
Abstract
Background
During two pivotal clinical trials of the infliximab biosimilar CT-P13 (PLANETAS and PLANETRA), antidrug antibodies (ADAs) and neutralising antibodies (NAbs) were detected in the sera of patients treated with CT-P13 and the reference product (RP; Remicade).
Objective
The aim was to assess the comparability of Remicade- and CT-P13-tagged immunoassays for the detection of ADAs and NAbs using data from these trials, in order to determine the cross-reactivity of CT-P13 and RP ADAs.
Methods
Sera from patients with rheumatoid arthritis and ankylosing spondylitis were analysed using an electrochemiluminescence (ECL) bridging assay or Gyros immunoassay, tagged with Remicade or CT-P13 at screening, weeks 14, 30 and 54, and the end of study visit. NAb titre was compared at screening and weeks 14 and 30. The proportion of cross-reactive samples was determined and an inter-rater agreement analysis performed to assess the concordance of results between assays.
Results
In PLANETAS, 93.1% (94/101) of RP ADA-positive samples and 93.0% (93/100) of RP NAb-positive samples cross-reacted with CT-P13; 99.0% (103/104) of CT-P13 ADA-positive and 98.0% (98/100) of CT-P13 NAb-positive samples cross-reacted with the RP. In PLANETRA, 94.7% (426/450) of RP ADA-positive samples and 94.3% (415/440) of RP NAb-positive samples cross-reacted with CT-P13, and 96.6% (458/474) of CT-P13 ADA-positive and 96.4% (452/469) of CT-P13 NAb-positive samples cross-reacted with the RP. In both studies, there was strong agreement in outcome between assays at all post-screening time points (PLANETAS: Cohen’s κ 0.89–0.98 for ADA, 0.86–0.98 for NAb; PLANETRA: 0.92–0.94 for both ADA and NAb, all p < 0.001). Significant concordance between assays was observed for NAb titre at weeks 14 and 30 (PLANETAS: Spearman’s ρ 0.73 and 0.74, respectively; PLANETRA: 0.61 and 0.72, respectively; all p < 0.001).
Conclusions
This study has demonstrated that ADAs and NAbs against CT-P13 and RP are cross-reactive, indicating that CT-P13 and RP share immunodominant epitopes.
Similar content being viewed by others


This study demonstrated that antibodies against the infliximab biosimilar CT-P13 and the infliximab reference product (RP; Remicade) recognise and bind RP and CT-P13, respectively. |
The cross-reactivity of CT-P13 and its RP indicate that the two products share immunodominant epitopes. |
The findings of this study are consistent with the previously reported high biosimilarity of CT-P13 to its RP and suggest that commercialised kits for the detection of antidrug antibodies against infliximab can be utilised for either CT-P13 or its RP. |
1 Background
The introduction of biological agents that target the immune response has had a dramatic effect on the treatment of immune-mediated inflammatory diseases (IMIDs), such as rheumatoid arthritis (RA), spondyloarthritis (SpA) and inflammatory bowel disease (IBD). For example, it has been consistently shown that agents targeting tumour necrosis factor (TNF) are effective and superior to placebo in treating IMIDs [1,2,3,4]. However, some patients may experience a loss of treatment efficacy over time. One of the primary reasons for this phenomenon is the development of antidrug antibodies (ADAs), which have been noted for all TNF inhibitors to varying degrees [5]. ADAs that inhibit the activity of the drug by binding to or near the active site are referred to as neutralising antibodies (NAbs), and it is thought that the majority (>90%) of ADAs generated against anti-TNF antibodies (adalimumab, certolizumab, golimumab and infliximab) are of this type [6]. Non-neutralising ADAs are also thought to contribute to a lack or loss of response by forming immune complexes with the drug, thereby reducing bioavailability [7]. There are safety concerns surrounding the development of ADAs, as they have been associated with an increased incidence of infusion-related reactions, injection site reactions, and hypersensitivity [5, 8]. The concomitant use of immunosuppressants such as methotrexate or azathioprine/mercaptopurine may delay or prevent the formation of ADAs [5, 9].
The European Medicines Agency (EMA) and the US Food and Drug Administration (FDA) have issued guidance on the assessment of immunogenicity of biological agents [10, 11]. Both stress the importance of developing valid and sensitive assays that are optimised for the ADAs being measured. The approval of biosimilars for the treatment of IMIDs has further highlighted the importance of using appropriate immunoassays for ADA testing. Differences in the manufacturing processes for biosimilars mean that they are never identical to their originator biological agents, or reference products (RPs), and they therefore may have different immunogenicity profiles. Thus, a comparative immunogenicity assessment of a biosimilar and its RP is a necessary step in the biosimilar approval process [12, 13]. A one-or two-assay approach may be taken for this assessment. In a one-assay approach, an assay tagged with the biosimilar is used to detect ADAs against the biosimilar and RP. Although information on the immunogenicity of the RP may be lost, this method minimises the potential impact of assay bias and ensures that all antibodies developed against the biosimilar are detected [10]. In a two-assay approach, an assay tagged with the RP is used to detect RP ADAs and a separate assay tagged with the biosimilar is used to detect biosimilar ADAs. This provides a comprehensive analysis reflecting the true immunogenicity for both drugs, but requires extensive validation as well as more resources and time [14].
CT-P13 (Remsima®; Inflectra®) was the first biosimilar of infliximab RP (Remicade®) to be approved by the EMA and the FDA. In the pivotal randomised controlled trials that led to its approval (PLANETAS and PLANETRA), ADAs and NAbs were assessed using electrochemiluminescence (ECL) and automated Gyros™ immunoassays, respectively. Each assay used EU-approved Remicade as the ‘tag’ to capture any antibodies present in the sera of CT-P13- or RP-treated patients [15,16,17,18]. In addition, in the PLANETRA study, ADA and NAb detection assays with CT-P13 as the tag were used in parallel with the Remicade-tagged assays. Comparison of immunogenicity results up to week 30 of PLANETRA indicated analytical agreement between the two assays. This suggests that RP ADAs have the ability to recognise and bind to similar antigenic sites on CT-P13, i.e. antibodies against the RP are cross-reactive with the biosimilar, and vice versa [17]. ADAs were detected in a proportion of patients throughout these trials [16, 18]. A higher proportion of patients with RA developed ADAs compared with patients with ankylosing spondylitis (AS). However, these proportions were comparable between those treated with CT-P13 and RP in each patient population [16, 18]. Furthermore, in the majority of patients (80–90%) in whom ADAs developed, these antibodies persisted to the end of the entire study period [19, 20].
The aim of the current study was to fully evaluate the comparability of Remicade- and CT-P13-tagged immunoassays for the detection of ADAs and NAbs by analysing all available data from the pivotal clinical studies of CT-P13, and to thus assess the cross-reactivity of ADAs against both CT-P13 and its RP.
2 Methods
2.1 Immunoassay Validation
The immunoassays used in the PLANETAS and PLANETRA trials were fully validated in accordance with criteria defined in the regulatory guidance and industry recommendations [11, 21]. To demonstrate that CT-P13 and infliximab RP were comparable in the immunoassays, a cross-inhibition test was performed. Here, immunocompetition controls [high positive control (HPC), 1000 ng/mL; low positive control (LPC), 150 ng/mL; and pooled negative control (PNC)] were analysed with and without spikes of CT-P13 (50 μg) or infliximab RP (50 μg), using CT-P13- or Remicade-tagged reagents.
2.2 Patient Population
Patient disease characteristics, interventions and study visits have previously been described in detail [15,16,17,18]. Briefly, PLANETAS included patients aged 18–75 years diagnosed with AS according to the 1984 modified New York classification criteria [22] for at least 3 months prior to screening for eligibility for the trial, with a Bath Ankylosing Spondylitis Disease Activity Index score of ≥4 and Visual Analogue Score for spinal pain of ≥4. PLANETRA included patients aged 18–75 years diagnosed with RA according to the revised 1987 American College of Rheumatology classification criteria [23] for at least 12 months prior to screening who had not responded adequately to methotrexate for at least 3 months. Patients were randomised 1:1 to receive CT-P13 or infliximab RP at a dose of 5 mg/kg (PLANETAS) or 3 mg/kg (PLANETRA) via 2-h intravenous infusion at weeks 0, 2 and 6 and every 8 weeks thereafter, up to week 54. Patients in PLANETRA received concomitant methotrexate and folic acid. Patients in both studies were permitted to receive low-dose oral glucocorticoid and nonsteroidal anti-inflammatory drugs.
2.3 Immunogenicity Evaluation
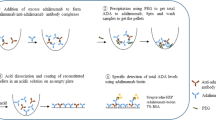
Samples for immunogenicity testing were drawn prior to drug administration to avoid potential drug interference. Sera from patients enrolled in PLANETAS/PLANETRA were analysed for levels of ADAs at the time of screening (before administration of study drug), at weeks 14, 30 and 54 and at the end of study (EOS) visit at week 62. Therefore, the maximum number of samples analysed per patient was five. The level of ADAs was assessed in 5% patient serum using a validated ECL bridging immunoassay tagged with either EU-approved Remicade (assay ADA-A) or CT-P13 (assay ADA-B). This involved a screening assay that categorised samples as positive or negative depending on the presence or absence of ADAs, followed by a confirmatory competitive/inhibition assay (Fig. 1). Relative sensitivity of the assay was 75 ng/mL in 100% human serum, using either rabbit anti-CT-P13 or anti-Remicade antibodies as surrogate positive controls.

Schematic of the bridging ECL immunoassay used to detect ADAs. In the screening assay, serum samples that potentially contain ADAs are acidified and then neutralised with labelled drug (biotinylated and SULFO-TAG™-labelled Remicade in assay ADA-A and biotinylated and SULFO-TAG™-labelled CT-P13 in assay ADA-B). ADAs present in the sample form immune complexes with the labelled drug. Samples are then loaded onto a streptavidin-coated electrode plate. During incubation, the biotin-containing antibody bridge binds to streptavidin on the plate. A reaction buffer is added, and SULFO-TAG™ labels near the electrode are electrochemically stimulated to emit light. The amount of luminescence is proportional to the amount of ADA in the sample. In the confirmatory assay, unlabelled drug is incubated with the sample and labelled drug. Unlabelled drug competes with the labelled drug to bind to ADAs in the sample. A reduction in the luminescence signal occurs if ADAs are present, thereby confirming the specificity of ADA and the positivity of the sample. Adapted from [30]. ADA antidrug antibody, ECL electrochemiluminescence, RP reference product
The neutralising activity of detected ADAs was assessed using a validated automated microfluidic Gyros™ immunoassay (Gyros AB, Uppsala, Sweden) tagged with either EU-approved Remicade (assay NAb-A) or CT-P13 (assay NAb-B). The assay is described in detail in Fig. 2. Relative sensitivity of the assay in 100% human serum, using rabbit anti-CT-P13 and rabbit anti-Remicade antibodies as surrogate positive controls, was 250 ng/mL and 631 ng/mL, respectively.

Schematic of automated Gyros™ immunoassay used to detect NAbs. The automated microfluidic Gyros immunoassay utilises capillary action and centrifugal force to load TNF-specific biotinylated capture antibodies and streptavidin-coated beads onto miniaturised affinity columns. Full-length recombinant TNF proteins are then added to the columns. Samples that have been pre-incubated with Alexa fluorophore (fluorescence)-labelled drug (EU-approved Remicade in assay NAb-A and CT-P13 in assay NAb-B) are added to the columns. If the sample contains no NAbs, the Alexa-labelled drug binds to the immobilised TNF, is retained during subsequent wash steps, and generates a fluorescent signal. In samples containing RP or CT-P13 NAbs, the NAb binds to the Alexa-labelled drug during pre-incubation, thereby preventing it from binding to the TNF proteins in the column, resulting in a reduction in the fluorescence signal. The greater the reduction in fluorescence, the greater the amount of NAb in the sample. Adapted from [31]. EU European Union, NAb neutralising antibody, RP reference product, TNF tumour necrosis factor
Assay ADA-B and assay NAb-B replicated the original assays (assay ADA-A and assay NAb-A, respectively) in all respects, with the exception of the CT-P13 tag in place of the Remicade tag. Assay ADA-B and assay NAb-B were applied to verify the original results for the presence of ADAs and NAbs up to week 54/EOS. NAb titre data were also compared up to week 30.
2.4 Statistical Analysis
The analyses were conducted in the safety populations of PLANETAS and PLANETRA, which included all patients who received at least one full or partial dose of study treatment during any dosing period.
2.4.1 Assay Concordance
In order to assess the concordance of results obtained from assay ADA-A versus assay ADA-B and assay NAb-A versus assay NAb-B, an inter-rater agreement analysis was performed based on Cohen’s κ coefficient up to the EOS visit and per treatment group. The 95% confidence interval (CI) and p value were calculated for κ, with p < 0.05 indicating a statistically significant agreement between the outcomes obtained from the differently tagged assays.
The comparison of NAb titre results was performed at screening, week 14 and week 30. Statistical analysis was performed using Spearman’s rank correlation coefficient (ρ) based on actual titre values.
2.4.2 Cross-reactivity of ADA/NAbs
To assess the cross-reactivity of RP ADAs and NAbs, the proportion of positive samples (assessed with assay ADA-A and assay NAb-A, respectively) from RP-treated patients that also gave a positive result using the CT-P13-tagged immunoassays was determined. Similarly, to assess the cross-reactivity of CT-P13 ADAs and NAbs, the proportion of positive samples (assessed with assay ADA-B and assay NAb-B, respectively) from CT-P13-treated patients that were also positive using the Remicade-tagged immunoassays was calculated.
3 Results
3.1 Cross-inhibition Test
The cross-inhibition test assesses the ability of the study drug (CT-P13 or RP) to compete with CT-P13- and Remicade-tagged reagents and bind to immunocompetition controls. This is determined by calculating the reduction in signal generated by CT-P13 and RP in the assay compared with that produced with buffer (i.e. no study drug) using ADA controls of varying concentrations. Results from the cross-inhibition test demonstrate that CT-P13 and RP were comparable in their ability to inhibit the signal across all controls (Table 1).
3.2 PLANETAS
The PLANETAS safety population (all patients who received at least one full or partial dose of study treatment during any dosing period) included all 250 patients with AS who were randomised to treatment in the study (n = 128 and n = 122 in the CT-P13 and RP treatment groups, respectively). The mean [standard deviation (SD)] total number of infusions received up to and including week 54 was 8.4 (1.7) and 8.5 (1.5) in the CT-P13 and RP groups, respectively. The mean (SD) total dose administered up to and including week 54 was also similar between treatment groups [3186.7 (969.1) mg and 3258.0 (861.5) mg, respectively]. Baseline patient characteristics in the safety population are shown in online resource 1 (see the electronic supplementary material).
3.2.1 Cross-reactivity of ADAs and NAbs
Of the 101 post-screening serum samples that tested positive for RP ADA in the RP-treated group, 94 (93.1%) were cross-reactive with CT-P13. A similar proportion of RP NAb-positive samples [93/100 (93.0%)] cross-reacted with the biosimilar (Fig. 3a). Almost all sera negative for RP ADA [345/346 (99.7%)] and NAb [345/347 (99.4%)] were also negative for CT-P13 ADA and NAb, respectively.

All positive and cross-reactive ADA/NAb samples from a PLANETAS, b PLANETRA and c pooled analysis of PLANETAS and PLANETRA. *Number of samples found to be positive for ADAs/NAbs using the assay tagged with the treatment drug, i.e. assay ADA-A/assay NAb-A in RP-treated patients and assay ADA-B/assay NAb-B in CT-P13-treated patients. **Number of samples found to be positive using both CT-P13- and Remicade-tagged assays. ADA antidrug antibody, NAb neutralising antibody, RP reference product
In the CT-P13 group, 104 samples tested positive for CT-P13 ADA. Of these, 103 (99.0%) were cross-reactive with RP. Similarly, 98 of the 100 samples (98.0%) containing CT-P13 NAb were also found to cross-react with RP (Fig. 3a). Of the 365 samples that were negative for CT-P13 ADA, 356 (97.5%) were negative for RP ADA. A similar proportion of CT-P13 NAb-negative samples were negative for RP NAb [356/367 (97.0%)]. The number of patients that developed cross-reactive ADAs and NAbs per study week is presented in Table 2.
3.2.2 Agreement of ADA Detection Assays
There was significant agreement in ADA detection between assay ADA-A and assay ADA-B at all post-screening visits [Cohen’s κ (95% CI): week 14 (n = 239), 0.98 (0.93–1.00); week 30 (n = 228), 0.95 (0.91–1.00); week 54 (n = 214), 0.97 (0.94–1.00); EOS (n = 235), 0.89 (0.83–0.95); all p < 0.001], with similar results observed for both treatment arms (Fig. 4a; online resource 2). Agreement of at least 86% was observed for both the positive and negative ADA subgroups at these time points (Table 3; online resource 2). Weaker agreement was demonstrated for samples taken at screening.

Agreement of immunogenicity testing between Remicade- and CT-P13-tagged immunoassays for the detection of a ADAs and b NAbs in PLANETAS. *Assay ADA-A = ADA detection immunoassay with EU-approved Remicade tag; assay ADA-B = ADA detection immunoassay with CT-P13 tag. **Assay NAb-A = NAb detection immunoassay with EU-approved Remicade tag; assay NAb-B = NAb detection immunoassay with CT-P13 tag. ADA antidrug antibody, CI confidence interval, EOS end of study, EU European Union, NAb neutralising antibody, NE not estimable, RP reference product
3.2.3 Agreement of NAb Detection Assays
The NAb detection assays showed strong agreement for the presence of NAb at all post-screening time points [Cohen’s κ (95% CI): week 14 (n = 239), 0.98 (0.93–1.00); week 30 (n = 228), 0.94 (0.89–0.99); week 54 (n = 214), 0.97 (0.94–1.00); EOS (n = 235), 0.86 (0.79–0.93); all p < 0.001]. Comparable results were observed for patients treated with CT-P13 and RP (Fig. 4b; online resource 3). Agreement between assays ranged from 81.40–100.00 for the positive and negative NAb subgroups at these time points (Table 3; online resource 3). Samples taken at screening demonstrated weaker agreement.
Significant concordance was observed between NAb titres in the total patient population at week 14 [Spearman’s ρ (95% CI) 0.73 (0.44–0.88), p < 0.001, n = 22] and at week 30 [0.74 (0.58–0.84), p < 0.001, n = 52]. Similar results were observed in both treatment groups [CT-P13 group: week 14, 0.82 (0.35–0.96), p = 0.004; week 30, 0.66 (0.38–0.83), p < 0.001; RP group: week 14, 0.76 (0.37–0.93), p = 0.001; week 30, 0.83 (0.64–0.92), p < 0.001]. Correlation at screening could not be assessed as there were no samples that gave a positive result for NAb using both detection assays at this time point.
3.3 PLANETRA
The safety population in PLANETRA (all patients who received at least one full or partial dose of study treatment during any dosing period) included 602 of 606 randomised patients with RA (n = 302 and n = 300 treated with CT-P13 or RP, respectively). The mean (SD) total number of doses received up to and including week 54 was 8.0 (2.1) and 7.9 (2.1) in the CT-P13 and RP groups, respectively. The mean (SD) total dose administered up to and including week 54 was also similar between treatment groups [1712.4 (608.3) mg and 1672.8 (595.1) mg, respectively]. Baseline patient characteristics are shown in online resource 4. Results for ADA and NAb detection up to week 30 using assay ADA-A and assay NAb-A, respectively, have been published previously [17].
3.3.1 Cross-reactivity of ADAs and NAbs
Of the 450 samples from patients in the RP-treated group that tested positive for RP ADA, 426 (94.7%) were cross-reactive with CT-P13. A similar proportion of RP NAb-positive samples [415/440 (94.3%)] cross-reacted with the biosimilar (Fig. 3b). Most of the RP ADA- [550/560 (98.2%)] and NAb-negative [556/568 (97.9%)] sera were also negative for CT-P13 ADAs and NAbs, respectively.
In the CT-P13 group, 474 samples tested positive for CT-P13 ADAs. Of these, 458 (96.6%) were cross-reactive with RP. Similarly, 452 of the 469 samples (96.4%) containing CT-P13 NAbs were also found to cross-react (Fig. 3b). Of the 555 samples that were negative for CT-P13 ADA, 540 (97.3%) were negative for RP ADA. A similar proportion of CT-P13 NAb-negative samples were negative for RP NAb [544/560 (97.1%)]. The number of patients that developed cross-reactive ADAs and NAbs per study week is presented in Table 2.
3.3.2 Agreement of ADA Detection Assays
There was significant and strong agreement in ADA detection between assay ADA-A and assay ADA-B at all study visits [Cohen’s κ (95% CI): screening (n = 599), 0.88 (0.76–1.00); week 14 (n = 543), 0.94 (0.91–0.97); week 30 (n = 505), 0.94 (0.92–0.97); week 54 (n = 455), 0.92 (0.88–0.95); EOS (n = 539), 0.92 (0.89–0.96); all p < 0.001], with similar results observed for both treatment arms (Fig. 5a; online resource 5). Agreement of at least 92% was observed for both the positive and negative ADA subgroups at the post-screening time points (Table 4; online resource 5).

Agreement of immunogenicity testing between Remicade- and CT-P13-tagged immunoassays for the detection of a ADAs and b NAbs in PLANETRA. *Assay ADA-A = ADA detection immunoassay with EU-approved Remicade tag; assay ADA-B = ADA detection immunoassay with CT-P13 tag. **Assay NAb-A = NAb detection immunoassay with EU-approved Remicade tag; assay NAb-B = NAb detection immunoassay with CT-P13 tag. ADA antidrug antibody, CI confidence interval, EOS end of study, EU European Union, NAb neutralising antibody, RP reference product
3.3.3 Agreement of NAb Detection Assays
The NAb detection assays showed high concordance for the presence of NAb at all post-screening time points [Cohen’s κ (95% CI): week 14 (n = 543), 0.94 (0.90–0.97); week 30 (n = 505), 0.93 (0.90–0.96); week 54 (n = 455), 0.92 (0.88–0.95); EOS (n = 539), 0.92 (0.89–0.95); all p < 0.001]. Comparable results were observed for patients treated with CT-P13 and those treated with RP (Fig. 5b; online resource 6). Agreement of at least 92% was observed for the positive and negative NAb subgroups at these time points (Table 4; online resource 6). Samples taken at screening demonstrated weaker agreement.
A statistically significant degree of concordance was observed between the NAb titre obtained using assay NAb-A and assay NAb-B in the total patient population at week 14 [Spearman’s ρ (95% CI) 0.61 (0.49–0.71), p < 0.001, n = 131] and at week 30 [0.72 (0.65–0.78), p < 0.001, n = 233]. Similar results were observed in both treatment groups [CT-P13 group: week 14, 0.64 (0.47–0.76); week 30, 0.69 (0.59–0.78); RP group: week 14, 0.59 (0.40–0.73); week 30, 0.74 (0.64–0.81); all p < 0.001]. Correlation at screening could not be determined due to small sample sizes.
3.4 Pooled PLANETAS and PLANETRA Study Data
3.4.1 Cross-reactivity of ADAs and NAbs
Of the 551 samples from patients treated with RP that tested positive for RP ADAs, 520 (94.4%) were cross-reactive with CT-P13. A similar proportion of RP NAb-positive samples [508/540 (94.1%)] cross-reacted with the biosimilar (Fig. 3c). Most of the RP ADA- [895/906 (98.8%)] and NAb-negative [901/915 (98.5%)] sera were also negative for CT-P13 ADA and NAb, respectively.
For patients treated with CT-P13, 578 samples tested positive for CT-P13 ADAs. Of these, 561 (97.1%) were cross-reactive with RP. Similarly, 550 of the 569 samples (96.7%) containing CT-P13 NAb were also found to cross-react (Fig. 3c). Of the 920 samples that were negative for CT-P13 ADA, 896 (97.4%) were negative for RP ADA. A similar proportion of CT-P13 NAb-negative samples were negative for RP NAb [900/927 (97.1%)].
3.4.2 Agreement of ADA Detection Assays
There was significant and strong agreement in ADA detection between assay ADA-A and assay ADA-B at all study visits [Cohen’s κ (95% CI): screening (n = 846), 0.83 (0.70–0.95); week 14 (n = 782), 0.95 (0.92–0.98); week 30 (n = 733), 0.95 (0.93–0.97); week 54 (n = 669), 0.94 (0.91–0.96); EOS (n = 774), 0.92 (0.89–0.95); all p < 0.001], with similar results observed for both treatment arms (all p < 0.001; Fig. 6a; online resource 7). Agreement of at least 92% was observed for both the positive and negative ADA subgroups at the post-screening time points (Table 5; online resource 7).

Agreement of immunogenicity testing between Remicade- and CT-P13-tagged immunoassays for the detection of a ADAs and b NAbs in PLANETAS and PLANETRA pooled analysis. *Assay ADA-A = ADA detection immunoassay with EU-approved Remicade tag; assay ADA-B = ADA detection immunoassay with CT-P13 tag. **Assay NAb-A = NAb detection immunoassay with EU-approved Remicade tag; assay NAb-B = NAb detection immunoassay with CT-P13 tag. ADA antidrug antibody, CI confidence interval, EOS end of study, EU European Union, NAb neutralising antibody, RP reference product
3.4.3 Agreement of NAb Detection Assays
The NAb detection assays showed strong agreement for the presence of NAb at all post-screening time points [Cohen’s κ (95% CI): week 14 (n = 782), 0.94 (0.92–0.97); week 30 (n = 733), 0.94 (0.91–0.96); week 54 (n = 669), 0.94 (0.91–0.96); EOS (n = 774), 0.91 (0.88–0.94); all p < 0.001]. Weaker agreement was demonstrated for samples taken at sampling [0.46 (0.12–0.80), p < 0.001]. Comparable results were observed for patients treated with CT-P13 and with RP at all post-screening time points (all p < 0.001; Fig. 6b; online resource 8). Agreement between assays ranged from 92.42 to 98.72 for the positive and negative NAb subgroups at the post-screening time points (Table 5; online resource 8).
Significant concordance was observed between NAb titres in the total patient population at week 14 [Spearman’s ρ (95% CI) 0.63 (0.52–0.72), p < 0.001, n = 153] and at week 30 [0.72 (0.66–0.77), p < 0.001, n = 285]. Similar results were observed in CT-P13 and RP treatment groups at week 14 [0.68 (0.53–0.78) vs. 0.59 (0.42–0.72), respectively, both p < 0.001] and week 30 [0.70 (0.60–0.77) vs. 0.74 (0.65–0.80), respectively, both p < 0.001]. Correlation at screening could not be determined due to small sample sizes.
4 Discussion
The pivotal PLANETAS and PLANETRA studies demonstrated that CT-P13 is equivalent to RP in terms of efficacy and pharmacokinetics (PK) and has comparable pharmacodynamics, safety and immunogenicity [15,16,17,18,19,20]. The immunoassays used during the main comparative immunogenicity testing of CT-P13 versus RP utilised EU-approved Remicade as the tag to detect ADAs to both RP and CT-P13. When a one-assay approach is used, the regulatory view is that the biosimilar should be used as both the capture and detector tag, as this ensures optimal detection of biosimilar ADA, making it the more conservative approach [10]. However, this study has clearly demonstrated that there is little difference between the Remicade-tagged and CT-P13-tagged immunoassays used for ADA and NAb detection, with similar results obtained from both assays at all post-screening time points. This was true for both treatment groups, suggesting a lack of bias in either CT-P13 or RP antibody detection. The use of Remicade-tagged assays for samples from both CT-P13- and RP-treated patients in the PLANETAS and PLANETRA studies was therefore not inappropriate. Furthermore, a subsequent comparative review of ADA assays revealed that the ECL immunoassay tagged with EU-approved Remicade used in the trials was sensitive and reliable for the detection of CT-P13 ADAs [24].
Weaker agreement between assays was observed for samples taken at screening in both treatment groups in both studies. This finding may be due to the over-sensitivity of the Cohen’s κ coefficient with the extremely low numbers of patients who were initially positive for ADAs or NAbs in these studies. As patients in PLANETAS and PLANETRA were naïve to biological treatment prior to study commencement, the positive results obtained from screening samples were not a result of the presence of ADAs or NAbs, but reflect background noise due to non-specific binding of molecules that can occur in some subjects. As ADA or NAb status at screening is not related to treatment with RP or CT-P13 during the trial, little emphasis should be placed on the concordance of data at this time point, with data from weeks 14 to 54 providing a more relevant assessment of immunogenicity of the drugs. Results of the statistical analysis support significant concordance for all treatment groups at all study visits after infusion of the test product. A limitation to the present study is that the concordance of ADA titres between immunoassays was not analysed. Furthermore, information on the kinetics of development of ADAs and NAbs has not been presented as this was outside the scope of the current study, which aimed to evaluate the concordance of results obtained from immunoassays using differently tagged reagents.
The present study provides further support for the cross-reactivity of RP ADAs in patients with rheumatic diseases, as recently reported elsewhere [25]. A comparison of three bridging enzyme-linked immunosorbent assays (ELISAs) that used Remicade, Remsima or Inflectra to detect ADAs in RP-treated patients with RA or SpA (n = 250) revealed highly comparable results between assays [25]. All patients that tested positive for RP ADAs were also found to be positive using the Remsima and Inflectra assays, giving positive and negative percentage agreements of 100% for all comparisons. Significant correlation was observed between ADA titres assessed by the three assays (ρ ≥ 0.99, p < 0.001), and there was no significant difference in bias, or between indications or concomitant immunosuppressant use. The study used a commercial ELISA for RP ADA detection (Promonitor-ANTI-IFX kit, Progenika-Grifols, Spain) and concluded that this could be used to monitor ADAs in biosimilar-treated patients [25]; however, this cannot be generalised to other biosimilars. Cross-reactivity of RP ADAs has also been demonstrated in patients with IBD treated with RP. Gils et al. [26] developed three bridging ELISAs to compare cross-reactivity of RP ADAs with Remsima and Inflectra in 36 serum samples from patients with IBD. The study showed excellent correlation of ADA titres between assays (ρ ≥ 0.975, p < 0.0001), although small but significant differences were observed between the titres obtained by the three ELISAs [26]. In another study in patients with IBD, all sera positive for RP ADAs were cross-reactive with Remsima, with strong correlation of ADA titres demonstrated for all experiments (ρ ≥ 0.92, p < 0.001) [27]. In addition, the study demonstrated that RP ADAs inhibited the TNF-binding capacity of RP and Remsima to a similar extent and found a higher background signal for Remsima in the negative controls compared with the RP [27]. The exact cause of the background signal was undetermined, but was not related to glycosylation patterns or the immunoglobulin G molecule itself. No such background signal was evident in our study or in any of the other cross-reactivity studies mentioned here [25, 26].
A limitation to the studies mentioned above is that they only demonstrate cross-reactivity of RP ADAs with CT-P13 and not the cross-reactivity of CT-P13 ADAs with RP. PLANETRA and PLANETAS offered the opportunity to address this issue, as head-to-head data from both RP- and CT-P13-treated patients were available. Here, we have shown that CT-P13 ADAs are equally cross-reactive with RP, and we have also demonstrated the cross-reactivity of NAbs against RP and CT-P13. Evidence to date has indicated that most ADAs against the RP are neutralising [6]. This study has shown that ADAs against CT-P13 were also largely neutralising and that the prevalence of ADAs and NAbs, as well as NAb titres, was similar in both treatment groups. Our data also suggest that in a small number of patients, CT-P13-induced ADAs may be directed towards epitopes not present in the originator, and that RP-treated patients may develop ADAs that are not able to recognise the biosimilar. In this circumstance, using a one-assay approach for comparative immunogenicity testing would be a disadvantage, as these differences between products would not be detected. However, as this only occurs in a small number of patients, the clinical relevance is debatable. It is likely that such differences represent stochastic effects and not a systematic difference. Furthermore, without titre information, it is not possible to determine whether these discordant assay results are due to ADAs against a novel antigenic epitope in the biosimilar, low ADA titres that are close to the assay cut point, or, alternatively, a result of background noise. One potential cause for this background signal could be the presence of naturally occurring autoantibodies against TNF that are known to exist in some healthy individuals and those with inflammatory diseases [28, 29].
Determining the impact of ADAs versus NAbs on efficacy and tolerance in PLANETAS and PLANETRA was outside the scope of this study, but is a matter of considerable interest for future research. However, the clinical impact of ADAs was assessed throughout the PLANETAS and PLANETRA studies. In PLANETRA, peak serum drug concentrations (C max) and clinical responses were lower in the ADA-positive versus ADA-negative patients at week 30 [17]. Similarly, in PLANETAS, lower C max and clinical response rates were evident in ADA-positive patients at week 54. There was also a trend for steady-state PK parameters to be lower at higher ADA titres; however, as the study was not powered for this analysis, no statistical inference was made [16]. In open-label extensions of these trials in which patients were maintained on CT-P13 or switched from RP to the biosimilar, ADA-positive patients had lower clinical responses, higher C-reactive protein levels and erythrocyte sedimentation rates, and a higher incidence of infusion-related reactions compared with ADA-negative patients. These ADA effects, however, were highly similar between treatment groups in both the main study and during the extension [19, 20].
5 Conclusions
A comprehensive evaluation of the immunogenicity data from the pivotal clinical trials of CT-P13 has demonstrated that ADAs against CT-P13 and its RP recognise and bind the RP and CT-P13, respectively, to a similar degree, indicating that the two biological drugs share immunodominant epitopes. This cross-reactivity suggests that there should be no concerns about using commercialised kits that are utilised for either CT-P13 or its RP for comparative immunogenicity assessment.
References
Aaltonen KJ, Virkki LM, Malmivaara A, Konttinen YT, Nordstrom DC, Blom M. Systematic review and meta-analysis of the efficacy and safety of existing TNF blocking agents in treatment of rheumatoid arthritis. PLoS One. 2012;7(1):e30275.
Corbett M, Soares M, Jhuti G, Rice S, Spackman E, Sideris E, et al. Tumour necrosis factor-alpha inhibitors for ankylosing spondylitis and non-radiographic axial spondyloarthritis: a systematic review and economic evaluation. Health Technol Assess. 2016;20(9):1–334.
Ford AC, Sandborn WJ, Khan KJ, Hanauer SB, Talley NJ, Moayyedi P. Efficacy of biological therapies in inflammatory bowel disease: systematic review and meta-analysis. Am J Gastroenterol. 2011;106(4):644–59.
Lemos LL, de Oliveira Costa J, Almeida AM, Junior HO, Barbosa MM, Kakehasi AM, et al. Treatment of psoriatic arthritis with anti-TNF agents: a systematic review and meta-analysis of efficacy, effectiveness and safety. Rheumatol Int. 2014;34(10):1345–60.
Thomas SS, Borazan N, Barroso N, Duan L, Taroumian S, Kretzmann B, et al. Comparative immunogenicity of TNF inhibitors: impact on clinical efficacy and tolerability in the management of autoimmune diseases. A systematic review and meta-analysis. BioDrugs. 2015;29(4):241–58.
van Schie KA, Hart MH, de Groot ER, Kruithof S, Aarden LA, Wolbink GJ, et al. The antibody response against human and chimeric anti-TNF therapeutic antibodies primarily targets the TNF binding region. Ann Rheum Dis. 2015;74(1):311–4.
Bendtzen K. Anti-TNF-α biotherapies: perspectives for evidence-based personalized medicine. Immunotherapy. 2012;4(11):1167–79.
Maneiro JR, Salgado E, Gomez-Reino JJ. Immunogenicity of monoclonal antibodies against tumor necrosis factor used in chronic immune-mediated inflammatory conditions: systematic review and meta-analysis. JAMA Intern Med. 2013;173(15):1416–28.
Garces S, Demengeot J, Benito-Garcia E. The immunogenicity of anti-TNF therapy in immune-mediated inflammatory diseases: a systematic review of the literature with a meta-analysis. Ann Rheum Dis. 2013;72(12):1947–55.
European Medicines Agency. Guideline on immunogenicity assessment of biotechnology-derived therapeutic proteins (draft). 2015. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/10/WC500194507.pdf. Accessed 21 Mar 2017.
US Food and Drug Administration. Assay development and validation for immunogenicity testing of therapeutic protein products. Guidance for industry. 2016. http://www.fda.gov/downloads/Drugs/…/Guidances/UCM192750.pdf. Accessed 21 Mar 2017.
European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. 2014. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/01/WC500180219.pdf. Accessed 21 Mar 2017.
US Food and Drug Administration. Scientific considerations in demonstrating biosimilarity to a reference product. Guidance for industry. 2015. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm291128.pdf. Accessed 21 Mar 2017.
Pineda C, Castaneda Hernandez G, Jacobs IA, Alvarez DF, Carini C. Assessing the immunogenicity of biopharmaceuticals. BioDrugs. 2016;30(3):195–206.
Park W, Hrycaj P, Jeka S, Kovalenko V, Lysenko G, Miranda P, et al. A randomised, double-blind, multicentre, parallel-group, prospective study comparing the pharmacokinetics, safety, and efficacy of CT-P13 and innovator infliximab in patients with ankylosing spondylitis: the PLANETAS study. Ann Rheum Dis. 2013;72(10):1605–12.
Park W, Yoo DH, Jaworski J, Brzezicki J, Gnylorybov A, Kadinov V, et al. Comparable long-term efficacy, as assessed by patient-reported outcomes, safety and pharmacokinetics, of CT-P13 and reference infliximab in patients with ankylosing spondylitis: 54-week results from the randomized, parallel-group PLANETAS study. Arthritis Res Ther. 2016;18:25.
Yoo DH, Hrycaj P, Miranda P, Ramiterre E, Piotrowski M, Shevchuk S, et al. A randomised, double-blind, parallel-group study to demonstrate equivalence in efficacy and safety of CT-P13 compared with innovator infliximab when coadministered with methotrexate in patients with active rheumatoid arthritis: the PLANETRA study. Ann Rheum Dis. 2013;72(10):1613–20.
Yoo DH, Racewicz A, Brzezicki J, Yatsyshyn R, Arteaga ET, Baranauskaite A, et al. A phase III randomized study to evaluate the efficacy and safety of CT-P13 compared with reference infliximab in patients with active rheumatoid arthritis: 54-week results from the PLANETRA study. Arthritis Res Ther. 2016;18:82.
Park W, Yoo DH, Miranda P, Brzosko M, Wiland P, Gutierrez-Urena S, et al. Efficacy and safety of switching from reference infliximab to CT-P13 compared with maintenance of CT-P13 in ankylosing spondylitis: 102-week data from the PLANETAS extension study. Ann Rheum Dis. 2017;76:346–54.
Yoo DH, Prodanovic N, Jaworski J, Miranda P, Ramiterre E, Lanzon A, et al. Efficacy and safety of CT-P13 (biosimilar infliximab) in patients with rheumatoid arthritis: comparison between switching from reference infliximab to CT-P13 and continuing CT-P13 in the PLANETRA extension study. Ann Rheum Dis. 2017;76:355–63.
Shankar G, Devanarayan V, Amaravadi L, Barrett YC, Bowsher R, Finco-Kent D, et al. Recommendations for the validation of immunoassays used for detection of host antibodies against biotechnology products. J Pharm Biomed Anal. 2008;48(5):1267–81.
van der Linden S, Valkenburg HA, Cats A. Evaluation of diagnostic criteria for ankylosing spondylitis. A proposal for modification of the New York criteria. Arthritis Rheum. 1984;27(4):361–8.
Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31(3):315–24.
Kim JS, Kim SH, Kwon B, Hong S. Comparison of immunogenicity test methods used in clinical studies of infliximab and its biosimilar (CT-P13). Expert Rev Clin Immunol. 2015;11(Suppl 1):S33–41.
Ruiz-Argüello MB, Maguregui A, Ruiz del Agua A, Pascual-Salcedo D, Martínez-Feito A, Jurado T, et al. Antibodies to infliximab in Remicade-treated rheumatic patients show identical reactivity towards biosimilars. Ann Rheum Dis. 2016;75:1693–6.
Gils A, Van Stappen T, Dreesen E, Storme R, Vermeire S, Declerck PJ. Harmonization of infliximab and anti-infliximab assays facilitates the comparison between originators and biosimilars in clinical samples. Inflamm Bowel Dis. 2016;22(4):969–75.
Ben-Horin S, Yavzori M, Benhar I, Fudim E, Picard O, Ungar B, et al. Cross-immunogenicity: antibodies to infliximab in Remicade-treated patients with IBD similarly recognise the biosimilar Remsima. Gut. 2016;65(7):1132–8.
Fomsgaard A, Svenson M, Bendtzen K. Auto-antibodies to tumour necrosis factor alpha in healthy humans and patients with inflammatory diseases and gram-negative bacterial infections. Scand J Immunol. 1989;30(2):219–23.
Watanabe M, Uchida K, Nakagaki K, Kanazawa H, Trapnell BC, Hoshino Y, et al. Anti-cytokine autoantibodies are ubiquitous in healthy individuals. FEBS Lett. 2007;581(10):2017–21.
Yoo DH, Suh C-H, Shim SC, Jeka S, Cons-Molina FF, Hrycaj P, et al. A multicentre randomised controlled trial to compare the pharmacokinetics, efficacy and safety of CT-P10 and innovator rituximab in patients with rheumatoid arthritis. Ann Rheum Dis. 2017;76:566–70.
CELLTRION. CT-P13 (infliximab). FDA advisory committee briefing document. 2016. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/ArthritisAdvisoryCommittee/UCM484860.pdf. Accessed 21 Mar 2017.
Author information
Authors and Affiliations
Contributions
DHY and WP were involved in the conception and design of the study, the acquisition, analysis and interpretation of data, drafting the manuscript and critically revising it for important intellectual content. DHY and WP had final responsibility for the decision to submit for publication. All authors contributed to the interpretation of the data, edited and reviewed drafts of the manuscript, and approved the final version.
Corresponding author
Ethics declarations
Funding
This work was funded by CELLTRION Inc. (Incheon, Republic of Korea). Medical writing support was provided by Joanna Chapman PhD, Alice Wareham PhD (Aspire Scientific Ltd, Bollington, UK) and SuYeon Kim (CELLTRION Healthcare Co., Ltd, Incheon, Republic of Korea) and funded by CELLTRION Healthcare Co., Ltd (Incheon, Republic of Korea).
Conflicts of interest
WR has served as a speaker, a consultant, or an advisory board member for Abbott Laboratories, Abbvie, Aesca, Amgen, AM Pharma, Aptalis, Astellas, AstraZeneca, Avaxia, Bioclinica, Biogen IDEC, Boehringer Ingelheim, Bristol-Myers Squibb, Celgene, Cellerix, CELLTRION, Centocor, ChemoCentryx, Covance, Danone Austria, Elan, Ernest & Young, Falk Pharma GmbH, Ferring, Galapagos, Genentech, Gilead, Grünenthal, ICON, Immundiagnostik, Index Pharma, Inova, Janssen, Johnson & Johnson, Kyowa Hakko Kirin Pharma, Lipid Therapeutics, Mallinckrodt, MedImmune, Millennium, Mitsubishi Tanabe Pharma Corporation, MSD, Nestle, Novartis, Ocera, Otsuka, PDL, Pfizer, Pharmacosmos, PLS Education, Procter & Gamble, Prometheus, Robarts Clinical Trial, Roland Berger GmbH, Schering-Plough, Second Genome, SetPoint Medical, Shire, Sigmoid, Takeda, Therakos, TiGenix, UCB Pharma, Vifor, Yakult, Zyngenia and 4SC, and has received research funding from Abbott Laboratories, Abbvie, Aesca, Centocor, Falk Pharma GmbH, Immundiagnostik, and MSD. JJ has served as a speaker, a consultant, or an advisory board member for Abbvie, Astro, CELLTRION, Hikma, Janssen, MSD, Mundipharma, Napp, Orion, Pfizer, Sandoz and Takeda. SS has received consulting fees from Abbvie, Biogen, CELLTRION, Janssen, MSD and Pfizer, and lecture fees from Abbvie, Biogen, CELLTRION, Mundipharma, Pfizer and UCB Pharma. SD has served as a speaker, a consultant, and an advisory board member for Abbvie, Allergan, Biogen, Boehringer Ingelheim, Celgene, CELLTRION, Ferring, Hospira, Janssen, Johnson & Johnson, Merck, MSD, Mundipharma, Pfizer, Sandoz, Takeda, TiGenix, UCB Pharma and Vifor. JP has received grant support from Abbvie, consulting fees from Abbvie, Boehringer Ingelheim, Celgene, CELLTRION, GSK, Janssen, MSD, Novartis, Pfizer, Roche, Second Genome, Takeda, TiGenix and TopiVert, and speaker fees from Abbvie, Biogen, Ferring, Janssen, MSD and Pfizer. AB has received consulting fees and/or non-restricted research grants from Abbvie, Biogen, BMS, CELLTRION, MSD, Novartis, Pfizer, Roche and UCB Pharma. WP has received consulting fees from CELLTRION and support for manuscript preparation. JSK is an employee of CELLTRION Inc. (Incheon, Republic of Korea). JUL is an employee of CELLTRION Healthcare Co., Ltd (Incheon, Republic of Korea). DHY is a scientific consultant for CELLTRION and has received a grant unrelated to this study.
Ethics approval and consent to participate
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all individual participants included in the study (please see [16, 18] for details). This article does not contain any studies with animals performed by any of the authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Reinisch, W., Jahnsen, J., Schreiber, S. et al. Evaluation of the Cross-reactivity of Antidrug Antibodies to CT-P13 and Infliximab Reference Product (Remicade): An Analysis Using Immunoassays Tagged with Both Agents. BioDrugs 31, 223–237 (2017). https://doi.org/10.1007/s40259-017-0219-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40259-017-0219-4