
Abstract
Biologic therapies have revolutionized treatment of a number of diseases. Patents and exclusivity for a number of biologics are expiring. This has created the opportunity for the development and approval of biosimilars. Biosimilars are biologic products developed using a step-wise approach to result in a biologic that demonstrates no clinically meaningful differences in terms of quality attributes, efficacy, safety, and immunogenicity compared with an existing licensed, originator biologic. As more biosimilars receive regulatory approval and reach the market, it is increasingly important for healthcare providers to understand the terminology about biosimilars. To help support healthcare providers, the aim of this manuscript is to (i) support understanding of the language of biosimilars, (ii) review the regulatory and manufacturing processes employed in developing a biosimilar, and (iii) provide information for clinical decisions about the use of biosimilars. Because biologics are large, structurally complex proteins, biosimilars cannot be considered generic equivalents to the originator. Biosimilars are developed and evaluated using rigorous processes involving detailed analytical and functional studies, nonclinical assessments, and clinical trials. Clinical studies evaluating the potential biosimilar are designed differently than those for approval of a novel biologic since the aim is merely to confirm similar efficacy and safety and not to demonstrate clinical benefit per se. Extrapolation of data may be used to grant approval of biosimilars in indications not directly evaluated in clinical studies using the biosimilar.
Similar content being viewed by others

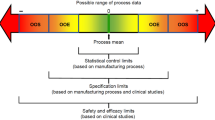

The potential exists for more biosimilars to become available over the next few years; therefore, it is important for healthcare providers to understand the terminology used to describe development and approval of biosimilars. |
Biosimilars undergo a rigorous evaluation using the criteria defined in the EMA, FDA, or WHO biosimilar guidelines before regulatory approval. |
Biosimilar approval is based on the totality of data demonstrating similarity between the biosimilar and the originator, including in terms of quality characteristics, biological activity, safety, and efficacy. |
1 Introduction
Biologic therapies have revolutionized the treatment of a variety of conditions, including cancers and autoimmune diseases. Patents and other periods of exclusivity for a number of biologics are nearing expiration or have already expired, leading to the development and approval of products (called ‘biosimilars’) that are similar to licensed biologic products [1]. The European Medicines Agency (EMA) has approved over 20 biosimilars since the development of specific guidelines and recommendations for evaluation of these agents [2, 3]. In 2015, the United States Food and Drug Administration (FDA) approved its first biosimilar (a biosimilar to filgrastim) under the Biologics Price Competition and Innovation Act of 2009 [4].
As a number of the highly utilized biologics will lose patent exclusivity by 2020, the potential exists for even more biosimilars to become available over the next few years [5]. As more biosimilars are approved and prescribed, it is increasingly important for healthcare providers to understand the terminology about biosimilars. To address this need, the aim of this manuscript is to (i) support understanding of the language of biosimilars, (ii) review the regulatory and manufacturing processes employed in developing a quality biosimilar, and (iii) provide information for clinical decisions about the use of biosimilars.
2 Defining a Biosimilar
Biosimilars are different from both originator biologic products and generic small molecule drugs in terms of their development and regulatory approval [1, 2]. As a result, there is a unique lexicon of terms used to describe the development, evaluation, and/or approval of biosimilars (Table 1). A biosimilar is a biological product that is approved based on the totality of evidence demonstrating that it is highly similar to an approved biological product (originator) in terms of structure, function, quality, and clinical efficacy and safety [1, 2, 6]. Biosimilars are developed such that there are “no clinically meaningful differences between the biological product and the reference [originator] product in terms of safety, purity, and potency” [6]. The EMA definition further clarifies that “a biosimilar demonstrates similarity to the [originator] in terms of quality characteristics, biological activity, safety, and efficacy based on a comprehensive comparability exercise” [2]. Unlike small molecule (chemical) drugs that can be fully defined structurally so generic versions can be produced, biologics are large, structurally complex products isolated from natural sources (including proteins, nucleic acids, or combinations of these, or living entities such as cells and tissues) and subject to post-translational structural modifications that lead to an intrinsic heterogeneity [1, 2, 7]. Thus, biosimilars cannot be considered generic equivalents to the originator and require additional characterization to confirm comparable clinical efficacy and safety [1, 2, 7].
The development and approval of a biosimilar is different from the process used for a new molecular entity. For one thing, the analyses and clinical trials for potential biosimilars compare the physicochemical and biological properties and short-term efficacy and safety to the originator; they do not re-establish the mechanism of action or proof of concept [1, 2]. In addition, because the manufacturing process for the originator is proprietary, the biosimilar developer must analyze the originator extensively and use reverse engineering to develop the biosimilar [8]. As described earlier, biologics are relatively large, complex proteins that are difficult to characterize, so regulatory processes for biosimilar approval are also not the same as those used for small-molecule generics (which usually just requires demonstration of bioequivalence of the generic medicine to the licensed originator small-molecule drug) [1].
3 Development and Regulatory Approval of Biosimilars
Because biosimilars have many specific and unique considerations related to regulatory approval, specific guidelines have been developed by the EMA, the FDA, and the World Health Organization (WHO) [1, 2, 6]. There are minor differences among guidelines, but all suggest following a step-wise approach to demonstrate biosimilarity with the originator [1, 2, 6].
Biosimilars are developed and evaluated in rigorous processes involving extensive analytical and functional studies; limited nonclinical assessments of pharmacokinetics (PK) and toxicity; and limited clinical evaluation of PK, efficacy, and safety [1, 2, 6]. Data supporting the demonstration of biosimilarity to the originator are based on a foundation of extensive analytical studies to compare the structural, physicochemical, and functional characteristics of the potential biosimilar and originator [1, 2, 6, 8, 9]. Minor differences in structure (such as glycosylation variants) may occur, yet the product may be considered a biosimilar as long as the structural differences are expected not to have a clinically meaningful impact on efficacy and/or safety [6]. After confirming a high degree of physicochemical and functional similarity, nonclinical studies may be conducted to demonstrate that the potential biosimilar has similar PK and toxicity to the originator [1, 2, 6, 8]. It should be noted that a paradigm shift has occurred in Europe in which the decision to conduct in vivo nonclinical studies is made as part of the step-wise approach and these studies are not performed by default [10]. The US guidelines also suggest a step-wise approach, although they do not specifically state that approval can be granted without nonclinical in vivo studies and have required at least one nonclinical in vivo study to date [11]. Finally, while the size and scope of the clinical program depends on data generated in the comparative analytical and nonclinical studies, the guidelines recommend including at least one human PK study and generally a minimum of one efficacy and safety study evaluating biosimilarity to the originator [1, 2, 6, 8].
Compared with the approval pathway for a novel biologic, the biosimilar approval pathway places more emphasis on data from comparative analytical studies and less on those from clinical trials. For example, biosimilars do not require studies to evaluate mechanism of action, determine optimal dosing, or demonstrate patient benefit because these were established by the originator [1, 2]. Instead, clinical studies evaluating a potential biosimilar serve to confirm similar efficacy and safety (including potency, PK, pharmacodynamics [PD], and immunogenicity) with respect to the originator [1, 2, 6].
4 Study Design
Clinical studies of potential biosimilars are conducted using a study population sensitive to detecting potential differences in efficacy, safety, or immunogenicity between the biosimilar and originator [1, 2, 6]. Thus, this population may include a different patient population than that employed in pivotal clinical trials of the originator [1, 2, 6]. In addition, the primary endpoints measured may or may not be the same as those used in those pivotal clinical trials [1, 2, 6]. Endpoints such as PD measures (a biomarker linked to efficacy so that it is considered an accepted surrogate; e.g., absolute neutrophil count and CD34+ cell count are PD measures for granulocyte colony stimulating factor) are considered sensitive clinical endpoints for evaluating biosimilarity and may be selected as the primary endpoints of a biosimilar clinical study to facilitate more precise efficacy comparisons to the originator [1, 2, 6]. Regulatory guidelines also recommend including some endpoints commonly used in the pivotal trials of the originator as secondary endpoints to enable comparisons across products [1, 2, 6].
Originator biologics were approved based upon the demonstration of clinically meaningful efficacy and safety versus a placebo, current therapeutic options, or current standard of care in clinical trials [12]. For regulatory approval, the biosimilar is expected to exhibit similar efficacy and safety profiles compared with the originator in clinical studies [1, 2, 6]. Regulatory guidelines recommend an equivalence trial since this should allow testing of the hypothesis that the treatments (the biosimilar and originator) result in no clinically meaningful differences [1, 6, 12, 13]. The exact nature of statistical analyses for biosimilars depends on the trial design employed [1, 12].
5 Extrapolation
One of the core concepts in the development and approval of biosimilars is the concept of extrapolation, which allows for the approval of a biosimilar for use in an indication held by the originator that was not directly studied in clinical trials using the biosimilar [1, 2, 6]. One misconception about extrapolation is that the decision to allow extrapolation of indications is based only on clinical data when, in fact, the decision is based on the totality of the evidence, including the structural, physicochemical, functional, and nonclinical data in addition to clinical evaluations, all of which must support similarity of the biosimilar to the originator [8]. Because of the possibility of extrapolation, the biosimilar may not need to be evaluated in clinical studies for each indication [1, 6]. This means that the entire clinical program of the originator does not have to be duplicated for the potential biosimilar. Thus, the number of clinical studies required for approval of the biosimilar is reduced compared with the number of studies conducted for approval of the originator [8].
To support extrapolation to other indications, sufficient data are necessary to provide scientific justification [1, 6, 14]. To be considered supportive of extrapolation, data should be based on studies using a sensitive clinical model to detect potential differences between the originator and the potential biosimilar [1, 6, 14]. The indications also should have the same molecular mechanism of action, involve the same receptors, have a similar binding dose-response and pattern of molecular signaling upon target binding, and have similar location and expression of the target [1, 6, 14]. The totality of the data should also include well characterized PK and biodistribution information as well as sufficient characterization of safety and immunogenicity to indicate that the potential biosimilar does not have unique or additional safety issues versus the originator [1, 6, 14].
Because the decision to allow extrapolation of data to indications is made on an agency-by-agency basis, not all regulatory agencies may come to the same conclusion about a given product [8]. This is demonstrated by the case of the recent approvals of biosimilars to infliximab, for which some regulatory agencies granted approval for the full range of indications of the originator whereas other regulatory agencies did not [8]. Health Canada initially determined that the data provided for the regulatory approval of infliximab biosimilars manufactured by Celltrion Healthcare Co. Ltd (Yeonsu-gu, Incheon, Republic of Korea) and distributed by Hospira Healthcare Corporation (Kirkland, Quebec, Canada) were indicative of structural and functional differences that were potentially clinically relevant and therefore excluded extrapolation to Crohn’s disease and ulcerative colitis [8, 15, 16]. Specifically, differences in antibody-dependent cell-mediated cytotoxicity (ADCC) assays suggested the biosimilars may differ from the originator in the ability to induce ADCC [15, 16]. Since ADCC may be involved in inflammatory bowel diseases such as Crohn’s disease and ulcerative colitis, extrapolation to these therapeutic indications was initially not recommended for the two biosimilars of infliximab [15, 16]. More recently, Health Canada approved both biosimilars for Crohn’s disease, fistulizing Crohn’s disease, and ulcerative colitis based on previously submitted clinical data that formed the basis for the initial approval (for rheumatoid arthritis, ankylosing spondylitis, psoriatic arthritis, and plaque psoriasis), newly submitted physicochemical and biological data (including non-comparative, observational clinical data in patients with inflammatory bowel diseases), and scientific rationales with respect to the molecular mechanism of action and approved indications for the originator [17, 18].
6 Interchangeability and Substitution
The review process and designation of interchangeability requires additional standards [19]. The designation of interchangeability may be granted after a biosimilar has been approved; interchangeability is not automatically granted upon regulatory approval of a biosimilar [19, 20]. The EMA does not regulate whether a biosimilar is considered interchangeable with the originator because interchangeability is regulated on the national level [20, 21]. Similarly, the WHO does not elaborate on interchangeability and substitution of biosimilars in their guidance documents [1]. Thus, the FDA is the only regulatory agency with a statutory definition of interchangeability and the authority to approve biosimilars that are interchangeable with the originator [20, 22]. Draft guidance on interchangeability was issued by the FDA in January 2017 [19]. These guidelines state that for a biosimilar to be considered interchangeable, it “is biosimilar to the reference product” and “can be expected to produce the same clinical result as the reference product in any given patient” and that “for a biological product that is administered more than once to an individual, the risk in terms of safety or diminished efficacy of alternating or switching between use of the biological product and the reference product is not greater than the risk of using the reference product without such alternation or switch” [19]. The designation of a biosimilar as interchangeable means “the biological product may be substituted for the reference product without the intervention of the healthcare provider who prescribed the reference product” [19].
One important consideration in the use of biosimilars is that multiple (independently developed) biosimilars could receive the designation of interchangeable with the originator. Since interchangeability is determined for a product versus the originator, it is unlikely that biosimilars will be compared to each other in formal head-to-head comparisons. There are some published comparisons of multiple biosimilars based on indirect evaluation of their properties. For example, a comparison of the properties of two epoetin biosimilars (HX575 [epoetin alfa] and SB309 [epoetin zeta]) indicated there were some differences in PK and dosing even though both were independently approved as biosimilars to the same originator epoetin by the EMA [24]. Specifically, for HX575, there were some PK differences versus the originator product epoetin alfa (Janssen-Cilag, New York, NY, USA)—such as a lower area under the curve (AUC)0–12h for HX575 versus epoetin alfa of 18% after a single dose; a steady state AUC0–36h that was approximately 10% lower; and a 10% reduction in exposure—although HX575 and the originator epoetin alfa were considered pharmacokinetically equivalent following multiple IV administrations [24]. Similarly, a crossover study comparing SB309 to epoetin alfa required modifications in dosing (switching from epoetin alfa to SB309 increased the dose required by approximately 10–15% and switching from SB309 to epoetin alfa reduced the dose required by around 10%), and modest changes in hemoglobin levels were observed [24]. Analysis of the total protein content showed that the amount of SB309 was lower than the amount of epoetin alfa so a correction based on the protein content was performed and the 90% CIs for the corrected values were well within the post-hoc defined equivalence [25]. Thus, SB309 met the bioequivalence endpoints required for approval in the EMA [24]. It is important to realize that this comparison was based on the published results supporting approval for each biosimilar and not on a clinical trial directly comparing the two biosimilars. This example demonstrates the need for clear and unambiguous guidance around interchangeability when multiple biosimilars of the same originator are available.
Once a biosimilar is designated as an ‘interchangeable biological product’ by the FDA, further regulation may be established at the state government level as to whether an interchangeable product may be substituted by the pharmacist [20]. When allowed, substitution may be done with or without the pharmacist notifying the prescriber (the latter is known as ‘automatic substitution’) [20].
7 Differences Between Biosimilars and Intended Copies
In some countries, there are commercially available agents that are copies of the originator but which have not been evaluated using the stringent, specifically defined criteria of the WHO guidelines for biosimilars [26]. These agents, called intended copies or biomimics, may have been approved prior to the development of approval guidelines or under guidelines considered less strict than those from the EMA, FDA, or WHO [26–28]. Intended copies have not undergone evaluation according to the stringent regulatory pathways used for biosimilars [26–28]. In addition, intended copies may have differences in formulation or dosages that could result in a clinically significant impact on efficacy and/or safety [26]. Thus, intended copies cannot be considered biosimilars of the originator.
8 Are Originators Biosimilars of Themselves?
For manufacturers of biologics, planned changes in manufacturing may occur over the life of the product. This is particularly true for changes in scale, manufacturing equipment, and improving efficiency, although changes to cell line also may (rarely) occur [9]. To address this, the FDA has generated guidance for industry for assessing biologics undergoing changes in manufacturing (by the same manufacturer; called the ‘comparability exercise’) [9, 29]. These guidelines indicate that quality data should be generated on the product prior to and after the manufacturing change and then analyzed using a comparison integrating all data [29]. The goal of the comparability exercise is to ensure the resulting product has consistent quality, safety, and efficacy using relevant data and that attributes of the biologic pre- and post-change are highly similar [29]. However, demonstration of comparability does not necessarily mean that pre- and post-change products are identical [29]. Ongoing, routine batch analyses, in-process control, process validation and/or evaluation data, characterization, and stability analyses are performed and compared to historic data to ensure batch variability and process changes have no adverse impact upon safety or efficacy [29]. It is important to note that for the assessments of a product undergoing a manufacturing change, manufacturers of an approved product know the product development history, cell line, entire production and purification process, and critical proprietary information [9]. As a result, changes in the manufacturing process can be conducted using extremely well controlled measures and internally available intermediates [9, 29].
In contrast, while the statistical approach to assessing a product pre-and post-change is similar to the analysis used to assess a potential biosimilar to an originator, the development process itself is very different because developers of potential biosimilars face a large knowledge gap in the manufacturing processes used for the originator [9]. As a result, developers of biosimilars must use reverse engineering to develop the product. This starts with the selection of a new cell line since they will not have access to the same cell line used by the manufacturer of the originator [9]. As a result, they will also need to establish a new production and purification process to reflect inherent differences in cell lines [9]. Although the concepts of comparability (evaluation of an original biologic after a manufacturing change) and biosimilarity (evaluation of a new biosimilar against the originator product) are related scientific and regulatory concepts (these terms are even used interchangeably in the EMA biosimilar guidelines), these are two very distinct processes from a developmental point of view [9]. As a result, originators undergoing manufacturing changes should not be considered biosimilars of themselves [9].
9 Future Perspectives and Challenges
Although guidance for the approval of biosimilars has been available for several years, some concepts still need to be addressed to ensure that efficient pharmacovigilance is performed to support the safe and effective treatment of patients. For example, regulatory approval usually requires the provision of appropriate ongoing, post-approval safety monitoring programs [1, 6]. It is important to note that currently there are no standard requirements for post-approval safety monitoring programs because they may depend on experience from the originator. Therefore, these programs are developed through discussions between the manufacturer and regulatory authorities to determine which appropriate study design or surveillance (e.g., patient registries) should be in place to evaluate any risks to safety [1, 6]. In addition, it is critical these programs have adequate mechanisms in place to differentiate between adverse events associated with a biosimilar and those associated with the originator [6]. Naming conventions for biosimilars currently are not consistent, with the WHO proposing a four-letter code (‘biological qualifier’) to the international non-proprietary name (INN) and the FDA using a four-letter suffix, whereas member states of the European Commission’s Pharmaceutical Committee indicate that biosimilars should use the same INN as the originator [30–32]. Identification of biosimilars will likely remain a challenge until harmonization of naming conventions occurs [33].
10 Conclusion
As biologic products lose patent exclusivity and more biosimilars receive regulatory approval, the terminology and definitions applied to biosimilars need to be clarified and used consistently. This is especially critical when multiple biosimilars of the same product are available. It is important to be clear about whether a specific product has been evaluated through a rigorous evaluation procedure based on the criteria defined in the EMA, FDA, or WHO biosimilar guidelines. It is also important for prescribers to understand what happens when a particular biosimilar receives a designation of ‘interchangeable’ with the originator and when substitution may occur, as these designations/policies may impact patient outcomes.
References
World Health Organization. Guidelines on evaluation of similar biotherapeutic products (SBPs) Expert Committee on Biological Standardization, Geneva, Switzerland. 2009 (last update 23 Oct 2009). http://www.who.int/biologicals/areas/biological_therapeutics/BIOTHERAPEUTICS_FOR_WEB_22APRIL2010.pdf Accessed 17 Mar 2016.
European Medicines Agency. Guideline on similar biological medicinal products (CHMP/437/04 Rev 1). European Medicines Agency, London. 2014. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/10/WC500176768.pdf Accessed 11 May 2016.
European Medicines Agency. European public assessment reports by type (biosimilars). European Medicines Agency, London. 2016. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/landing/epar_search.jsp&mid=WC0b01ac058001d124&searchTab=searchByAuthType&keyword=Enter%20keywords&searchType=name&alreadyLoaded=true&status=Authorised&status=Withdrawn&status=Suspended&status=Refused&jsenabled=false&searchGenericType=biosimilars&orderBy=status&pageNo=1. Accessed 26 Apr 2016.
US Food and Drug Administration. FDA approves first biosimilar product Zarxio [media release]. US Department of Health and Human Services, Food and Drug Administration, Silver Spring, MD. 2015 (last update 6 Mar 2015). http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm436648.htm. Accessed 11 May 2016.
Generics and Biosimilars Initiative (GaBi). US$67 billion worth of biosimilar patents expiring before 2020. Pro Pharma Communications International, Mol, Belgium. 2012 (last update 20 Jan 2014). http://www.gabionline.net/Biosimilars/General/US-67-billion-worth-of-biosimilar-patents-expiring-before-2020. Accessed 11 May 2016.
US Food and Drug Administration. Scientific considerations in demonstrating biosimilarity to a reference product. Guidance for industry. US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER), Silver Spring, MD. 2015. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm291128.pdf. Accessed 20 May 2015.
Ventola CL. Evaluation of biosimilars for formulary inclusion: factors for consideration by P&T committees. P T. 2015;40:680–9.
Socinski MA, Curigliano G, Jacobs I, Gumbiner B, MacDonald J, Thomas D. Clinical considerations for the development of biosimilars in oncology. MAbs. 2015;7:286–93. doi:10.1080/19420862.2015.1008346.
Declerck P, Farouk-Rezk M, Rudd PM. Biosimilarity versus manufacturing change: two distinct concepts. Pharm Res. 2016;33:261–8. doi:10.1007/s11095-015-1790-3.
van Aerts LA, De Smet K, Reichmann G, van der Laan JW, Schneider CK. Biosimilars entering the clinic without animal studies. A paradigm shift in the European Union. MAbs. 2014;6:1155–62. doi:10.4161/mabs.29848.
Chapman K, Adjei A, Baldrick P, da Silva A, De Smet K, DiCicco R, et al. Waiving in vivo studies for monoclonal antibody biosimilar development: national and global challenges. MAbs. 2016;8:427–35. doi:10.1080/19420862.2016.1145331.
Isakov L, Jin B, Jacobs IA. Statistical primer on biosimilar clinical development. Am J Ther. 2016;23:e1903–10. doi:10.1097/MJT.0000000000000391.
US Food and Drug Administration. Guidance for industry: E9 statistical principles for clinical trials. US Department of Health and Human Services, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER), Rockville, MD. 1998 (last update September 1998). http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm073137.pdf. Accessed 17 March 2016.
Weise M, Kurki P, Wolff-Holz E, Bielsky MC, Schneider CK. Biosimilars: the science of extrapolation. Blood. 2014;124:3191–6. doi:10.1182/blood-2014-06-583617.
Health Canada. Summary basis of decision (SBD) for Remsima. Office of Regulatory Affairs, Health Canada, Ottawa, Ontario, Canada. 2014 (last update Apr 2015). http://www.hc-sc.gc.ca/dhp-mps/prodpharma/sbd-smd/drug-med/sbd_smd_2014_remsima_160195-eng.php#sbd. Accessed 11 May 2016.
Health Canada. Summary basis of decision (SBD) for Inflectra. Office of Regulatory Affairs, Health Canada, Ottawa, Ontario, Canada. 2014 (last update 8 Jun 2015). http://www.hc-sc.gc.ca/dhp-mps/prodpharma/sbd-smd/drug-med/sbd_smd_2014_inflectra_159493-eng.php. Accessed 11 May 2016.
Health Canada. Regulatory decision summary for REMSIMA (Control number 184568). 2016. Available at: http://www.hc-sc.gc.ca/dhp-mps/prodpharma/rds-sdr/drug-med/rds-sdr-remsima-184568-eng.php. Accessed 10 Oct 2016.
Health Canada. Regulatory decision summary INFLECTRA. 2016. Available at: http://www.hc-sc.gc.ca/dhp-mps/prodpharma/rds-sdr/drug-med/rds-sdr-infectra-184564-eng.php. Accessed 10 Oct 2016.
US Food and Drug Administration. Considerations in demonstrating interchangeability with a reference product. Guidance for Industry. DRAFT GUIDANCE. 2017. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM537135.pdf. Accessed 31 Jan 2017.
Tothfalusi L, Endrenyi L, Chow SC. Statistical and regulatory considerations in assessments of interchangeability of biological drug products. Eur J Health Econ. 2014;15(Suppl 1):S5–11. doi:10.1007/s10198-014-0589-1.
Declerck P, Mellstedt H, Danese S. Biosimilars—terms of use. Curr Med Res Opin. 2015;31:2325–30. doi:10.1185/03007995.2015.1098601.
Abraham J. Developing oncology biosimilars: an essential approach for the future. Semin Oncol. 2013;40(Suppl 1):S5–24. doi:10.1053/j.seminoncol.2013.09.015.
Christl L. FDA’s overview of the regulatory guidance for the development and approval of biosimilar products in the US. http://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/UCM428732.pdf. Accessed 27 June 2016.
Mikhail A, Farouk M. Epoetin biosimilars in Europe: five years on. Adv Ther. 2013;30:28–40. doi:10.1007/s12325-012-0072-2.
European Medicines Agency. Retacrit: EPAR—Scientific Discussion. 2008. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Scientific_Discussion/human/000872/WC500054374.pdf. Accessed 11 Jan 2017.
Mysler E, Pineda C, Horiuchi T, Singh E, Mahgoub E, Coindreau J, et al. Clinical and regulatory perspectives on biosimilar therapies and intended copies of biologics in rheumatology. Rheumatol Int. 2016;36:613–25. doi:10.1007/s00296-016-3444-0.
Castaneda-Hernandez G, Gonzalez-Ramirez R, Kay J, Scheinberg MA. Biosimilars in rheumatology: what the clinician should know. RMD Open. 2015;1:e000010. doi:10.1136/rmdopen-2014-000010.
Dorner T, Kay J. Biosimilars in rheumatology: current perspectives and lessons learnt. Nat Rev Rheumatol. 2015;11:713–24. doi:10.1038/nrrheum.2015.110.
US Food and Drug Administration. Guidance for Industry. Q5E comparability of biotechnological/biological products subject to changes in their manufacturing process. U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER), Rockville, MD. 2005 (last update Jun 2005). http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm073476.pdf Accessed 11 May 2016.
US Food and Drug Administration. Nonproprietary Naming of Biological Products. Guidance for Industry. 2017. http://www.fda.gov/downloads/drugs/guidances/ucm459987.pdf. Accessed 31 Jan 2017.
World Health Organization. Biological Qualifier An INN Proposal. 2015. http://www.who.int/medicines/services/inn/WHO_INN_BQ_proposal_2015.pdf. Accessed 7 Feb 2017.
Generics and Biosimilars Initiative (GaBi). EU majority says same INNs for biosimilars. 2014. http://www.gabionline.net/Biosimilars/General/EU-majority-says-same-INNs-for-biosimilars. Accessed 31 Jan 2017.
Danese S, Bonovas S, Peyrin-Biroulet L. Biosimilars in IBD: from theory to practice. Nat Rev Gastroenterol Hepatol. 2017;14:22–31. doi:10.1038/nrgastro.2016.155.
US Food and Drug Administration. Drugs@FDA Glossary of Terms. 2012. http://www.fda.gov/drugs/informationondrugs/ucm079436.htm#B. Accessed 18 July 2016.
Generics and Biosimilars Initiative (GaBi) Online. Small molecule versus biological drugs. 2012. http://www.gabionline.net/Biosimilars/Research/Small-molecule-versus-biological-drugs. Accessed 18 July 2016.
Acknowledgements
Medical writing support was provided by Christina McManus, PhD, of Engage Scientific Solutions and funded by Pfizer Inc.
Author contributions
All authors made substantial contributions to the conception or design of the work and/or the acquisition, analysis, or interpretation of data.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Funding for medical writing support was provided by Pfizer Inc.
Conflict of interest
PD participated in advisory board meetings for AbbVie, Amgen, and Hospira and received speakers honoraria from AbbVie, Celltrion, Hospira, Merck Serono, Pfizer, and Roche. RD has received honoraria for speaking at symposia for Amgen, Bayer, Celgene, Lilly, Novartis, Pfizer, and Sanofi, holds positions on advisory boards for Amgen, Bayer, Lilly, Novartis, Pfizer, and Sanofi, and has received research funding from Novartis. DP and IJ are full-time employees of and hold stock or stock options in Pfizer.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Declerck, P., Danesi, R., Petersel, D. et al. The Language of Biosimilars: Clarification, Definitions, and Regulatory Aspects. Drugs 77, 671–677 (2017). https://doi.org/10.1007/s40265-017-0717-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-017-0717-1