels can be used in cancer and immune diseases.
PPARγ are expressed in various cancer tissues, including the breast, colon, lung, prostate and bladder (Campbell et al. 2008). Many investigations are now aimed at determining whether PPARγ and its ligands could be used as therapeutics for cancer treatment. Jeong et al. showed that PPARγ-positive lung cancer cell lines are more sensitive to the anti-cancerous effects of its ligands, implicating its possible usage for cancer therapy (Jeong et al. 2012). In a phase 2 clinical trial, troglitazone showed that it could inhibit the growth of prostate cancer cells (Mueller et al. 2000). Troglitazone also had a negative effect on renal cell carcinoma by inducing p38 MAPK-mediated-cell cycle arrest (Fujita et al. 2011). Rosiglitazone, in a phase II trial, raised the radioiodine uptake in thyroid cancer patients (Kebebew et al. 2006). Recently, many trials have been conducted regarding the combination treatment of PPARγ ligands with other putative cancer drugs such as RXRα agonists, chemotherapeutic agents, statins, or some of cell-to-cell signaling molecules to synergistically cure cancers (Skelhorne-Gross and Nicol 2012). For example, IFN-β-treated pancreatic cancer cells were more affected by co-treatment with troglitazone, which represses the NF-κB-related survival pathway (Vitale et al. 2012). A PPARγ agonist, DIM-C, makes bladder cancer cells more susceptible to EGFR inhibition (Mansure et al. 2013). The combination treatment of troglitazone and the RXR agonist RA inhibits the proliferation of gastric cancer cells by increasing apoptosis (Liu et al. 2013). These results indicate that targeting PPARγ alone or with combination treatments could be beneficial for curing cancer in the future.
Future directions
In this review, the pleiotropic effects of PPARγ in multiple cell types were discussed (Fig. 1). PPARγ is one of the most important lipid sensors that plays a communicative role at the crossroads between metabolism and immunity or cancer. An increasing number of publications have addressed the roles of PPARγ in various settings. However, it is clear that several issues, as noted below, remain to be addressed.
As described above, TZDs are the principal class of drugs available for the improvement of insulin sensitivity in obese diabetic subjects. However, from a therapeutic standpoint of diabetes, treatment with TZD is hampered by many side effects. Thus, the limitation of TZDs requires other therapeutic approaches. To develop alternatives, identification of novel chemicals followed by structural optimization and assignments of new molecular targets need to be performed. New pharmacological targets can be elucidated by dissecting the molecular mechanisms of bioactive small molecules. Studies on resveratrol, an anti-obese and anti-oxidant bioactive compound, have identified the novel targets Sirtuin 1, AMP-activated protein kinase, and phosphodiesterase 4 (Picard et al. 2004; Park et al. 2012). The anti-diabetic small molecule harmine has led to the identification of Id2 as a new factor in adipogenesis (Park et al. 2008b). Similarly, phytochemicals from various herbal products can also be used for the elucidation of new metabolic players (Wang et al. 2014b). Therefore, such studies have revealed potential chemicals for the treatment of insulin resistance, have expanded our knowledge of certain unidentified pathways in biology, and have offered new molecular targets for insulin resistance. Based on these studies, efforts to screen for novel chemical regulators, along with the elucidation of their molecular pathways, should be investigated more extensively in the future.
PPARγ agonists exert anti-inflammatory effects in macrophages. Transrepression is primarily responsible for the anti-inflammatory actions of PPARγ activation. However, the detailed mechanistic explanation regarding how PPARγ mediates anti-inflammatory effects in macrophages remains elusive. Transrepression, as well as the related PPARγ-dependent and -independent effects, in innate immune systems need to be further clarified.
Given the complexity of adaptive immunity, it is not surprising that activation of PPARγ in subsets of T cells and B cells produces inconsistent results in proliferation and activation. Pharmacological interventions targeted at a single cell type or molecule are thus prone to be ineffective for autoimmune diseases. Regarding the alteration of immune responses toward the genesis of a specific subset of immune cells such as Treg or Th17 through the modulation of metabolic sensors, PPARγ can be an alternative or can be used in new combinatory strategies.
Concerning cancer therapy, using of PPARγ has numerous merits. However, some reports have suggested that PPARγ or its agonists could accelerate tumorigenesis in colon and bladder cancer. For example, in APCmin/+ mice, troglitazone-mediated PPARγ activation could increase polyp formation in colon cancer (Saez et al. 1998). Additionally, rosiglitazone displayed more eruption of urinary bladder cancer induced by OH-BBN (Lubet et al. 2008). Based on these observations, the roles of PPARγ in various physiological contexts should be pursued extensively. One other problem in cancer therapy targeting PPARγ is the side effects caused by its ligands such as heart failure, bone fractures, and obesity (Erdmann et al. 2009; Govindarajan et al. 2007). To circumvent these problems, putative therapeutic drugs with fewer side effects are actively being investigated and pursued worldwide. Several recent reports have highlighted the new development of non-TZD chemicals and natural extracts developed for cancer therapy.
In conclusion, it is obvious that the metabolic sensor PPARγ interprets information from various cellular environments of available growth factors, lipid metabolites, and synthetic ligands to control metabolic gene expression in metabolic, immune, and cancer cells. Figure 1 summarizes the functions of PPARγ and further delineates PPARγ as a key integrator of cellular signals in various tissues. Due to the multifaceted roles of PPARγ, complicated and inconsistent biological actions of PPARγ in different settings are somewhat expected. Once we clearly understand the cellular and molecular complexity of PPARγ, the context-dependent pleiotropic roles of PPARγ will be better resolved. Subsequently, more defined strategies targeting PPARγ in clinical trials that explore possible treatments using PPARγ for various associated human diseases will be proposed in the future.
References
Appel, S., V. Mirakaj, A. Bringmann, M.M. Weck, F. Grunebach, and P. Brossart. 2005. PPAR-gamma agonists inhibit toll-like receptor-mediated activation of dendritic cells via the MAP kinase and NF-kappaB pathways. Blood 106(12): 3888–3894. doi:10.1182/blood-2004-12-4709.
Bae, M.A., and B.J. Song. 2003. Critical role of c-Jun N-terminal protein kinase activation in troglitazone-induced apoptosis of human HepG2 hepatoma cells. Molecular Pharmacology 63(2): 401–408.
Balint, B.L., and L. Nagy. 2006. Selective modulators of PPAR activity as new therapeutic tools in metabolic diseases. Endocrine, Metabolic & Immune Disorders: Drug Targets 6(1): 33–43.
Barak, Y., M.C. Nelson, E.S. Ong, Y.Z. Jones, P. Ruiz-Lozano, K.R. Chien, A. Koder, and R.M. Evans. 1999. PPAR gamma is required for placental, cardiac, and adipose tissue development. Molecular Cell 4(4): 585–595. doi:10.1016/S1097-2765(00)80209-9.
Bartelt, A., and J. Heeren. 2014. Adipose tissue browning and metabolic health. Nature Reviews Endocrinology 10(1): 24–36. doi:10.1038/nrendo.2013.204.
Battaglia, S., O. Maguire, J.L. Thorne, L.B. Hornung, C.L. Doig, S. Liu, L.E. Sucheston, et al. 2010. Elevated NCOR1 disrupts PPARalpha/gamma signaling in prostate cancer and forms a targetable epigenetic lesion. Carcinogenesis 31(9): 1650–1660. doi:10.1093/carcin/bgq086.
Bonofiglio, D., S. Aquila, S. Catalano, S. Gabriele, M. Belmonte, E. Middea, H. Qi, et al. 2006. Peroxisome proliferator-activated receptor-gamma activates p53 gene promoter binding to the nuclear factor-kappaB sequence in human MCF7 breast cancer cells. Molecular Endocrinology 20(12): 3083–3092. doi:10.1210/me.2006-0192.
Bren-Mattison, Y., A.M. Meyer, V. Van Putten, H. Li, K. Kuhn, R. Stearman, M. Weiser-Evans, R.A. Winn, L.E. Heasley, and R.A. Nemenoff. 2008. Antitumorigenic effects of peroxisome proliferator-activated receptor-gamma in non-small-cell lung cancer cells are mediated by suppression of cyclooxygenase-2 via inhibition of nuclear factor-kappaB. Molecular Pharmacology 73(3): 709–717. doi:10.1124/mol.107.042002.
Campbell, M.J., C. Carlberg, and H.P. Koeffler. 2008. A role for the PPARgamma in cancer therapy. PPAR Research 2008: 314974. doi:10.1155/2008/314974.
Cesario, R.M., J. Stone, W.C. Yen, R.P. Bissonnette, and W.W. Lamph. 2006. Differentiation and growth inhibition mediated via the RXR:PPARgamma heterodimer in colon cancer. Cancer Letters 240(2): 225–233. doi:10.1016/j.canlet.2005.09.010.
Chawla, A., W.A. Boisvert, C.H. Lee, B.A. Laffitte, Y. Barak, S.B. Joseph, D. Liao, et al. 2001. A PPAR gamma-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Molecular Cell 7(1): 161–171.
Chen, F., and L.E. Harrison. 2005. Ciglitazone-induced cellular anti-proliferation increases p27kip1 protein levels through both increased transcriptional activity and inhibition of proteasome degradation. Cellular Signalling 17(7): 809–816. doi:10.1016/j.cellsig.2004.11.002.
Choi, J.H., A.S. Banks, J.L. Estall, S. Kajimura, P. Bostrom, D. Laznik, J.L. Ruas, et al. 2010. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature 466(7305): 451–456. doi:10.1038/nature09291.
Choi, J.H., A.S. Banks, T.M. Kamenecka, S.A. Busby, M.J. Chalmers, N. Kumar, D.S. Kuruvilla, et al. 2011. Antidiabetic actions of a non-agonist PPARgamma ligand blocking Cdk5-mediated phosphorylation. Nature 477(7365): 477–481. doi:10.1038/nature10383.
Cristancho, A.G., and M.A. Lazar. 2011. Forming functional fat: A growing understanding of adipocyte differentiation. Nature Reviews Molecular Cell Biology 12(11): 722–734. doi:10.1038/nrm3198.
da Rocha Junior, L.F., A.T. Dantas, A.L. Duarte, M.J. de Melo Rego, R. Pitta Ida, and M.G. Pitta. 2013. PPARgamma agonists in adaptive immunity: What do immune disorders and their models have to tell us? PPAR Research 2013: 519724. doi:10.1155/2013/519724.
DePaoli, A.M., L.S. Higgins, R.R. Henry, C. Mantzoros, and F.L. Dunn. 2014. Can a selective PPARgamma modulator improve glycemic control in patients with type 2 diabetes with fewer side effects compared with pioglitazone? Diabetes Care 37(7): 1918–1923. doi:10.2337/dc13-2480.
Duan, S.Z., C.Y. Ivashchenko, S.E. Whitesall, L.G. D’Alecy, D.C. Duquaine, F.C. Brosius, F.J. Gonzalez, et al. 2007. Hypotension, lipodystrophy, and insulin resistance in generalized PPAR gamma-deficient mice rescued from embryonic lethality. Journal of Clinical Investigation 117(3): 812–822. doi:10.1172/Jci28859.
Erdmann, E., B. Charbonnel, and R. Wilcox. 2009. Thiazolidinediones and cardiovascular risk—a question of balance. Current Cardiology Reviews 5(3): 155–165. doi:10.2174/157340309788970333.
Farrow, B., and B.M. Evers. 2003. Activation of PPARgamma increases PTEN expression in pancreatic cancer cells. Biochem Biophys Res Commun 301(1): 50–53.
Feldman, P.L., M.H. Lambert, and B.R. Henke. 2008. PPAR modulators and PPAR pan agonists for metabolic diseases: The next generation of drugs targeting peroxisome proliferator-activated receptors? Current Topics in Medicinal Chemistry 8(9): 728–749.
Ferrante Jr, A.W. 2013. Macrophages, fat, and the emergence of immunometabolism. The Journal of Clinical Investigation 123(12): 4992–4993. doi:10.1172/JCI73658.
Fisher, B.L., and P. Schauer. 2002. Medical and surgical options in the treatment of severe obesity. American Journal of Surgery 184(6B): 9S–16S.
Floyd, Z.E., and J.M. Stephens. 2004. Control of peroxisome proliferator-activated receptor gamma2 stability and activity by SUMOylation. Obesity Research 12(6): 921–928. doi:10.1038/oby.2004.112.
Fredenrich, A., and P.A. Grimaldi. 2005. PPAR delta: An uncompletely known nuclear receptor. Diabetes & Metabolism 31(1): 23–27.
Fujita, M., T. Yagami, M. Fujio, C. Tohji, K. Takase, Y. Yamamoto, K. Sawada, M. Yamamori, and N. Okamura. 2011. Cytotoxicity of troglitazone through PPARgamma-independent pathway and p38 MAPK pathway in renal cell carcinoma. Cancer Letters 312(2): 219–227. doi:10.1016/j.canlet.2011.08.010.
Gavrilova, O., M. Haluzik, K. Matsusue, J.J. Cutson, L. Johnson, K.R. Dietz, C.J. Nicol, C. Vinson, F.J. Gonzalez, and M.L. Reitman. 2003. Liver peroxisome proliferator-activated receptor gamma contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass. Journal of Biological Chemistry 278(36): 34268–34276. doi:10.1074/jbc.M300043200M300043200.
Govindarajan, R., L. Ratnasinghe, D.L. Simmons, E.R. Siegel, M.V. Midathada, L. Kim, P.J. Kim, R.J. Owens, and N.P. Lang. 2007. Thiazolidinediones and the risk of lung, prostate, and colon cancer in patients with diabetes. Journal of Clinical Oncology 25(12): 1476–1481. doi:10.1200/JCO.2006.07.2777.
Graham, D.J., R. Ouellet-Hellstrom, T.E. MaCurdy, F. Ali, C. Sholley, C. Worrall, and J.A. Kelman. 2010. Risk of acute myocardial infarction, stroke, heart failure, and death in elderly medicare patients treated with rosiglitazone or pioglitazone. JAMA 304(4): 411–418. doi:10.1001/jama.2010.920.
Guri, A.J., S.K. Mohapatra, W.T. Horne 2nd, R. Hontecillas, and J. Bassaganya-Riera. 2010. The role of T cell PPAR gamma in mice with experimental inflammatory bowel disease. BMC Gastroenterol 10: 60. doi:10.1186/1471-230X-10-60.
Hammad, H., H.J. de Heer, T. Soullie, V. Angeli, F. Trottein, H.C. Hoogsteden, and B.N. Lambrecht. 2004. Activation of peroxisome proliferator-activated receptor-gamma in dendritic cells inhibits the development of eosinophilic airway inflammation in a mouse model of asthma. American Journal of Pathology 164(1): 263–271.
Hauser, S., G. Adelmant, P. Sarraf, H.M. Wright, E. Mueller, and B.M. Spiegelman. 2000. Degradation of the peroxisome proliferator-activated receptor gamma is linked to ligand-dependent activation. Journal of Biological Chemistry 275(24): 18527–18533. doi:10.1074/jbc.M001297200M001297200.
He, W., Y. Barak, A. Hevener, P. Olson, D. Liao, J. Le, M. Nelson, E. Ong, J.M. Olefsky, and R.M. Evans. 2003. Adipose-specific peroxisome proliferator-activated receptor gamma knockout causes insulin resistance in fat and liver but not in muscle. Proceedings of the National Academy of Sciences USA 100(26): 15712–15717. doi:10.1073/pnas.2536828100.
Hevener, A.L., W.M. He, Y. Barak, J. Le, G. Bandyopadhyay, P. Olson, J. Wilkes, R.M. Evans, and J. Olefsky. 2003. Muscle-specific Pparg deletion causes insulin resistance. Nature Medicine 9(12): 1491–1497. doi:10.1038/Nm956.
Hill, J.O., H.R. Wyatt, G.W. Reed, and J.C. Peters. 2003. Obesity and the environment: where do we go from here? Science 299(5608): 853–855. doi:10.1126/science.1079857299/5608/853.
Hong, J.W., and K.W. Park. 2010. Further understanding of fat biology: lessons from a fat fly. Experimental & Molecular Medicine 42(1): 12–20. doi:10.3858/emm.2010.42.1.007.
Hontecillas, R., and J. Bassaganya-Riera. 2007. Peroxisome proliferator-activated receptor gamma is required for regulatory CD4+ T cell-mediated protection against colitis. The Journal of Immunology 178(5): 2940–2949.
Hwa, J.S., L. Mun, H.J. Kim, H.G. Seo, J.H. Lee, J.H. Kwak, D.U. Lee, and K.C. Chang. 2011. Genipin selectively inhibits TNF-alpha-activated VCAM-1 but not ICAM-1 expression by upregulation of PPAR-gamma in human endothelial cells. Korean Journal of Physiology and Pharmacology 15(3): 157–162. doi:10.4196/kjpp.2011.15.3.157.
Jeong, Y., Y. Xie, W. Lee, A.L. Bookout, L. Girard, G. Raso, C. Behrens, et al. 2012. Research resource: Diagnostic and therapeutic potential of nuclear receptor expression in lung cancer. Molecular Endocrinology 26(8): 1443–1454. doi:10.1210/me.2011-1382.
Jones, J.R., C. Barrick, K.A. Kim, J. Lindner, B. Blondeau, Y. Fujimoto, M. Shiota, R.A. Kesterson, B.B. Kahn, and M.A. Magnuson. 2005. Deletion of PPARgamma in adipose tissues of mice protects against high fat diet-induced obesity and insulin resistance. The Proceedings of the National Academy USA 102(17): 6207–6212. doi:10.1073/pnas.0306743102.
Kebebew, E., Peng, M., Reiff, E., Treseler, P., Woeber, K., Clark, O.H., Greenspan, F.S., Lindsay, S., Duh, Q.Y., and Morita, E. 2006. A phase II trial of rosiglitazone in patients with thyroglobulin-positive and radioiodine-negative differentiated thyroid cancer. Surgery 140 (6):960–966. Discussion 966–967. doi:10.1016/j.surg.2006.07.038.
Kidani, Y., and S.J. Bensinger. 2012. Liver X receptor and peroxisome proliferator-activated receptor as integrators of lipid homeostasis and immunity. Immunological Reviews 249(1): 72–83. doi:10.1111/j.1600-065X.2012.01153.x.
Kilroy, G.E., X. Zhang, and Z.E. Floyd. 2009. PPAR-gamma AF-2 domain functions as a component of a ubiquitin-dependent degradation signal. Obesity (Silver Spring) 17(4): 665–673. doi:10.1038/oby.2008.616.
Kilroy, G., H. Kirk-Ballard, L.E. Carter, and Z.E. Floyd. 2012. The ubiquitin ligase Siah2 regulates PPARgamma activity in adipocytes. Endocrinology 153(3): 1206–1218. doi:10.1210/en.2011-1725.
Kim, J.H., K.W. Park, E.W. Lee, W.S. Jang, J. Seo, S. Shin, K.A. Hwang, and J. Song. 2014. Suppression of PPARgamma through MKRN1-mediated ubiquitination and degradation prevents adipocyte differentiation. Cell Death and Differentiation 21(4): 594–603. doi:10.1038/cdd.2013.181.
Kim, K.Y., S.S. Kim, and H.G. Cheon. 2006. Differential anti-proliferative actions of peroxisome proliferator-activated receptor-gamma agonists in MCF-7 breast cancer cells. Biochemical Pharmacology 72(5): 530–540. doi:10.1016/j.bcp.2006.05.009.
Kim, Y., N. Suh, M. Sporn, and J.C. Reed. 2002. An inducible pathway for degradation of FLIP protein sensitizes tumor cells to TRAIL-induced apoptosis. Journal of Biological Chemistry 277(25): 22320–22329. doi:10.1074/jbc.M202458200.
Kiss, M., Z. Czimmerer, and L. Nagy. 2013. The role of lipid-activated nuclear receptors in shaping macrophage and dendritic cell function: From physiology to pathology. The Journal of Allergy and Clinical Immunology 132(2): 264–286. doi:10.1016/j.jaci.2013.05.044.
Klotz, L., S. Burgdorf, I. Dani, K. Saijo, J. Flossdorf, S. Hucke, J. Alferink, et al. 2009. The nuclear receptor PPAR gamma selectively inhibits Th17 differentiation in a T cell-intrinsic fashion and suppresses CNS autoimmunity. Journal of Experimental Medicine 206(10): 2079–2089. doi:10.1084/jem.20082771.
Koeffler, H.P. 2003. Peroxisome proliferator-activated receptor gamma and cancers. Clinical Cancer Research 9(1): 1–9.
Kopelman, P.G. 2000. Obesity as a medical problem. Nature 404(6778): 635–643. doi:10.1038/35007508.
Lehmann, J.M., L.B. Moore, T.A. Smith-Oliver, W.O. Wilkison, T.M. Willson, and S.A. Kliewer. 1995. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma). Journal of Biological Chemistry 270(22): 12953–12956.
Liu, Y., Z.A. Zhu, S.N. Zhang, J. Mou, L. Liu, T. Cui, and D.S. Pei. 2013. Combinational effect of PPARgamma agonist and RXR agonist on the growth of SGC7901 gastric carcinoma cells in vitro. Tumour Biology 34(4): 2409–2418. doi:10.1007/s13277-013-0791-2.
Lubet, R.A., S.M. Fischer, V.E. Steele, M.M. Juliana, R. Desmond, and C.J. Grubbs. 2008. Rosiglitazone, a PPAR gamma agonist: potent promoter of hydroxybutyl(butyl)nitrosamine-induced urinary bladder cancers. International Journal of Cancer 123(10): 2254–2259. doi:10.1002/ijc.23765.
Manolagas, S.C. 1998. Cellular and molecular mechanisms of osteoporosis. Aging (Milano) 10(3): 182–190.
Mansure, J.J., R. Nassim, S. Chevalier, K. Szymanski, J. Rocha, S. Aldousari, and W. Kassouf. 2013. A novel mechanism of PPAR gamma induction via EGFR signalling constitutes rational for combination therapy in bladder cancer. PLoS ONE 8(2): e55997. doi:10.1371/journal.pone.0055997.
Matsusue, K., M. Haluzik, G. Lambert, S.H. Yim, O. Gavrilova, J.M. Ward, B. Brewer Jr, M.L. Reitman, and F.J. Gonzalez. 2003. Liver-specific disruption of PPARgamma in leptin-deficient mice improves fatty liver but aggravates diabetic phenotypes. Journal of Clinical Investigation 111(5): 737–747. doi:10.1172/JCI17223.
McAlpine, C.A., Y. Barak, I. Matise, and R.T. Cormier. 2006. Intestinal-specific PPARgamma deficiency enhances tumorigenesis in ApcMin/+ mice. International Journal of Cancer 119(10): 2339–2346. doi:10.1002/ijc.22115.
McKinnon, B., N.A. Bersinger, and M.D. Mueller. 2012. Peroxisome proliferating activating receptor gamma-independent attenuation of interleukin 6 and interleukin 8 secretion from primary endometrial stromal cells by thiazolidinediones. Fertility and Sterility 97(3): 657–664. doi:10.1016/j.fertnstert.2011.12.001.
Moerman, E.J., K. Teng, D.A. Lipschitz, and B. Lecka-Czernik. 2004. Aging activates adipogenic and suppresses osteogenic programs in mesenchymal marrow stroma/stem cells: the role of PPAR-gamma2 transcription factor and TGF-beta/BMP signaling pathways. Aging Cell 3(6): 379–389. doi:10.1111/j.1474-9728.2004.00127.x.
Motomura, W., T. Okumura, N. Takahashi, T. Obara, and Y. Kohgo. 2000. Activation of peroxisome proliferator-activated receptor gamma by troglitazone inhibits cell growth through the increase of p27KiP1 in human pancreatic carcinoma cells. Cancer Research 60(19): 5558–5564.
Mueller, E., M. Smith, P. Sarraf, T. Kroll, A. Aiyer, D.S. Kaufman, W. Oh, et al. 2000. Effects of ligand activation of peroxisome proliferator-activated receptor gamma in human prostate cancer. The Proceedings of the National Academy USA 97(20): 10990–10995. doi:10.1073/pnas.180329197.
Nagy, L., P. Tontonoz, J.G. Alvarez, H. Chen, and R.M. Evans. 1998. Oxidized LDL regulates macrophage gene expression through ligand activation of PPARgamma. Cell 93(2): 229–240.
Nencioni, A., F. Grunebach, A. Zobywlaski, C. Denzlinger, W. Brugger, and P. Brossart. 2002. Dendritic cell immunogenicity is regulated by peroxisome proliferator-activated receptor gamma. Journal of Immunology 169(3): 1228–1235.
Nesto, R.W., D. Bell, R.O. Bonow, V. Fonseca, S.M. Grundy, E.S. Horton, M. Le Winter, et al. 2004. Thiazolidinedione use, fluid retention, and congestive heart failure: a consensus statement from the American Heart Association and American Diabetes Association. Diabetes Care 27(1): 256–263.
Nguyen, M.T., A. Chen, W.J. Lu, W. Fan, P.P. Li, D.Y. Oh, and D. Patsouris. 2012. Regulation of chemokine and chemokine receptor expression by PPARgamma in adipocytes and macrophages. PLoS ONE 7(4): e34976. doi:10.1371/journal.pone.0034976.
Nissen, S.E., and K. Wolski. 2007. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. New England Journal of Medicine 356(24): 2457–2471. doi:10.1056/NEJMoa072761.
Norris, A.W., L. Chen, S.J. Fisher, I. Szanto, M. Ristow, A.C. Jozsi, M.F. Hirshman, et al. 2003. Muscle-specific PPARgamma-deficient mice develop increased adiposity and insulin resistance but respond to thiazolidinediones. Journal of Clinical Investigation 112(4): 608–618. doi:10.1172/JCI17305.
Odegaard, J.I., and A. Chawla. 2011. Alternative macrophage activation and metabolism. Annual Review of Pathology: Mechanisms of Disease 6: 275–297. doi:10.1146/annurev-pathol-011110-130138.
Odegaard, J.I., R.R. Ricardo-Gonzalez, M.H. Goforth, C.R. Morel, V. Subramanian, L. Mukundan, A. Red Eagle, et al. 2007. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature 447(7148): 1116–1120. doi:10.1038/nature05894.
Ogino, S., K. Shima, Y. Baba, K. Nosho, N. Irahara, S. Kure, L. Chen, et al. 2009. Colorectal cancer expression of peroxisome proliferator-activated receptor gamma (PPARG, PPARgamma) is associated with good prognosis. Gastroenterology 136(4): 1242–1250. doi:10.1053/j.gastro.2008.12.048.
Park, K.W., D.S. Halperin, and P. Tontonoz. 2008a. Before they were fat: adipocyte progenitors. Cell Metabolism 8(6): 454–457. doi:10.1016/j.cmet.2008.11.001.
Park, K.W., H. Waki, S.P. Choi, K.M. Park, and P. Tontonoz. 2010. The small molecule phenamil is a modulator of adipocyte differentiation and PPARgamma expression. Journal of Lipid Research 51(9): 2775–2784. doi:10.1194/jlr.M008490.
Park, K.W., H. Waki, C.J. Villanueva, L.A. Monticelli, C. Hong, S. Kang, O.A. MacDougald, A.W. Goldrath, and P. Tontonoz. 2008b. Inhibitor of DNA binding 2 is a small molecule-inducible modulator of peroxisome proliferator-activated receptor-gamma expression and adipocyte differentiation. Molecular Endocrinology 22(9): 2038–2048. doi:10.1210/me.2007-0454.
Park, S.J., F. Ahmad, A. Philp, K. Baar, T. Williams, H. Luo, H. Ke, et al. 2012. Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell 148(3): 421–433. doi:10.1016/j.cell.2012.01.017.
Pascual, G., A.L. Fong, S. Ogawa, A. Gamliel, A.C. Li, V. Perissi, D.W. Rose, T.M. Willson, M.G. Rosenfeld, and C.K. Glass. 2005. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature 437(7059): 759–763. doi:10.1038/nature03988.
Peirce, V., S. Carobbio, and A. Vidal-Puig. 2014. The different shades of fat. Nature 510(7503): 76–83. doi:10.1038/nature13477.
Pi-Sunyer, X. 2003. A clinical view of the obesity problem. Science 299(5608): 859–860. doi:10.1126/science.1082319299/5608/859.
Picard, F., M. Kurtev, N. Chung, A. Topark-Ngarm, T. Senawong, R. Machado De Oliveira, M. Leid, M.W. McBurney, and L. Guarente. 2004. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature 429(6993): 771–776. doi:10.1038/nature02583nature02583.
Qiu, Y., K.D. Nguyen, J.I. Odegaard, X. Cui, X. Tian, R.M. Locksley, R.D. Palmiter, and A. Chawla. 2014. Eosinophils and type 2 cytokine signaling in macrophages orchestrate development of functional beige fat. Cell 157(6): 1292–1308. doi:10.1016/j.cell.2014.03.066.
Quintanilla, R.A., E. Utreras, and F.A. Cabezas-Opazo. 2014. Role of PPAR gamma in the differentiation and function of neurons. PPAR Research 2014: 768594. doi:10.1155/2014/768594.
Ramkalawan, H., Y.Z. Wang, A. Hurbungs, Y.F. Yang, F.F. Tian, W.B. Zhou, J. Li, H. Yang, B. Xiao, and W. Zhang. 2012. Pioglitazone, PPARgamma agonist, attenuates experimental autoimmune neuritis. Inflammation 35(4): 1338–1347. doi:10.1007/s10753-012-9447-4.
Ramos-Nino, M.E., C.D. MacLean, and B. Littenberg. 2007. Association between cancer prevalence and use of thiazolidinediones: results from the vermont diabetes information system. BMC Medicine 5: 17. doi:10.1186/1741-7015-5-17.
Ricote, M., A.C. Li, T.M. Willson, C.J. Kelly, and C.K. Glass. 1998. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 391(6662): 79–82. doi:10.1038/34178.
Saez, E., P. Tontonoz, M.C. Nelson, J.G. Alvarez, U.T. Ming, S.M. Baird, V.A. Thomazy, and R.M. Evans. 1998. Activators of the nuclear receptor PPARgamma enhance colon polyp formation. Nature Medicine 4(9): 1058–1061. doi:10.1038/2042.
Sharma, C., A. Pradeep, L. Wong, A. Rana, and B. Rana. 2004. Peroxisome proliferator-activated receptor gamma activation can regulate beta-catenin levels via a proteasome-mediated and adenomatous polyposis coli-independent pathway. Journal of Biological Chemistry 279(34): 35583–35594. doi:10.1074/jbc.M403143200.
Shiau, C.W., C.C. Yang, S.K. Kulp, K.F. Chen, C.S. Chen, J.W. Huang, and C.S. Chen. 2005. Thiazolidenediones mediate apoptosis in prostate cancer cells in part through inhibition of Bcl-xL/Bcl-2 functions independently of PPARgamma. Cancer Research 65(4): 1561–1569. doi:10.1158/0008-5472.CAN-04-1677.
Shoelson, S.E., J. Lee, and M. Yuan. 2003. Inflammation and the IKK beta/I kappa B/NF-kappa B axis in obesity- and diet-induced insulin resistance. International Journal of Obesity and Related Metabolic Disorders 27(Suppl 3): S49–S52. doi:10.1038/sj.ijo.08025010802501.
Sikka, S., L. Chen, G. Sethi, and A.P. Kumar. 2012. Targeting PPARgamma signaling cascade for the prevention and treatment of prostate cancer. PPAR Research 2012: 968040. doi:10.1155/2012/968040.
Sims, N.A., and J.H. Gooi. 2008. Bone remodeling: Multiple cellular interactions required for coupling of bone formation and resorption. Seminars in Cell & Developmental Biology 19(5): 444–451. doi:10.1016/j.semcdb.2008.07.016.
Skelhorne-Gross, G., and C.J. Nicol. 2012. The key to unlocking the chemotherapeutic potential of ppargamma ligands: Having the right combination. PPAR Research 2012: 946943. doi:10.1155/2012/946943.
Skelhorne-Gross, G., A.L. Reid, A.J. Apostoli, M.A. Di Lena, R.E. Rubino, N.T. Peterson, M. Schneider, S.K. SenGupta, F.J. Gonzalez, and C.J. Nicol. 2012. Stromal adipocyte PPARgamma protects against breast tumorigenesis. Carcinogenesis 33(7): 1412–1420. doi:10.1093/carcin/bgs173.
Straus, D.S., and C.K. Glass. 2007. Anti-inflammatory actions of PPAR ligands: New insights on cellular and molecular mechanisms. Trends in Immunology 28(12): 551–558. doi:10.1016/j.it.2007.09.003.
Szanto, A., and L. Nagy. 2008. The many faces of PPARgamma: Anti-inflammatory by any means? Immunobiology 213(9–10): 789–803. doi:10.1016/j.imbio.2008.07.015.
Szatmari, I., E. Rajnavolgyi, and L. Nagy. 2006. PPARgamma, a lipid-activated transcription factor as a regulator of dendritic cell function. Annals of the New York Academy of Sciences 1088: 207–218. doi:10.1196/annals.1366.013.
Tontonoz, P., L. Nagy, J.G. Alvarez, V.A. Thomazy, and R.M. Evans. 1998. PPARgamma promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell 93(2): 241–252.
Tontonoz, P., and B.M. Spiegelman. 2008. Fat and beyond: The diverse biology of PPARgamma. Annual Review of Biochemistry 77: 289–312. doi:10.1146/annurev.biochem.77.061307.091829.
Turner, N., G.J. Cooney, E.W. Kraegen, and C.R. Bruce. 2014. Fatty acid metabolism, energy expenditure and insulin resistance in muscle. Journal of Endocrinology 220(2): T61–T79. doi:10.1530/JOE-13-0397.
van Beekum, O., V. Fleskens, and E. Kalkhoven. 2009. Posttranslational modifications of PPAR-gamma: Fine-tuning the metabolic master regulator. Obesity (Silver Spring) 17(2): 213–219. doi:10.1038/oby.2008.473.
van Raalte, D.H., M. Li, P.H. Pritchard, and K.M. Wasan. 2004. Peroxisome proliferator-activated receptor (PPAR)-alpha: A pharmacological target with a promising future. Pharmaceutical Research 21(9): 1531–1538.
Vitale, G., S. Zappavigna, M. Marra, A. Dicitore, S. Meschini, M. Condello, G. Arancia, et al. 2012. The PPAR-gamma agonist troglitazone antagonizes survival pathways induced by STAT-3 in recombinant interferon-beta treated pancreatic cancer cells. Biotechnology Advances 30(1): 169–184. doi:10.1016/j.biotechadv.2011.08.001.
Waki, H., K.W. Park, N. Mitro, L. Pei, R. Damoiseaux, D.C. Wilpitz, K. Reue, E. Saez, and P. Tontonoz. 2007. The small molecule harmine is an antidiabetic cell-type-specific regulator of PPARgamma expression. Cell Metabolism 5(5): 357–370. doi:10.1016/j.cmet.2007.03.010.
Waki, H., and P. Tontonoz. 2007. Endocrine functions of adipose tissue. Annual Review of Pathology: Mechanisms of Disease 2: 31–56. doi:10.1146/annurev.pathol.2.010506.091859.
Wang, C.Z., Y. Zhang, X.D. Li, Y. Hu, Z.G. Fang, D.J. Lin, R.Z. Xiao, et al. 2011. PPARgamma agonist suppresses TLR4 expression and TNF-alpha production in LPS stimulated monocyte leukemia cells. Cell Biochemistry and Biophysics 60(3): 167–172. doi:10.1007/s12013-010-9136-6.
Wang, F., S.E. Mullican, J.R. DiSpirito, L.C. Peed, and M.A. Lazar. 2013. Lipoatrophy and severe metabolic disturbance in mice with fat-specific deletion of PPARgamma. The Proceedings of the National Academy USA 110(46): 18656–18661. doi:10.1073/pnas.1314863110.
Wang, L., B. Waltenberger, E.M. Pferschy-Wenzig, M. Blunder, X. Liu, C. Malainer, T. Blazevic, et al. 2014a. Natural product agonists of peroxisome proliferator-activated receptor gamma (PPARgamma): A review. Biochemical Pharmacology. doi:10.1016/j.bcp.2014.07.018.
Wang, S., N. Moustaid-Moussa, L. Chen, H. Mo, A. Shastri, R. Su, P. Bapat, I. Kwun, and C.L. Shen. 2014b. Novel insights of dietary polyphenols and obesity. Journal of Nutritional Biochemistry 25(1): 1–18. doi:10.1016/j.jnutbio.2013.09.001.
Wei, S., L.F. Lin, C.C. Yang, Y.C. Wang, G.D. Chang, H. Chen, and C.S. Chen. 2007. Thiazolidinediones modulate the expression of beta-catenin and other cell-cycle regulatory proteins by targeting the F-box proteins of Skp1-Cul1-F-box protein E3 ubiquitin ligase independently of peroxisome proliferator-activated receptor gamma. Molecular Pharmacology 72(3): 725–733. doi:10.1124/mol.107.035287.
Weisberg, S.P., D. McCann, M. Desai, M. Rosenbaum, R.L. Leibel, and A.W. Ferrante Jr. 2003. Obesity is associated with macrophage accumulation in adipose tissue. The Journal of Clinical Investigation 112(12): 1796–1808. doi:10.1172/JCI19246112/12/1796.
Wernstedt Asterholm, I., C. Tao, T.S. Morley, Q.A. Wang, F. Delgado-Lopez, Z.V. Wang, and P.E. Scherer. 2014. Adipocyte inflammation is essential for healthy adipose tissue expansion and remodeling. Cell Metabolism 20(1): 103–118. doi:10.1016/j.cmet.2014.05.005.
Xu, H., G.T. Barnes, Q. Yang, G. Tan, D. Yang, C.J. Chou, J. Sole, et al. 2003. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. The Journal of Clinical Investigation 112(12): 1821–1830. doi:10.1172/JCI19451112/12/1821.
Yach, D., D. Stuckler, and K.D. Brownell. 2006. Epidemiologic and economic consequences of the global epidemics of obesity and diabetes. Nature Medicine 12(1): 62–66. doi:10.1038/nm0106-62.
Yan, K.H., C.J. Yao, H.Y. Chang, G.M. Lai, A.L. Cheng, and S.E. Chuang. 2010. The synergistic anticancer effect of troglitazone combined with aspirin causes cell cycle arrest and apoptosis in human lung cancer cells. Molecular Carcinogenesis 49(3): 235–246. doi:10.1002/mc.20593.
Yin, F., S. Wakino, Z. Liu, S. Kim, W.A. Hsueh, A.R. Collins, A.J. Van Herle, and R.E. Law. 2001. Troglitazone inhibits growth of MCF-7 breast carcinoma cells by targeting G1 cell cycle regulators. Biochemical and Biophysical Research Communications 286(5): 916–922. doi:10.1006/bbrc.2001.5491.
Yu, S., K. Matsusue, P. Kashireddy, W.Q. Cao, V. Yeldandi, A.V. Yeldandi, M.S. Rao, F.J. Gonzalez, and J.K. Reddy. 2003. Adipocyte-specific gene expression and adipogenic steatosis in the mouse liver due to peroxisome proliferator-activated receptor gamma1 (PPARgamma1) overexpression. Journal of Biological Chemistry 278(1): 498–505. doi:10.1074/jbc.M210062200M210062200.
Zander, T., J.A. Kraus, C. Grommes, U. Schlegel, D. Feinstein, T. Klockgether, G. Landreth, J. Koenigsknecht, and M.T. Heneka. 2002. Induction of apoptosis in human and rat glioma by agonists of the nuclear receptor PPARgamma. Journal of Neurochemistry 81(5): 1052–1060.
Acknowledgments
This study was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science, and Technology (NRF-2013R1A1A2060447), by the National Research Foundation of Korea (NRF) funded by the Korean government (MEST) (2010-0017787), by the National Cancer Center, Korea (NCC-1420300), and by the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI) funded by the Ministry of Health & Welfare of the Republic of Korea (HI12C1280).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Kim, JH., Song, J. & Park, K.W. The multifaceted factor peroxisome proliferator-activated receptor γ (PPARγ) in metabolism, immunity, and cancer. Arch. Pharm. Res. 38, 302–312 (2015). https://doi.org/10.1007/s12272-015-0559-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-015-0559-x