
Abstract
Infertility is a relatively common health condition, affecting nearly 7 % of all couples. Clinically, it is a highly heterogeneous pathology with a complex etiology that includes environmental and genetic factors. It has been estimated that nearly 50 % of infertility cases are due to genetic defects. Hundreds of studies with animal knockout models convincingly showed infertility to be caused by gene defects, single or multiple. However, despite enormous efforts, progress in translating basic research findings into clinical studies has been challenging. The genetic causes remain unexplained for the vast majority of male or female infertility patients. A particular difficulty is the huge number of candidate genes to be studied; there are more than 2,300 genes expressed in the testis alone, and hundreds of those genes influence reproductive function in humans and could contribute to male infertility. At present, there are only a handful of genes or genetic defects that have been shown to cause, or to be strongly associated with, primary infertility. Yet, with completion of the human genome and progress in personalized medicine, the situation is rapidly changing. Indeed, there are 10–15 new gene tests, on average, being added to the clinical genetic testing list annually.
Similar content being viewed by others

Introduction
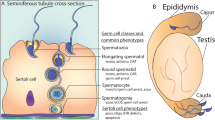
Infertility is a highly complex disorder of the reproductive system. There are two forms of male or female sterility: primary and secondary. The primary form affects germ cell structure or physiology, causing arrest of germ cell development and ultimately cell death. Primary female infertility includes premature ovarian failure (POF), polycystic ovary syndrome (PCOS), endometriosis, and leiomyoma. Primary male sterility disrupts spermatogenesis and is associated with abnormal semen (i.e., abnormal sperm count, morphology, or motility), but often the semen is normal (idiopathic infertility).
Secondary infertility arises because of systemic or syndromic genetic defects, including developmental, endocrine, and metabolic defects. Genetic syndromes that manifest male or female infertility are fragile X syndrome, Kartagener’s syndrome, myotonic dystrophy, Noonan syndrome, Fanconi anemia, sickle cell anemia, β-thalassemia, etc. Other notable conditions include disorders of sex development (DAX1, CBX2, SRY, SOX9, RSPO1) [1–6], reproductive dysgenesis disorders (AMH, AMHR2, ARX, DHH, NR5A1, WNT4, WT1) [6], hypogonadotrophic hypogonadism and Kallmann syndrome (KAL1, FGFR1, PROKR2, GNRH1, TAC3, LEP, NSMF, CHD7, DAX1, KISS1) [7], ambiguous genitalia and androgen insensitivity (AR) [8], and congenital bilateral absence of the vas deferens (CFTR); (Tables 1, 2) [9, 10].
Endocrine defects comprise disruption of steroid synthesis and metabolism, and are caused by CYP17, CYP21, and CYP21A2 mutations [6, 11, 12]. Also, various metabolic defects (e.g., galactosemia) [13] and mutations in mitochondrial energy pathways (POLG1 and mitochondrial DNA genes) cause toxic effects and lead to secondary female or male infertility [14]. All genetic defects can be divided into the following categories: chromosome aberrations, DNA copy number variants (micro deletions and duplications), single-gene disorders, complex conditions, and epigenetic disorders.
Chromosomal Defects in Male Infertility
Constitutional chromosome aberrations are the most frequent cause of male infertility, detected in up to 20 % of infertile men with semen defects; i.e., azoospermia and oligozoospermia [6, 11, 12, 15]. The aberrations include numerical defects, such as the XXY karyotype in Klinefelter syndrome or its variants and structural rearrangements, Robertsonian translocations, balanced reciprocal translocations, and inversions. Rarely, infertile men with normal karyotypes have chromosome aberrations in sperm [16]. Increased germ cell defects have been reported for chromosomes 21, 22, X, and Y [15, 16].
Klinefelter Syndrome
Klinefelter syndrome (KS, karyotype 47,XXY) is the most common chromosomal aberration, detected in up to 14 % of infertile patients with azoospermia [17]. Klinefelter patients manifest language delay and learning and behavioral problems [18]. Their testis histology shows germ cell degeneration, while serum levels of hormones are abnormal, with a decline in testosterone level and elevated follicle-stimulating hormone (FSH) and luteinizing hormone (LH) [12, 17, 18]. The 47,XXY karyotype accounts for nearly 90 % of the patients, while other variants are rare [17]. Usually, the extra X is a result of chromosome nondisjunction in male or female meiosis [18]. Nearly 10 % of KS patients are mosaic 47,XXY/46,XY. Although the sperm of Klinefelter men usually have a normal 23,X or 23,Y haploid genome, an increased rate of autosomal and sex chromosome aneuploidies was reported in KS men’s offspring [19].
The 47,XYY Syndrome
This syndrome occurs in 1:1,000 men, but is more common among infertile males [15, 17]. Infertile men with the 47,XYY karyotype are otherwise healthy. While semen analyses of 47,XYY males frequently indicate oligozoospermia or azoospermia, the majority of them are fertile, with normal semen parameters [16]. It has been shown that germ cells with an extra Y chromosome from men with the 47,XYY karyotype have abnormal meiotic pairing, suggesting disrupted meiosis, eventual sperm apoptosis, and subsequent oligozoospermia and infertility [15, 16].
Structural Chromosomal Abnormalities (SCAs)
SCAs include deletions, duplications, translocations (balanced, imbalanced, and Robertsonian), and inversions. Overall, SCAs occur in nearly 5 % of infertile men (0.5 % in the general population) [11, 12, 17]. Most frequently, SCAs are found in patients with azoospermia and oligozoospermia. Interestingly, while autosomal defects (3 %) are more common in oligozoospermia, structural aberrations involving sex chromosomes are associated with azoospermia (12.6 %) [20, 21]. There are two alternative models that explain the aberration effect: (1) it blocks spermatogenesis via abnormal chromosome synapsis in crossover and meiosis arrest; (2) the aberration disrupts a dosage-sensitive gene, resulting in spermatogenesis arrest and infertility [15, 16, 20, 21].
Y Chromosome Microdeletions
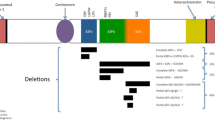
There are three frequent microdeletions of the azoospermia factor (AZF) region. The AZF deletions are detected in up to 15 % of azoospermic and about 5 % of oligozoospermic patients [17, 22, 23]. The deletions affect three distinct regions: AZFa, AZFb, and AZFc. The AZFa is a nearly 0.8-Mb region that maps to proximal region Yq11.21. AZFb and AZFc map to regions Yq11.22 and Yq11.23, respectively. The AZF deletions influence, overall, 27 unique genes or gene families [24, 25].
AZFa deletions are uncommon and associated with Sertoli cell-only syndrome; testicular histology shows complete germ cell loss and degeneration of seminiferous tubules [15, 17]. The AZFa deletion is a result of nonallelic homologous recombination (NAHR) between two nearly identical DNA repeats. The general view is that the deletion of two genes, USP9Y and DBY (DDX3Y), cause the pathology [22, 24].
AZFb deletions cause spermatogenesis arrest at the spermatocyte stage, loss of mature sperm, and milder azoospermia [17, 22, 24]. The AZFb region has a complex genomic structure and high-identity repeats (palindromic amplicons) that spread in opposite directions up to AZFc [25]. This structure is prone to NAHR between AZFb and AZFc, resulting in two frequent deletions, 6.2 and 7.7 Mb [24, 25]. The 6.2-Mb AZFb deletions remove multiple copies of testis-specific CDY, HSFY, RBMY, and PRY genes. The 7.7-Mb deletion spans an extra 1.5 Mb in the AZFc region (AZFb+c deletion).
AZFc deletions are found in around 12 % of infertile azoospermic men and about 6 % with severe oligozoospermia [24, 25]. The AZFc 3.5-Mb region contains multiple copies of five large repeats—b1, b2, b3, b4, and gr—that are placed in opposite directions and predispose to various partial deletions [25]. Loss of four dosage-sensitive, germ-specific gene families—BPY2, PRY2, DAZ, and CDY1—is associated with full AZFc deletions [22, 23, 25]. A common view is that a low dosage of DAZ (Deleted in Azoospermia) causes oligozoospermia. The DAZ1-4 genes encode germ cell-specific RNA-binding proteins, and their loss affects translation in germ cells and leads to meiosis arrest [25]. Among smaller AZFc deletions, only gr/gr deletions are associated with increased risk of semen defects. The majority of AZFc deletions occur de novo, while partial gr/gr deletions can be passed from father to son [17, 25]. It was shown that inherited gr/gr deletion increases the risk of a complete AZFc deletion in the offspring [11, 12].
Robertsonian Translocations
Robertsonian translocations are the most common SCAs in humans, resulting in a derivative chromosome composed of two long arms from two acrocentric chromosomes (13, 14, 15, 21, and 22). Commonly, RT carriers are healthy but have a high risk of infertility, aberrations in offspring, and spontaneous abortions. The aberrations are found in nearly 1.6 % of infertile male patients. The most frequent RTs are der(13;14) and der(14;21), with incidences of 1:1,000 and 1:5,000, respectively [12, 15, 16, 26, 27]. Nearly 20 % of male carriers of der(13;14) are infertile [27]. Most mature spermatozoa with RT are normal or balanced (75–90 %) as a result of alternate segregation. However, these translocations are associated with disrupted pairing in crossingover, lead to trivalent formation and subsequent meiotic arrest, and result in oligozoospermia or azoospermia [26, 27]. Male carriers of RTs involving chromosome 21 are more likely to have the disomic gametes likely to produce embryos with trisomy 21. However, male RT carriers are often subfertile, lessening that likelihood [15].
Balanced Translocations
Balanced translocations predispose to the formation of a quadrivalent structure between aberrant chromosomes and their normal homologues in meiotic germ cells. This leads to a high proportion (up to 80 %) of unbalanced spermatozoa [20, 21]. Carriers of balanced aberrations are otherwise healthy but often present with infertility [15, 20, 21]. This infertility is due to fertilization by an abnormal gamete with an unbalanced SCA, leading to an unbalanced embryo unlikely to survive. Thus, preimplantation genetic diagnosis benefits carriers of balanced SCAs by identifying embryos with normal or balanced chromosomes. Although carriers with chromosome inversions are generally healthy, they often present with recurrent pregnancy loss and offspring with abnormal chromosomes [15, 20, 21]. Since inverted segments pair and form an inversion loop in meiosis, its position and inversion size determine the outcome of meiotic pairing. In general, large pericentric inversions (spanning over half of the chromosome) are likely to produce unbalanced SCAs and are frequently observed in infertile men [16, 21].
Testicular Disorder of Sex Development (DSD)
The DSD, known as 46,XX male syndrome, is a rare pathology seen in nearly 1/25,000 newborn males [12, 17]. Most DSD patients have an X;Y translocation with Y-linked gene SRY (sex region Y chromosome) placed on one of the X chromosomes [1, 28]. The 46,XX males with SRY and testicular DSD have normal male genitalia, but show spermatogenesis arrest and develop severe testicular atrophy and azoospermia [29]. The SRY encodes the critical testis-determining transcription factor that activates a number of downstream transcription factors involved in testes formation. SOX9 is a direct target of SRY, and its overexpression can mimic male development without SRY. Mutations and small duplications of the SOX9 upstream regulatory region were demonstrated in SRY-negative XX [3, 29] males. Alternatively, increased expression of SOX9 can be induced by steroidogenic factor 1, NR5A1, and SOX3 [29]. Recently, R-spondin 1 (RSPO1) mutations were shown to cause an XX male condition [2].
Complex Structural Chromosome Aberrations (CSAs)
CSAs involve at least three regions in an exchange. Balanced CSAs are rare in individuals with a history of recurrent abortions and infertility. Molecular studies of CSA breakpoints indicate that the complexity and number of breaks cannot predict fertility status. Risk of spontaneous abortion for CSA carriers is estimated at around 50 %. Intracytoplasmic sperm injection is not recommended for male CSA carriers because of the low amount of balanced sperm produced [21, 30].
Genomic Copy Number Variants (CNVs)
CNVs are submicroscopic aberrations of 0.05–3 Mb that cannot be detected by regular chromosome testing. To date, two microarray technologies are used to detect loss or gain CNVs: single nucleotide polymorphism (SNP) array and the comparative genomic hybridization (CGH) array that detects specific probes in comparison to a control specimen. Recent whole-genome and X chromosome CGH studies detected multiple CNVs, with loss and gain that probably affect critical dosage-sensitive genes on sex or autosomal chromosomes [31•, 32, 33]. These studies note that infertile men usually do not have an increased number of CNVs, suggesting that their infertility is likely due to specific defects in single or multiple gene(s). Other studies, using genome-wide SNP arrays, revealed multiple CNVs and regions of homozygosity that could worsen the effect of multiple recessive mutations present in the human genome [34••, 35, 36]. A recent study of CNVs via SNP array demonstrated various individual CNVs in multiple male germ cell–specific genes [34••]. Of interest are multiple loss-of-function deletions of the DMRT1 in patients with various types of azoospermia (e.g., Sertoli cell-only syndrome, testicular failure, or maturation arrest). These genomic aberrations range from 0.132 to 2 Mb, deleting a few exons or the entire gene. The gene encodes an ortholog of an avian sex determination factor and was implicated in nonsyndromic gonadal dysgenesis [34••]. Two recent studies showed that 200-Kb deletions on the 12q14 chromosome remove testis-specific DPY19L2. The deletions are a major cause of globozoospermia (abnormally round sperm head morphology) and detected in nearly 67 % of patients [37, 38••]. The gene encodes a C. elegans ortholog involved in initial stages of round spermatid polarization [38••].
Single-Gene Disorders
The study of male infertility caused by single-gene mutations is a rapidly changing field. In the past decade, we have seen a number of reports newly demonstrating the association of various genes with male infertility. Here we present a list of noteworthy single-gene disorders, while a larger list of single-gene mutations with initial reports is shown in Table 1. The best known single-gene disorder is congenital bilateral aplasia of the vas deferens (CBAVD) with obstructive azoospermia. Mutations in cystic fibrosis transmembrane conductance regulator (CFTR) were reported in more than 90 % of CBVAD patients [10, 39]. The gene encodes the ATP-binding chloride channel and regulates chloride secretion and sodium ion absorption [40]. A general view is that a pair of CFTR mutations, IVS8-5T in intron 8 and another gene-coding mutation, causes isolated CBAVD. However, if the patient has two coding CFTR mutations in both alleles, he will develop classical cystic fibrosis syndrome, including multiple-organ clinical symptoms and male infertility [10].
AR (androgen receptor) encodes testis-specific testosterone receptor. The gene has been associated with androgen insensitivity (AIS) disorder, which ranges from females with testes (complete AIS) to normal and otherwise healthy infertile males (partial AIS) [8]. To date, more than 300 gene mutations have been reported, with multiple forms of testosterone insensitivity and abnormal development of internal and external genitalia. Recently, two studies using conventional and Cre/Lox conditional Ar-null male mice recreated human disorders [41, 42]. They showed female external sex development and testis atrophy with spermatocyte-stage arrest, resembling AIS human pathology.
AURKC (aurora kinase C) is highly expressed in testis and encodes a protein that is probably involved in cytokinesis during germ cell mitosis and meiosis [43, 44]. Mutations in the gene were reported in a number of infertile males with entirely abnormal sperm morphology and sperm polyploidy [45]. The initial report identified a founder loss-of-function mutation; however, subsequent studies have expanded the list of gene mutations, supporting the importance of AURKC for sperm morphology [46].
HSF2 encodes a testis-expressed heat-shock transcription factor 2. Recent study of Hsf2-null male mice demonstrated embryonic lethality, neuronal defects, and a reduced spermatogenesis that includes meiotic disruption and increased sperm apoptosis with seminiferous tubule dysgenesis [47, 48]. A subsequent study of HSF2 in 766 azoospermic males revealed missense mutations in approximately 1 % of patients, suggesting high heterogeneity of the azoospermia [49].
KLHL10 (kelch-like 10) is exclusively expressed in elongating and elongated spermatids. The gene encodes a sperm-specific substrate-targeting module of Cullin 3 ubiquitin ligase and regulates protein ubiquitination and sperm maturation [50]. Null Klhl10 in male mice causes haploinsufficiency with meiotic arrest, absence of mature spermatozoa in semen and male infertility [50]. A study of human oligozoospermia identified missense and splicing mutations in KLHL10 in nearly 2 % of patients that were not observed in controls [51]. Further functional analysis reported a damaging effect of missense mutations. Some gene mutations showed variable effects, ranging from severe to mild oligozoospermia.
NR5A1 encodes steroidogenic factor 1 (SF1), crucial in male and female gonadal development and steroidogenesis. Nr5a1-null male mice present with adrenal agenesis, female internal genitalia, and gonadal absence [52]. Initial human studies of the gene reported mutations associated with female sex development and infertility in males. However, two recent studies of male infertility found different NR5A1 missense mutations associated with 1–4 % of men with oligozoospermia [53, 54••].
PRM1 encodes the testis-specific nuclear protein protamine 1 that replaces histones in postmeiotic sperm. Null PRM1 mutations in male mice showed a haploinsufficiency effect and had a chromatin compaction defect, sperm DNA damage, and severe teratozoospermia [55]. A study of PRM1 in men with similar semen defects identified missense mutations in 10 % of infertile patients [56, 57]. However, later studies used insufficient patient and/or control populations and led to conflicting results [57–59]. Recently, the mutation search was expanded to PRM2, PRM3, and TNP2 located adjacent to the protamine gene cluster, reporting novel mutations [60].
SLC26A8 encodes a sperm-specific sulfate exchange channel (also known as testis anion transporter, TAT1) that regulates CFTR and is vital for sperm motility [61]. Male mice with null SLC26A8 show sperm with abnormal heads, no motility, and male sterility [62]. Recently, two studies revealed that nearly 2–5 % of patients with asthenozoospermia (reduced sperm motility) have various gene mutations with deleterious effects [63, 64•].
SOHLH1 (spermatogenesis- and oogenesis-specific basic helix-loop-helix 1) encodes a critical testis-specific transcription factor essential for spermatogonial differentiation [65]. Male mice with homozygous deletion of Sohlh1 demonstrated spermatocyte arrest, testicular failure, and azoospermia [65]. A subsequent study of the gene in male patients with testicular failure and nonobstructive azoospermia (NOA) supported this notion [66•]. Two missense and one splicing mutation in the SOHLH1 were detected in about 3 % of patients with NOA. Further in vitro functional assay provided evidence that the mutations had a damaging effect [66•].
SPATA16 (spermatogenesis-associated protein 16) encodes a testis-specific Golgi apparatus protein with a tetratricopeptide motif. The original study of one consanguineous family with male infertility and globozoospermia (abnormally round-headed sperm) reported homozygous mutations in the autosomal gene, SPATA16, in three affected brothers [67]. It was shown that the sperm head defects were due to SPATA16 protein involvement in acrosome formation.
ZPBP1 encodes zona pellucida binding protein 1. The protein is located in the acrosomal extracellular matrix of mature sperm and is involved in the initial steps of oocyte fertilization. Zpbp1-null male mice show acrosome fragmentation and abnormal sperm head morphology resembling globozoospermia [68]. A study of human ZPBP1 revealed that nearly 4 % of patients with abnormal sperm-head morphology have missense and splicing gene mutations [69].
Epigenetic and Posttranscriptional Modifications
Epigenetic abnormalities were recently reported in male infertility. One study tested genome-wide methylation defects in 27,000 CpG dinucleotides in sperm from men with abnormal sperm chromatin packaging and patients displaying defective spermatogenesis [70]. It described altered DNA methylation patterns in 3 out of 43 patients, suggesting that a systemic methylation defect contributes to male infertility. A second study identified novel posttranscriptional UBE2B mRNA mutations in approximately 4 % of patients with severe oligozoospermia [71•]. This report is consistent with results of earlier study of Ube2b-null male mice with spermatogenic meiotic disruption, increased apoptosis, and male sterility and suggests that such modifications substantially contribute to an abnormal protein load and sperm disruption [72].
Female Infertility
Reproductive defects and genital tract developmental defects are not uncommon in women, yet little is known about the genetics behind them. During sexual development, congenital malformations of the reproductive tract may occur that affect fertility; these include anatomical abnormalities of the Mullerian ducts, uterus, endometrium, Fallopian tubes, and ovaries [6]. Premature menopause in women of reproductive age and the increasing prevalence of intentionally delayed pregnancy, in Western countries at least, also contribute to female infertility [73].
Chromosomal Aberrations
The 47,XXX Syndrome
The 47,XXX syndrome, also known as trisomy X, is one of the most common causes of premature ovarian insufficiency (POI); it occurs in 1 in 1,000 female births. While the majority of women with trisomy X present as normal, some suffer from POI or from malformations of the genitourinary tract [74].The aberration happens because of chromosome nondisjunction errors in meiosis I or II in oogenesis.
Turner Syndrome
Turner syndrome, also known as monosomy X or 45,XO, chromosome disorder in females, has an incidence of 1:2,000 births. The loss of chromosome X in the oocyte is a result of chromosome nondisjunction in meiosis. The 45,XO females display skeletal abnormalities, congenital heart defects, and physical attributes such as short stature, a webbed neck, low hairline, flat chest, and gonadal dysgenesis with signs of amenorrhea or ovarian failure. Females mosaic for Turner syndrome present with a milder form, often noted because of infertility. 45,X/47,XXX is not a common mosaic presentation, but does present similarly to 45,X/46,XX, with ovarian function declining quickly and leading to infertility [75].
Genomic Aberrations, Copy Number Variants (CNVs)
Recently, a number of microarray studies reported genomic regions associated with female infertility, including the complex disorders endometriosis and primary ovarian failure (POF). They provided stepping stones toward in-depth whole-genome analysis and the discovery of novel genes that would not have been detected with the use of older technologies. One SNP array study identified 6 novel microdeletions affecting at least 1 gene each from a total of 198 autosomal CNVs (microdeletions and microduplications) in 89 women with POF [76•]. Among deleted genes, two previously described POF genes, SYCE1 and CPEB1, were found. Null mutations in these two genes were responsible for ovarian failure in female mice [77, 78]. Another CGH study tested genomic CNVs in 74 patients with POF and ovarian dysfunction [79•]. Multiple genes, such as PLCB1, RB1CC1, MAP4K4, RBBP8, IMMP2L, FER1L6, and MEIG1, involved in meiosis, DNA repair, or folliculogenesis, were identified as possible candidate genes for POF and ovarian dysfunction. Following CGH studies identified new infertility-associated candidate genes and unveiled new details of the pathophysiology [79•, 80].
Single-Gene Disorders
Fragile X Syndrome
Fragile X syndrome is a disorder characterized mainly by mental retardation, long faces, large ears, and prominent jaws. The syndrome was first reported in 1969 with constriction of the long arm on the X chromosome [81]. It has an incidence of 1:5,161 [82]. The critical gene for fragile X is FMR1, fragile X mental retardation gene, located at Xq27.3. The pathology is caused by expansion of the CGG repeat in the gene’s 5′ untranslated region to a premutation state of between 56 and 199 repeats (a complete mutation has over 200 repeats). Generally, FMR1 premutations are found in 16 % of POF females, with about 2 % of the cases being sporadic and 14–21 % familial [82, 83]. Carrier screening of FMR1 premutation is recommended for women of advanced maternal age or with a family history of fragile X.
Galactosemia
Many women with galactosemia manifest hypergonadotropic hypogonadism, presenting with secondary amenorrhea [84, 85]. Premature ovarian failure is independent of age. A candidate gene that has been shown to be associated with galactosemia and endometriosis is the GALT gene, although conflicting results in various studies have reduced its likelihood of being causal [13, 84]. Early genetic counseling about the related risk of infertility and pediatric endocrinologist assistance greatly improve the prognosis of ovarian failure for girls with galactosemia [13, 86]. Since most women with galactosemia are infertile, pregnancy is achieved by oocyte or pre-embryo donation.
Primary Ovarian Failure (POF)
POF is defined as either complete or incomplete failure of the ovaries (also known as primary ovarian insufficiency, POI). Recently, several genes have been discovered that show incomplete loss of function of the ovary, leading to the term “primary ovarian insufficiency” (POI). A number of genes have been associated with POF/POI. X-linked genes include FMR1 and bone morphogenetic protein 15 (BMP15) located at the Xp11.2 region [87, 88]. Among autosomal gene mutations often found in women with POF/POI are AR, CDKN1B, CYP19A1, GDF9, FIGLA, FOXL2, FOXO1a, FOXO3a, INHA, LHX8, NOBOX, NANOS3, FSHR, and SALL4 (Table 2).
FOXL2 encodes an ovarian development-specific transcription factor with forkhead box L2. The forkhead box is a DNA-binding domain that plays a key role in the protein function. Dominant mutations in FOXL2 cause premature ovarian failure 3 [89]. The gene is also responsible for the blepharophimosis, ptosis, and epicanthus syndrome, which may include POF [90].
GDF9 (growth/differentiation factor 9) and GDF9B (BMP15) are necessary for ovarian folliculogenesis and somatic cell function in both mice and humans. Initial genetic studies in women with POF discovered multiple missense mutations in 3–4 % of patients [91]. Subsequent studies of POF in women of various ethnic backgrounds reported more mutations in these two genes.
FIGLA (factor in germline alpha) is located at 2p13.3 and encodes a germ cell-specific basic helix-loop-helix transcription factor. It regulates the expression of the zona pellucida- and oocyte-specific genes. A knockout of FIGLA in female mice prevents the formation of primordial follicles, and oocyte numbers drop rapidly after birth [92]. The initial study of FIGLA identified mutations in approximately 4 % of POF patients [93]. Two women had missense mutations and two had other gene deletions that resulted in a frameshift and haploinsufficiency [93].
NOBOX (newborn ovary homeobox) is located at 7q35 and encodes a transcriptional regulator with a homeobox motif. Study of Nobox-null female mice indicates that this gene is critical for early folliculogenesis [94], and an independent human study found NOBOX defects in POF patients [95]. Later, two missense mutations in the homeobox domain were found in 6.2 % of patients of Caucasian or African descent [96•].
SALL4 (SAL-like 4) is located at 20q13.2 and encodes putative zinc finger transcription factor and plays a role in the pluripotency of oocytes. In Nobox-deficient mouse ovaries, SALL4 is downregulated, suggesting it is activated by NOBOX [94]. In a study of 100 Han Chinese women with nonsyndromic POF, two probable gene mutations were discovered in POF subjects and not in the control group [97]. Remaining known genes associated with POF have been recessive, producing a variety of phenotypes and causing idiopathic infertility (Table 2) [6].
Leiomyomas
Leiomyomas, also referred to as fibroids, are benign tumors found in the smooth muscle layers of the uterus. It is not uncommon to have irregular bleeding and pain, sometimes necessitating a hysterectomy. The number of tumors varies among those affected and can change in conjunction with hormones. During pregnancy, the tumor lesions have a tendency to increase in size. There is a prevalence of lesions among black women, who undergo hysterectomies twice as often as whites [98••, 99]. Somatic chromosomal rearrangements or deletions are found in the 12q and 7q regions, respectively [100]. Deletions within 7q are observed in about 20 % of leiomyomas [100]. Whole genome sequencing implicated CUX1, ZNHIT1, and CUL1 as key genes within 7q deletions [101••]. In addition, mutations affecting oncogenes, metabolism, and follicle-stimulating hormone signaling, and changes in COL4A5-COL4A6, HMGA2 and RAD51B were identified [101••]. New studies using conventional and next-generation sequencing techniques identified mutations in the MED12 gene as a major contributor to leiomyoma [102, 103••, 104]. Nearly 60 % of patients with the pathology have MED12 mutations.
Polygenic, Complex Female Infertility
Endometriosis
Endometriosis is a complex disease, characterized by the inflammation and bleeding of the endometrium. It affects 5–10 % of females. Often there is infertility and pain due to endometrial tissue in the pelvic region outside of the uterus [98••, 105]. Those with affected first degree relatives have five to eight times increased risk for the disease. Genetic association and linkage studies have identified some candidate genes, SNPs, and CNVs, but follow-up studies are needed to replicate the studies and narrow down critical regions (Table 2). One study determined a significant linkage to 10q26, but the linkage peak was too broad to identify a single gene as a causative factor of endometriosis [106]. Among two genome-wide association (GWAS) studies conducted in Australia and Japan in 2011 and 2010 [107, 108], only one common locus was found—in the 1p36 region—which contains WNT4, a gene responsible for cell proliferation and that plays a key role in embryogenesis [109]. Prior studies have also noted this region’s involvement in endometriosis [98••].
Polycystic Ovarian Syndrome (PCOS)
PCOS is a complex endocrine disorder with heterogeneous genetic causes. It is found in about 7 % of women of reproductive age. Aside from metabolic issues that may be associated with PCOS, such as obesity and type 2 diabetes mellitus, common symptoms are irregularities in the menstrual cycle, polycystic ovaries, secondary amenorrhea, anovulation, hirsutism, and reduced fertility due to oversized, dysfunctional follicles [99, 110–112]. The adrenal-form adult-onset disorder is due primarily to enzyme deficiencies, often causing pseudo-hermaphroditism and hirsutism. Nonadrenal PCOS phenotypes differ in how they manifest and in their pathophysiology. There appears to be dominant inheritance, yet no specific gene(s) has been identified as the cause for PCOS, rendering it idiopathic [99]. Studies of multiple candidate genes including FBN3, FST, INS, INSR, TCF7L2, CAPN10, FTO, SHBG, PCOS1, SRD5A1, SRD5A2, and CYP11A1 have shown association with PCOS [98••, 111–122]. However, these genes have also shown significant associations with other disorders such as obesity, diabetes, and insulin resistance, which are commonly associated with PCOS. These results suggest the complex nature of PCOS. To date, a direct correlation with PCOS has been shown for one gene, INSR. The insulin receptor gene has shown the highest likelihood as a susceptibility gene in the Han Chinese after being identified in a GWAS study, which also identified three additional PCOS loci (2p16.3, 2p21, 9q33.3) [114]. Two studies were able to replicate the results for one identified gene of interest, DENND1A, a guanine nucleotide exchange factor [114, 123]. As yet, the cause of PCOS is unknown; both, genetic and environmental factors must be taken into consideration. Current treatment and management of the symptoms of PCOS include ovulation induction [112].
Conclusion
Many men and women affected by infertility or disorders that lead to decreased fertility have turned to assisted reproductive technology (ART). Recent estimates show nearly 5 million newborns have been assisted by ART [124]. It is known that a woman’s fecundity decreases with advancing age. Also, the modern tendency to delay childbirth contributes to the increased use of ART. Yet, there are several concerns about the safety and possible negative outcomes of the ICSI treatment, such as pregnancy complication or termination, risk of various birth defects, and childhood developmental and mental defects [124]. It is important that all couples undergoing infertility treatment should be informed of the risks, benefits, and possible outcomes through the use of genetic testing and counseling. As more genes are discovered, and the etiology of infertility disorders become better understood, the management and treatment of infertility will improve as well.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Sinclair AH, Berta P, Palmer MS, et al. A gene from the human sex-determining region encodes a protein with homology to a conserved DNA-binding motif. Nature. 1990;346(6281):240–4.
Parma P, Radi O, Vidal V, et al. R-spondin1 is essential in sex determination, skin differentiation and malignancy. Nat Genet. 2006;38(11):1304–9.
Wagner T, Wirth J, Meyer J, et al. Autosomal sex reversal and campomelic dysplasia are caused by mutations in and around the SRY-related gene SOX9. Cell. 1994;79(6):1111–20.
Foster JW, Dominguez-Steglich MA, Guioli S, et al. Campomelic dysplasia and autosomal sex reversal caused by mutations in an SRY-related gene. Nature. 1994;372(6506):525–30.
Cameron FJ, Sinclair AH. Mutations in SRY and SOX9: testis-determining genes. Hum Mutat. 1997;9(5):388–95.
Matzuk MM, Lamb DJ. The biology of infertility: research advances and clinical challenges. Nat Med. 2008;14(11):1197–213.
Semple RK, Topaloglu AK. The recent genetics of hypogonadotrophic hypogonadism: novel insights and new questions. Clin Endocrinol (Oxf). 2010;72(4):427–35.
McPhaul MJ, Marcelli M, Zoppi S, Wilson CM, Griffin JE, Wilson JD. Mutations in the ligand-binding domain of the androgen receptor gene cluster in two regions of the gene. J Clin Invest. 1992;90(5):2097–101.
Costes B, Girodon E, Ghanem N, et al. Frequent occurrence of the CFTR intron 8 (TG)n 5T allele in men with congenital bilateral absence of the vas deferens. Eur J Hum Genet. 1995;3(5):285–93.
Riordan JR, Rommens JM, Kerem B, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245(4922):1066–73.
McLachlan RI, O’Bryan MK. Clinical review: state of the art for genetic testing of infertile men. J Clin Endocrinol Metab. 2010;95(3):1013–24.
O’Flynn O’Brien KL, Varghese AC, Agarwal A. The genetic causes of male factor infertility: a review. Fertil Steril. 2010;93(1):1–12.
Preece MA, Green A. Pregnancy and inherited metabolic disorders: maternal and fetal complications. Ann Clin Biochem. 2002;39(Pt 5):444–55.
Rovio AT, Marchington DR, Donat S, et al. Mutations at the mitochondrial DNA polymerase (POLG) locus associated with male infertility. Nat Genet. 2001;29(3):261–2.
Harton GL, Tempest HG. Chromosomal disorders and male infertility. Asian J Androl. 2012;14(1):32–9.
Martin RH. Cytogenetic determinants of male fertility. Hum Reprod Update. 2008;14(4):379–90.
Walsh TJ, Pera RR, Turek PJ. The genetics of male infertility. Semin Reprod Med. 2009;27(2):124–36.
Oates RD. The natural history of endocrine function and spermatogenesis in Klinefelter syndrome: what the data show. Fertil Steril. 2012;98(2):266–73.
Hennebicq S, Pelletier R, Bergues U, Rousseaux S. Risk of trisomy 21 in offspring of patients with Klinefelter’s syndrome. Lancet. 2001;357(9274):2104–5.
Hann MC, Lau PE, Tempest HG. Meiotic recombination and male infertility: from basic science to clinical reality? Asian J Androl. 2011;13(2):212–8.
Marchetti F, Wyrobek AJ. Mechanisms and consequences of paternally-transmitted chromosomal abnormalities. Birth Defects Res C Embryo Today. 2005;75(2):112–29.
Reijo R, Lee TY, Salo P, et al. Diverse spermatogenic defects in humans caused by Y chromosome deletions encompassing a novel RNA-binding protein gene. Nat Genet. 1995;10(4):383–93.
Reijo R, Alagappan RK, Patrizio P, Page DC. Severe oligozoospermia resulting from deletions of azoospermia factor gene on Y chromosome. Lancet. 1996;347(9011):1290–3.
Vogt PH, Edelmann A, Kirsch S, et al. Human Y chromosome azoospermia factors (AZF) mapped to different subregions in Yq11. Hum Mol Genet. 1996;5(7):933–43.
Kuroda-Kawaguchi T, Skaletsky H, Brown LG, et al. The AZFc region of the Y chromosome features massive palindromes and uniform recurrent deletions in infertile men. Nat Genet. 2001;29(3):279–86.
Roux C, Tripogney C, Morel F, et al. Segregation of chromosomes in sperm of Robertsonian translocation carriers. Cytogenet Genome Res. 2005;111(3–4):291–6.
Engels H, Eggermann T, Caliebe A, et al. Genetic counseling in Robertsonian translocations der(13;14): frequencies of reproductive outcomes and infertility in 101 pedigrees. Am J Med Genet A. 2008;146A(20):2611–6.
Andersson M, Page DC, de la Chapelle A. Chromosome Y-specific DNA is transferred to the short arm of X chromosome in human XX males. Science. 1986;233(4765):786–8.
Kousta E, Papathanasiou A, Skordis N. Sex determination and disorders of sex development according to the revised nomenclature and classification in 46,XX individuals. Hormones (Athens). 2010;9(3):131–218.
Madan K. Balanced complex chromosome rearrangements: reproductive aspects. A review. Am J Med Genet A. 2012;158A(4):947–63.
• Tuttelmann F, Simoni M, Kliesch S, et al. Copy number variants in patients with severe oligozoospermia and Sertoli-cell-only syndrome. PLoS One. 2011;6(4):e19426. First array-CGH study of male infertility. Discovered a number of candidate genes and CNVs associated with spermatogenic failure and Sertoli cell only syndrome.
Krausz C, Giachini C, Lo Giacco D, et al. High resolution X chromosome-specific array-CGH detects new CNVs in infertile males. PLoS One. 2012;7(10):e44887.
Jorgez CJ, Weedin JW, Sahin A, et al. Aberrations in pseudoautosomal regions (PARs) found in infertile men with Y-chromosome microdeletions. J Clin Endocrinol Metab. 2011;96(4):E674–9.
•• Lopes AM, Aston KI, Thompson E, et al. Human spermatogenic failure purges deleterious mutation load from the autosomes and both sex chromosomes, including the gene DMRT1. PLoS Genet. 2013;9(3):e1003349. Large number of cases and controls studied; only cases with spermatogenic impairment or idiopathic azoospermia had the DMRT1 gene deletion. This study discovered the DMRT1 loss of function mutation, implicating the gene as a cause of spermatogenic failure.
Aston KI, Krausz C, Laface I, Ruiz-Castane E, Carrell DT. Evaluation of 172 candidate polymorphisms for association with oligozoospermia or azoospermia in a large cohort of men of European descent. Hum Reprod. 2010;25(6):1383–97.
Aston KI, Carrell DT. Genome-wide study of single-nucleotide polymorphisms associated with azoospermia and severe oligozoospermia. J Androl. 2009;30(6):711–25.
Elinati E, Kuentz P, Redin C, et al. Globozoospermia is mainly due to DPY19L2 deletion via non-allelic homologous recombination involving two recombination hotspots. Hum Mol Genet. 2012;21(16):3695–702.
•• Koscinski I, Elinati E, Fossard C, et al. DPY19L2 deletion as a major cause of globozoospermia. Am J Hum Genet. 2011;88(3):344–350. This study consisted of patients of three different origins diagnosed with complete globozoospermia. A discovery of a deletion on chromosome 12 that encompassed the DPY19L2 gene suggests that recurrent deletions are caused by genomic structure of the locus and are not a result of founder effect. The DPY19L2 gene is strongly associated with globozoospermia.
Grangeia A, Sa R, Carvalho F, et al. Molecular characterization of the cystic fibrosis transmembrane conductance regulator gene in congenital absence of the vas deferens. Genet Med. 2007;9(3):163–72.
Schwiebert EM, Egan ME, Hwang TH, et al. CFTR regulates outwardly rectifying chloride channels through an autocrine mechanism involving ATP. Cell. 1995;81(7):1063–73.
Sato T, Matsumoto T, Yamada T, Watanabe T, Kawano H, Kato S. Late onset of obesity in male androgen receptor-deficient (AR KO) mice. Biochem Biophys Res Commun. 2003;300(1):167–71.
Yeh S, Tsai MY, Xu Q, et al. Generation and characterization of androgen receptor knockout (ARKO) mice: an in vivo model for the study of androgen functions in selective tissues. Proc Natl Acad Sci USA. 2002;99(21):13498–503.
Kimura M, Matsuda Y, Yoshioka T, Okano Y. Cell cycle-dependent expression and centrosome localization of a third human aurora/Ipl1-related protein kinase, AIK3. J Biol Chem. 1999;274(11):7334–40.
Tang CJ, Lin CY, Tang TK. Dynamic localization and functional implications of aurora-C kinase during male mouse meiosis. Dev Biol. 2006;290(2):398–410.
Dieterich K, Soto Rifo R, Faure AK, et al. Homozygous mutation of AURKC yields large-headed polyploid spermatozoa and causes male infertility. Nat Genet. 2007;39(5):661–5.
Ben Khelifa M, Coutton C, Blum MG, et al. Identification of a new recurrent aurora kinase C mutation in both European and African men with macrozoospermia. Hum Reprod. 2012;27(11):3337–46.
Wang G, Zhang J, Moskophidis D, Mivechi NF. Targeted disruption of the heat shock transcription factor (hsf)-2 gene results in increased embryonic lethality, neuronal defects, and reduced spermatogenesis. Genesis. 2003;36(1):48–61.
Wang G, Ying Z, Jin X, et al. Essential requirement for both hsf1 and hsf2 transcriptional activity in spermatogenesis and male fertility. Genesis. 2004;38(2):66–80.
Mou L, Wang Y, Li H, et al. A dominant-negative mutation of HSF2 associated with idiopathic azoospermia. Hum Genet. 2013;132(2):159–65.
Yan W, Ma L, Burns KH, Matzuk MM. Haploinsufficiency of kelch-like protein homolog 10 causes infertility in male mice. Proc Natl Acad Sci USA. 2004;101(20):7793–8.
Yatsenko AN, Roy A, Chen R, et al. Non-invasive genetic diagnosis of male infertility using spermatozoal RNA: KLHL10 mutations in oligozoospermic patients impair homodimerization. Hum Mol Genet. 2006;15(23):3411–9.
Luo X, Ikeda Y, Parker KL. A cell-specific nuclear receptor is essential for adrenal and gonadal development and sexual differentiation. Cell. 1994;77(4):481–90.
Ropke A, Tewes AC, Gromoll J, Kliesch S, Wieacker P, Tuttelmann F. Comprehensive sequence analysis of the NR5A1 gene encoding steroidogenic factor 1 in a large group of infertile males. Eur J Hum Genet. 2013;21(9):1012–5.
•• Bashamboo A, Ferraz-de-Souza B, Lourenco D, et al. Human male infertility associated with mutations in NR5A1 encoding steroidogenic factor 1. Am J Hum Genet. 2010;87(4):505–512. The study demonstrates that NR5A1 is a cause of male infertility in men with idiopathic spermatogenetic failure. Missense mutations were only seen in cases and not controls, and functional studies showed how the mutations impair the gene’s activity, indicating the importance of this gene with unexplained severe spermatogenic failure in 4 % of the men.
Cho C, Willis WD, Goulding EH, et al. Haploinsufficiency of protamine-1 or -2 causes infertility in mice. Nat Genet. 2001;28(1):82–6.
Iguchi N, Yang S, Lamb DJ, Hecht NB. An SNP in protamine 1: a possible genetic cause of male infertility? J Med Genet. 2006;43(4):382–4.
Ravel C, Chantot-Bastaraud S, El Houate B, et al. Mutations in the protamine 1 gene associated with male infertility. Mol Hum Reprod. 2007;13(7):461–4.
Kichine E, Msaidie S, Bokilo AD, et al. Low-frequency protamine 1 gene transversions c.102G->T and c.-107G->C do not correlate with male infertility. J Med Genet. 2008;45(4):255–6.
Tanaka H, Miyagawa Y, Tsujimura A, Matsumiya K, Okuyama A, Nishimune Y. Single nucleotide polymorphisms in the protamine-1 and -2 genes of fertile and infertile human male populations. Mol Hum Reprod. 2003;9(2):69–73.
Imken L, Rouba H, El Houate B, et al. Mutations in the protamine locus: association with spermatogenic failure? Mol Hum Reprod. 2009;15(11):733–8.
Toure A, Morin L, Pineau C, Becq F, Dorseuil O, Gacon G. Tat1, a novel sulfate transporter specifically expressed in human male germ cells and potentially linked to rhogtpase signaling. J Biol Chem. 2001;276(23):20309–15.
Toure A, Lhuillier P, Gossen JA, et al. The testis anion transporter 1 (Slc26a8) is required for sperm terminal differentiation and male fertility in the mouse. Hum Mol Genet. 2007;16(15):1783–93.
Makela S, Eklund R, Lahdetie J, Mikkola M, Hovatta O, Kere J. Mutational analysis of the human SLC26A8 gene: exclusion as a candidate for male infertility due to primary spermatogenic failure. Mol Hum Reprod. 2005;11(2):129–32.
• Dirami T, Rode B, Jollivet M, et al. Missense mutations in SLC26A8, encoding a sperm-specific activator of CFTR, are associated with human asthenozoospermia. Am J Hum Genet. 2013;92(5):760–766. SCL26A8 protein works with CFTR to stimulate sperm motility and capacitation. Missense mutations in SLC26A8, present in 146 men with asthenozoospermia, are shown to be associated with asthenozoospermia at a power of more than 95 %. The SLC26A8-CFTR complex formation is impaired, and therefore activation of sperm is affected in those individuals.
Ballow D, Meistrich ML, Matzuk M, Rajkovic A. Sohlh1 is essential for spermatogonial differentiation. Dev Biol. 2006;294(1):161–7.
• Choi Y, Jeon S, Choi M, et al. Mutations in SOHLH1 gene associate with nonobstructive azoospermia. Hum Mutat. Jul 2010;31(7):788–793. SOHLH1, a candidate gene for nonobstructive azoospermia, and essential for spermatogonial differentiation, is shown to become nonfunctional when three novel mutations occur. The mutations in the protein cause a lack of normal spermatogenesis.
Dam AH, Koscinski I, Kremer JA, et al. Homozygous mutation in SPATA16 is associated with male infertility in human globozoospermia. Am J Hum Genet. 2007;81(4):813–20.
Lin YN, Roy A, Yan W, Burns KH, Matzuk MM. Loss of zona pellucida binding proteins in the acrosomal matrix disrupts acrosome biogenesis and sperm morphogenesis. Mol Cell Biol. 2007;27(19):6794–805.
Yatsenko AN, O’Neil DS, Roy A, et al. Association of mutations in the zona pellucida binding protein 1 (ZPBP1) gene with abnormal sperm head morphology in infertile men. Mol Hum Reprod. 2012;18(1):14–21.
Hammoud SS, Nix DA, Hammoud AO, Gibson M, Cairns BR, Carrell DT. Genome-wide analysis identifies changes in histone retention and epigenetic modifications at developmental and imprinted gene loci in the sperm of infertile men. Hum Reprod. 2011;26(9):2558–69.
• Yatsenko AN, Georgiadis AP, Murthy LJ, Lamb DJ, Matzuk MM. UBE2B mRNA alterations are associated with severe oligozoospermia in infertile men. Mol Hum Reprod. 2013;19(6):388–394. This is the first paper that demonstrates post-transcriptional mRNA editing and splicing defects associated with oligozoospermia and suggested that alterations in UBE2B could contribute to a significant fraction of oligozoospermia.
Roest HP, van Klaveren J, de Wit J, et al. Inactivation of the HR6B ubiquitin-conjugating DNA repair enzyme in mice causes male sterility associated with chromatin modification. Cell. 1996;86(5):799–810.
Matthews TJ, Hamilton BE. Delayed childbearing: more women are having their first child later in life. NCHS Data Brief. 2009;21:1–8.
Tartaglia NR, Howell S, Sutherland A, Wilson R, Wilson L. A review of trisomy X (47, XXX). Orphanet J Rare Dis. 2010;5:8.
Simpson JL, Rajkovic A. Ovarian differentiation and gonadal failure. Am J Med Genet. 1999;89(4):186–200.
• McGuire MM, Bowden W, Engel NJ, Ahn HW, Kovanci E, Rajkovic A. Genomic analysis using high-resolution single-nucleotide polymorphism arrays reveals novel microdeletions associated with premature ovarian failure. Fertil Steril. 2011;95(5):1595–1600. The SNP-array study found 198 autosomal CNVs, microdeletions, and microduplications in women with POF. Two of the deletions were seen to cause haploinsufficiency in SYCE1 and CPEB1 genes that cause ovarian failure in KO mouse models.
Tay J, Richter JD. Germ cell differentiation and synaptonemal complex formation are disrupted in CPEB knockout mice. Dev Cell. 2001;1(2):201–13.
Bolcun-Filas E, Hall E, Speed R, et al. Mutation of the mouse Syce1 gene disrupts synapsis and suggests a link between synaptonemal complex structural components and DNA repair. PLoS Genet. 2009;5(2):e1000393.
• Ledig S, Ropke A, Wieacker P. Copy number variants in premature ovarian failure and ovarian dysgenesis. Sex Dev. Sep 2010;4(4–5):225–232. Array-CGH was performed in 74 patients with POF and OD and discovered 44 CNVs that could be causative factors for POF. A number of the genes affected were related in some way to fertility (meiosis, DNA repair, folliculogenesis, or male fertility).
Matsuzaki S. DNA microarray analysis in endometriosis for development of more effective targeted therapies. Front Biosci (Elite Ed). 2011;3:1139–53.
Lubs HA. A marker X chromosome. Am J Hum Genet. 1969;21(3):231–44.
Coffee B, Keith K, Albizua I, et al. Incidence of fragile X syndrome by newborn screening for methylated FMR1 DNA. Am J Hum Genet. 2009;85(4):503–14.
Sherman S, Pletcher BA, Driscoll DA. Fragile X syndrome: diagnostic and carrier testing. Genet Med. 2005;7(8):584–7.
Fridovich-Keil JL, Gubbels CS, Spencer JB, Sanders RD, Land JA, Rubio-Gozalbo E. Ovarian function in girls and women with GALT-deficiency galactosemia. J Inherit Metab Dis. 2011;34(2):357–66.
Kaufman FR, Kogut MD, Donnell GN, Goebelsmann U, March C, Koch R. Hypergonadotropic hypogonadism in female patients with galactosemia. N Engl J Med. 1981;304(17):994–8.
Guerrero NV, Singh RH, Manatunga A, Berry GT, Steiner RD, Elsas LJ 2nd. Risk factors for premature ovarian failure in females with galactosemia. J Pediatr. 2000;137(6):833–41.
Di Pasquale E, Beck-Peccoz P, Persani L. Hypergonadotropic ovarian failure associated with an inherited mutation of human bone morphogenetic protein-15 (BMP15) gene. Am J Hum Genet. 2004;75(1):106–11.
Cordts EB, Christofolini DM, Dos Santos AA, Bianco B, Barbosa CP. Genetic aspects of premature ovarian failure: a literature review. Arch Gynecol Obstet. 2011;283(3):635–43.
Harris SE, Chand AL, Winship IM, Gersak K, Aittomaki K, Shelling AN. Identification of novel mutations in FOXL2 associated with premature ovarian failure. Mol Hum Reprod. 2002;8(8):729–33.
De Baere E, Dixon MJ, Small KW, et al. Spectrum of FOXL2 gene mutations in blepharophimosis-ptosis-epicanthus inversus (BPES) families demonstrates a genotype–phenotype correlation. Hum Mol Genet. 2001;10(15):1591–600.
Zhao H, Qin Y, Kovanci E, Simpson JL, Chen ZJ, Rajkovic A. Analyses of GDF9 mutation in 100 Chinese women with premature ovarian failure. Fertil Steril. 2007;88(5):1474–6.
Soyal SM, Amleh A, Dean J. FIGalpha, a germ cell-specific transcription factor required for ovarian follicle formation. Development. 2000;127(21):4645–54.
Zhao H, Chen Z-J, Qin Y, et al. Transcription factor FIGLA is mutated in patients with premature ovarian failure. Am J Hum Genet. 2008;82(6):1342–8.
Rajkovic A, Pangas SA, Ballow D, Suzumori N, Matzuk MM. NOBOX deficiency disrupts early folliculogenesis and oocyte-specific gene expression. Science. 2004;305(5687):1157–9.
Qin Y, Choi Y, Zhao H, Simpson JL, Chen ZJ, Rajkovic A. NOBOX homeobox mutation causes premature ovarian failure. Am J Hum Genet. 2007;81(3):576–81.
• Bouilly J, Bachelot A, Broutin I, Touraine P, Binart N. Novel NOBOX loss-of-function mutations account for 6.2 % of cases in a large primary ovarian insufficiency cohort. Hum Mutat. 2011;32(10):1108–1113. The study suggests that NOBOX haploinsufficiency is responsible for POI. One nonsense and four missense mutations identified in the study reduced the ability of NOBOX to transactivate the GDF9 promoter. NOBOX is the first autosmal disease gene for POI.
Wang B, Li L, Ni F, et al. Mutational analysis of SAL-Like 4 (SALL4) in Han Chinese women with premature ovarian failure. Mol Hum Reprod. 2009;15(9):557–62.
•• Layman LC. The genetic basis of female reproductive disorders: etiology and clinical testing. Mol Cell Endocrinol. 2013;370(1–2):138–148. The paper provides a thorough overview of the different causes of female infertility and the genes affecting it. It presents the most up-to-date information on the genetics of female reproductive disorders.
Simpson J. Genetic factors in common disorders of female infertility. Reprod Med Rev. 2000;8:173–202.
Nilbert M, Heim S, Mandahl N, Floderus UM, Willen H, Mitelman F. Characteristic chromosome abnormalities, including rearrangements of 6p, del(7q), +12, and t(12;14), in 44 uterine leiomyomas. Hum Genet. 1990;85(6):605–11.
•• Mehine M, Kaasinen E, Makinen N, et al. Characterization of Uterine Leiomyomas by Whole-Genome Sequencing. N Engl J Med. 2013. Whole-genome sequencing and gene-expression profiling was done on leiomyomas of 30 women. The study discovered identical variants, more specifically chromosome rearrangements, translocation, and aberrations in the tumors of the different women.
McGuire MM, Yatsenko A, Hoffner L, Jones M, Surti U, Rajkovic A. Whole exome sequencing in a random sample of North American women with leiomyomas identifies MED12 mutations in majority of uterine leiomyomas. PLoS One. 2012;7(3):e33251.
•• Makinen N, Mehine M, Tolvanen J, et al. MED12, the mediator complex subunit 12 gene, is mutated at high frequency in uterine leiomyomas. Science. 2011;334(6053):252–255. Study of MED12 tumor-specific mutations, discovered as the gene altered in 70 % of leiomyomas. Mutations were clustered in exon 2, supporting the conclusion that changes in this region contribute to tumorigenesis.
Perot G, Croce S, Ribeiro A, et al. MED12 alterations in both human benign and malignant uterine soft tissue tumors. PLoS One. 2012;7(6):e40015.
Simpson JL. Molecular approach to common causes of female infertility. Best Pract Res Clin Obstet Gynaecol. 2002;16(5):685–702.
Treloar SA, Wicks J, Nyholt DR, et al. Genomewide linkage study in 1,176 affected sister pair families identifies a significant susceptibility locus for endometriosis on chromosome 10q26. Am J Hum Genet. 2005;77(3):365–76.
Painter JN, Anderson CA, Nyholt DR, et al. Genome-wide association study identifies a locus at 7p15.2 associated with endometriosis. Nat Genet. 2011;43(1):51–4.
Uno S, Zembutsu H, Hirasawa A, et al. A genome-wide association study identifies genetic variants in the CDKN2BAS locus associated with endometriosis in Japanese. Nat Genet. 2010;42(8):707–10.
Vainio S, Heikkila M, Kispert A, Chin N, McMahon AP. Female development in mammals is regulated by Wnt-4 signalling. Nature. 1999;397(6718):405–9.
Kosova G, Urbanek M. Genetics of the polycystic ovary syndrome. Mol Cell Endocrinol. 2013;373(1–2):29–38.
Murphy BD. Revisiting reproduction: what a difference a gene makes. Nat Med. 2010;16(5):527–9.
Norman RJ, Dewailly D, Legro RS, Hickey TE. Polycystic ovary syndrome. Lancet. 2007;370(9588):685–97.
Urbanek M, Legro RS, Driscoll DA, et al. Thirty-seven candidate genes for polycystic ovary syndrome: strongest evidence for linkage is with follistatin. Proc Natl Acad Sci USA. 1999;96(15):8573–8.
Chen ZJ, Zhao H, He L, et al. Genome-wide association study identifies susceptibility loci for polycystic ovary syndrome on chromosome 2p16.3, 2p21 and 9q33.3. Nat Genet. 2011;43(1):55–9.
Urbanek M, Sam S, Legro RS, Dunaif A. Identification of a polycystic ovary syndrome susceptibility variant in fibrillin-3 and association with a metabolic phenotype. J Clin Endocrinol Metab. 2007;92(11):4191–8.
Waterworth DM, Bennett ST, Gharani N, et al. Linkage and association of insulin gene VNTR regulatory polymorphism with polycystic ovary syndrome. Lancet. 1997;349(9057):986–90.
Goodarzi MO, Louwers YV, Taylor KD, et al. Replication of association of a novel insulin receptor gene polymorphism with polycystic ovary syndrome. Fertil Steril. 2011;95(5):1736–1741. e1731–1711.
Gonzalez A, Abril E, Roca A, et al. Specific CAPN10 gene haplotypes influence the clinical profile of polycystic ovary patients. J Clin Endocrinol Metab. 2003;88(11):5529–36.
Christopoulos P, Mastorakos G, Gazouli M, et al. Genetic variants in TCF7L2 and KCNJ11 genes in a Greek population with polycystic ovary syndrome. Gynecol Endocrinol. 2008;24(9):486–90.
Barber TM, Bennett AJ, Groves CJ, et al. Association of variants in the fat mass and obesity associated (FTO) gene with polycystic ovary syndrome. Diabetologia. 2008;51(7):1153–8.
Ewens KG, Stewart DR, Ankener W, et al. Family-based analysis of candidate genes for polycystic ovary syndrome. J Clin Endocrinol Metab. 2010;95(5):2306–15.
Urbanek M, Woodroffe A, Ewens KG, et al. Candidate gene region for polycystic ovary syndrome on chromosome 19p13.2. J Clin Endocrinol Metab. 2005;90(12):6623–9.
Yoshimura S-I, Gerondopoulos A, Linford A, Rigden DJ, Barr FA. Family-wide characterization of the DENN domain Rab GDP–GTP exchange factors. J Cell Biol. 2010;191(2):367–81.
Davies MJ, Moore VM, Willson KJ, et al. Reproductive technologies and the risk of birth defects. N Engl J Med. 2012;366(19):1803–13.
Compliance with Ethics Guidelines
Conflict of Interest
M. Zorrilla and A.N. Yatsenko have received research grants and travel reimbursements from the NIH.
Human and Animal Rights and Informed Consent
This article contains no studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zorrilla, M., Yatsenko, A.N. The Genetics of Infertility: Current Status of the Field. Curr Genet Med Rep 1, 247–260 (2013). https://doi.org/10.1007/s40142-013-0027-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40142-013-0027-1