
Abstract
The health effects of ambient air pollution are well-described, but less is known about the biological mechanisms mediating these health effects. In recent years, the hypothesis that epigenetics may play a role in driving exposure-disease associations has gained traction, in part because epigenetic modifications are labile and may respond to environmental exposures in ways that directly affect gene transcription and disease risk. The purpose of this review is to provide an understanding of the latest evidence to support this hypothesis. The studies selected for this review were collected through a PubMed search for articles published between January 2010 and July 2013 using the following epigenetics keywords: epigenetics, histone, microRNA, and DNA methylation; along with air pollutant keywords: air pollution, benzo[a]pyrene, nitrogen dioxide, ozone, particulate matter 1.0 (PM1), particulate matter 2.5 (PM2.5), particulate matter 10 (PM10) and polycyclic aromatic hydrocarbon (PAH). A total of 38 original research articles were identified for our review. The scientific studies to date provide evidence that PAHs and PM2.5 may have modest effects on methylation levels of certain CpG sites within candidate genes of interest in cardiovascular and respiratory disease as well as cancer. However, the data remain too sparse to draw any meaningful conclusions with regard to histone modifications, miRNAs, or effects of other pollutants such as NO2, O3, and SO4.
Similar content being viewed by others

Introduction
Ambient air pollution is associated with numerous adverse health outcomes, the most notable of which are allergic, respiratory, and cardiovascular diseases [1–3]. The biological mechanisms driving these exposure-disease associations are thought to be largely through oxidative stress or inflammatory pathways or endothelial or autonomic dysfunction [4]. Importantly, genetic variation in genes involved in these biological pathways can alter an individual’s susceptibility to air pollution health effects [5], providing further evidence for their involvement. In recent years, the notion that epigenetics may play a role in driving exposure-disease associations has emerged, adding to the complexity of teasing out causal associations in population-based studies of air pollution.
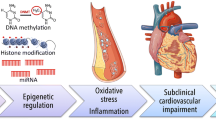
Epigenetics – the study of alterations to gene function that do not change the DNA sequence itself – provides an attractive target when investigating the biological mechanisms through which air pollution might act. Alterations to epigenetic patterns have been implicated in numerous diseases [6–10], but most extensively in cancer [11, 12]. CpG methylation, histone modifications, and non-coding RNAs are all types of epigenetic marks that are labile [13] and thus may respond to environmental exposures in ways that directly affect gene transcription and disease risk (Fig. 1).

Overview of mechanisms through which air pollutants may act on the epigenome and subsequent disease risk
The purpose of this review is to summarize the latest scientific evidence supporting or refuting the hypothesis that ambient air pollutants can alter epigenetic patterns. We focus primarily on epidemiologic studies in the past three years, as several good reviews exist summarizing the scientific evidence prior to 2010 (Table 1) [14–17]. We include discussion of supporting experimental data when appropriate, although an exhaustive review of experimental data is beyond the scope of this review. The air pollutants investigated are polycyclic aromatic hydrocarbons (PAHs), including Benzo[a]pyrene (B[a]P), particulate matter (PM), ozone (O3), nitrogen dioxide (NO2), and sulfates (SO4). The majority of data available investigates the effects of these pollutants on global DNA methylation or gene-specific CpG methylation, with limited investigations on microRNAs and histone modifications. The studies selected for this review were collected through a PubMed search for articles published between January 2010 and July 2013 using the following epigenetics keywords: epigenetics, histone, microRNA, and DNA methylation; along with air pollutant keywords: air pollution, benzo[a]pyrene, nitrogen dioxide, ozone, particulate matter 1.0 (PM1), particulate matter 2.5 (PM2.5), particulate matter 10 (PM10), and PAH. A total of 38 original research articles were identified for our review.
PAH, B[a]P and DNA Methylation
PAHs are a byproduct of incomplete fossil fuel combustion or burning of organic elements from a variety of sources [18–20]. PAHs are ubiquitous environmental pollutants that are classified as Group 1 carcinogens by the International Agency for Research on Cancer (IARC) [21, 22]. When metabolized, PAHs can form DNA adducts and induce global hypomethylation and hypermethylation of specific genes associated with increased risk of neural impairments, neoplasia, and carcinogenesis [18, 20, 23, 24]. Epigenetic modifications are also increasingly linked to processes related to cardiovascular disease, atherosclerosis, and endothelial function [25]. Within the PAH mixtures, B[a]P is considered one of the most potent carcinogens [20, 24] and is often used as a tracer of PAH mixtures in studies to observe the health effects on humans and animals.
PAHs are associated with DNA methylation levels of repeated elements as well as with changes in levels of gene-specific DNA methylation (Table 2). Of the repeated elements, the long interspersed nucleotide element-1 (LINE-1) and the short interspersed nucleotide element (Alu) are the most-studied in epidemiological studies. In normal tissues, LINE–1 is typically highly methylated [26], whereas hypomethylation of LINE-1 is commonly observed in cancerous tissues and is believed to affect widespread alterations in gene expression and chromatin packaging control as well as higher genomic instability [27]. Demethylation of LINE-1 and Alu can increase their activity as transposable sequences, which may induce genomic alterations associated with unfavorable chronic health outcomes [26, 28]. Such epigenetic alterations are increasingly recognized as potential important mediators of health effects in the context of environmental or occupational settings that have high levels of PAHs. Several occupational studies, including studies of coke oven workers and steel, oil refinery, and petrochemical complexes, have observed associations between high levels of PAH exposure and LINE-1 hypomethylation [21, 22, 27].
Gene-specific DNA methylation has also been investigated, largely by taking a candidate-gene approach. In a study of smoke exposure among firefighters compared to non-firefighters, the firefighters had a lower level of methylation in dual specificity phosphatase 22-promoter (DUSP22), a gene suspected of playing a key role in inflammatory and proliferative disorders. In vitro experiments that challenged cell lines with B[a]P showed similar results, lending support to the evidence that B[a]P itself, not other constituents in smoke, can cause DNA methylation changes [30]. In a study of coke oven workers, PAH exposure was associated with hypomethylation in the promoters of O6-methylguanine-DNA methyltransferase (MGMT), p53, and interleukin-6 (IL-6) in workers of the largest steel, oil refinery, and petrochemical complex in southeastern Asia (Rayong, Thailand) [27]. These genes are involved in DNA repair, cell cycle arrest and cell death, and inflammation, respectively [21, 22, 27, 28]. Glutathione S-transferase Mu 2 (GSTM2) is another important gene in detoxification of PAHs. Tang et al investigated GSTM2 methylation in lung cancer cell lines and tissue samples, and found that aberrant hypermethylation of GSTM2 down-regulated GSTM2 expression and that tumor tissues had low or absent levels of GSTM2 expression, whereas adjacent non-malignant lung tissues had high levels of GSTM2 [31]. Lastly, one study of children in the Czech Republic evaluated effects of living in communities with high versus low ambient air pollution, including B[a]P levels that were sixfold higher in the highly polluted area, on DNA methylation using the Illumina Human Methylation 27k BeadChip [32]. The authors identified 58 CpG loci in different genes for which the difference in methylation level was greater than 10 %. In all loci, the DNA methylation level was lower in the highly polluted community. B[a]P-induced epigenetic effects have also been associated with immunosuppression, teratogenicity, and hormonal changes [20]. The preponderance of evidence to date suggests that PAH – or specifically B[a]P – exposure leads to hypomethylation in the promoters of genes involved in oxidative stress, inflammation, DNA repair, and cell cycle regulation, suggesting general up-regulation of these gene pathways that may have negative consequences for health.
Conversely, B[a]P exposure that causes DNA hypermethylation of tumor suppressor genes leads to their silencing in many human cancers. In vitro experiments of esophageal cancer cell lines treated with benzo[a]pyrene diol epoxide (BPDE) demonstrate that BPDE induces methylation of the retinoic acid receptor beta2 (RAR-β2) gene promoter [33]. RAR-β2 expression is suppressed in premalignant and malignant esophageal cells, and is associated with lung carcinogenesis [33]. Hypermethylation of HIC1, a tumor suppressor gene that is inactivated by DNA methylation in cancer tissues such as lung cancer, has also been observed [27].
While all of the above studies were conducted in adults, previous research suggests that early developmental periods are particularly vulnerable to epigenetic dysregulation in response to environmental exposures [18, 34]. Both human and animal models of prenatal exposure provide evidence that increased DNA adducts and global hypomethylation induce genomic and chromosomal instability [18, 34]. Herbtsman et al found prenatal exposure to PAHs was associated with lower global DNA methylation levels measured in umbilical cord white blood cell DNA [18]. Similarly, an animal study of zebrafish embryos focusing on early exposure to high concentrations of B[a]P observed significant global loss of DNA methylation and decreased DNA methylation in the germ cell-specific vasa promoter, whose altered gene expression can compromise developmental success and affect fertility [35, 36]. Taken together, the studies to date support the hypothesis that PAH exposure affects DNA methylation levels both globally and in specific genes involved in an array of disease processes.
Particulate Matter and DNA Methylation
The majority of human studies of PM and DNA methylation (Table 3) have evaluated PM2.5. PM2.5 has been inconsistently associated with DNA methylation in repeated elements, such as LINE-1 and Alu, or with other assays of global methylation. In an occupational cohort of boilermakers, personal PM2.5 measured over the work shift was not associated with LINE-1 or Alu methylation measured in blood [37]. However, short-term black carbon (BC) exposure was associated with hypomethylation in Alu [26], and workplace industrial exposure from a steel, oil refinery, and petrochemical complex was associated with LINE-1 hypomethylation [27]. PM2.5 during pregnancy, and particularly during the first trimester, was associated with global DNA hypomethylation in placental tissue [34]. DNA methylation in LINE-1 or Alu may play another role as well, which is to increase individual susceptibility to health effects of air pollution. At least one study to date has shown that, while pollutants such as black carbon, NO2, CO, PM2.5, and sulfates were associated with endothelial adhesion molecules ICAM-1 and VCAM-1 blood levels, the effect was stronger among subjects with higher Alu or lower LINE-1 methylation status [29]. Given the scant number of studies, the various pollutants evaluated, and the likelihood of different PM constituents across these occupational and environmental settings, the impact of particulate matter on repeated element methylation or global DNA methylation remains uncertain.
Investigations of PM2.5 exposure and DNA methylation have also focused on candidate genes of interest, primarily in the context of cardiovascular or respiratory disease. For example, several studies have evaluated the effect of PM on regulation of the inducible nitric oxide gene (iNOS), a gene responsible for production of nitric oxide, which is important for both cardiovascular and respiratory health. Recently, chronic exposure to PM2.5 generated from welding activities was associated with a modest change in DNA methylation of the iNOS gene [37]. This work builds upon earlier evidence from three other cohorts suggesting that PM2.5 can affect DNA methylation in iNOS [23, 38, 39•, 40]. In a cohort of steelworkers, 3 days of work exposure was associated with a decrease in iNOS promoter methylation [40]. In a cohort of elderly men, acute exposure to BC and PM2.5 was associated with a decrease in iNOS methylation [23]. Similarly, in our own study of a cohort of children from Southern California, increased levels of PM2.5 and PM10 were associated with decreased methylation in the iNOS promoter but increased methylation within the CpG island [38]. We also observed higher DNA methylation in endothelial nitric oxide synthase (eNOS). Moreover, the regulation of nitric oxide is quite complex, and we demonstrated that iNOS promoter haplotypes, in conjunction with PM exposure and DNA methylation levels, synergistically affected exhaled nitric oxide levels [39•]. The evidence to date, which spans acute and chronic exposures from occupationally and environmentally exposed subjects both young and old, consistently demonstrates that PM is associated with iNOS promoter methylation and affects production of nitric oxide. The extent that these modest effects alter cardiovascular and respiratory health is still an active area of investigation.
Other candidate genes of interest are those that may affect asthma pathogenesis. In one study that took a pathway-based approach, exposure to BC was significantly associated with the methylation patterns in 31 genes previously identified as part of the asthma pathway [41•]. These pollutant exposures had the greatest effect on methylation in genes that were either part of the Th2/B-cell signaling pathway or contributed to controlling eosinophils and airway inflammation, obstruction, and hyper-responsiveness [41•]. Increased exposure to ambient air pollution, comparing communities with high and low levels of air pollutants, is associated with hypermethylation of the Forkhead box transcription factor 3 (Foxp3), a key transcription factor in regulatory T- cells [42•]. These changes are hypothesized to lead to impaired Treg-cell function and increased asthma morbidity. A further study in the same population demonstrated similar effects of ambient air pollution on hypermethylation of interferon-gamma (IFN-gamma), another gene important to asthma, in effector T cells [43].
There have been very few human exposure trials of air pollution for the purpose of investigating DNA methylation. However, in one small crossover trial of exposure to concentrated ambient particles (CAPs), 15 healthy adult participants were exposed for 130 minutes to fine CAPs, coarse CAPs, or HEPA-filtered medical air [28]. Fine CAPs exposure lowered Alu methylation, whereas coarse CAPs exposure lowered Toll-like receptor 4 (TLR4) methylation, another gene of interest in asthma pathogenesis. Interestingly, hypomethylation in these loci was associated with increased blood pressure, providing some of the first evidence that methylation in certain genes may mediate PM exposures on blood pressure [28].
A handful of studies have also evaluated air pollution effects on genes involved in cancer pathogenesis. In vivo and in vitro experiments demonstrate that direct exposure to PM2.5 can induce methylation of the p16 promoter, a tumor suppressor gene important in cancer in mice and in epithelial cell lines [44]. Expression of DNA methyltransferase 1 (DNMT1) was also elevated in these studies, suggesting that increased DNMT1 may be the mechanism through which the promoter is methylated.
In some cases, DNA methylation may alter individual susceptibility to pollutant exposures as opposed to being directly affected by the exposure. For instance, the effects of several pollutants with ICAM-1 and VCAM-1 blood levels were stronger among subjects having higher tissue factor (F3) or Toll-like receptor 2 (TLR-2) methylation status [29].
Not all candidate genes demonstrate associations. DNA methylation in the glucocorticoid receptor (GCR), a gene implicated in stress response, as well as in interleukins 6 and 12 (IL-6, IL-12) genes involved in immune response, have not been associated with PM2.5 to date [23, 28]. Nevertheless, there is emerging literature that suggests PM2.5 may affect DNA methylation in certain genes involved in asthma pathogenesis. These effects are complicated by the knowledge that the DNA methylation levels can vary by cell type, and therefore the response to pollutant exposure may also vary.
The data on PM other than PM2.5 are sparse. A handful of studies from one cohort evaluated PM10 or PM1 [45, 46]. Short-term exposure to metal-rich PM in an occupational cohort of steel workers was associated with leukocyte telomere length – an inflammatory and cardiovascular risk factor. However, no association was observed with DNA methylation level in the telomerase catalytic enzyme gene (hTERT), the gene responsible for telomere length [45]. Additional studies in the same cohort provide evidence that PM exposure may also affect blood coagulation through altered methylation [46]. PM10 and PM1, as well as zinc and iron exposures, were associated with decreased eNOS methylation, whereas only zinc exposure was associated with endothelin-1 (EDN1) methylation. This hypomethylation in both eNOS and EDN1 mediated the PM-induced coagulation effects as measured by the endogenous thrombin potential [46].
Other Air Pollutants and DNA Methylation
Very few studies have investigated the effects of other ambient pollutants, such as O3, NO2, or SO4, on epigenetic modification. No associations have been reported for O3 thus far [39•]. One study in asthmatic children found that NO2 exposure in combination with high levels of DNA methylation, the beta-2 adrenergic receptor (ADRB2) gene, was associated with severe asthma [47]. ADRB2 is a gene expressed on airway smooth muscles important in reducing inflammatory cytokine production, stimulating relaxation, and clearing lung fluid. SO4 was associated with decreased LINE-1 methylation in a cohort of elderly men [26]. Additionally, in the same cohort, the effects of SO4 on fibrinogen were modified by LINE-1 methylation level, whereas the effects of NO2 on ICAM-1 and VCAM-1 blood levels were stronger in subjects with higher Alu and lower LINE-1 methylation [29]. SO4 has also been linked to altered methylation levels in several genes in the asthma pathway, and these are largely different genes than those associated with BC [41•].
Air Pollutants and miRNAs
MicroRNAs (miRNAs) are highly conserved noncoding small RNAs that regulate the expression of broad gene networks at the posttranscriptional level. Although there is debate over whether to classify miRNAs as epigenetic marks, we have chosen to include them in this review for the following reasons. Over 1,000 miRNAs have been discovered, and they regulate nearly two-thirds of human genes [48]. Because miRNAs control numerous cellular processes, they may be critical for mediating biological responses to air pollutants [17]. To this end, a few studies have begun investigating the effects of air pollution exposure on selected miRNAs of interest.
In one of the first studies to evaluate miRNA response, human airway epithelial cells were challenged with diesel exhaust particles (DEP) for 24 hours, and expression changes were measured using a miRNA microarray [49•]. Jardim et al found 197 detectable miRNAs that were either up- or down-regulated in response to exposure, of which the top 12 were primarily associated with inflammatory and tumorigenic processes [49•]. Candidate miRNAs putatively involved in oxidative stress and inflammation – specifically miR-222, miR-21, and miR-146a – were subsequently investigated in a cohort of steelworkers [50]. Pre- and post- workday exposures to PM1, coarse PM, and PM metal components (chromium, lead, cadmium, arsenic, nickel, manganese) were evaluated, and miR-222 and miR-146a expression were correlated with lead exposure, although none were correlated with PM size fractions [50]. Polymorphisms in miRNA-processing genes were also found to modify the effects of ambient pollution on cardiovascular disease biomarkers [51]. Lastly, a recent study comparing subjects living in an area contaminated with electronic waste to those living in an unpolluted area found a total of 182 differentially expressed miRNAs [52]. The contaminated area likely contained high levels of heavy metals, atmospheric polychlorodibenzo-p-dioxins (PCDDs), polychlorodibenzofurans (PCDFs), and polybrominated diphenyl ether (PBDE), along with thousands of other pollutants.
While the emerging literature on the associations between ambient air pollutants and miRNAs is nascent, this avenue of investigation holds promise, considering that small changes to a single miRNA have the potential to affect broad downstream gene networks involved in multiple disease processes.
Air Pollutants and Histones
Histones are proteins that form one of the fundamental building blocks in chromatin organization. Modification of histones can therefore affect chromatin organization as well as gene regulation [53]. Examples of such modifications include lysine acetylation, lysine methylation, arginine methylation, serine phosphorylation, lysine ubiquitination, and many others [54, 55]. Histone modifications provide yet another potential mechanism by which air pollution exposure may affect downstream regulation of genes and gene pathways. However, due primarily to the laborious nature of the laboratory assays and the associated costs, measurement of histone modifications in large population studies has not been feasible.
To date, limited information exists on the effect of ambient air pollutants on histone modifications. In one human study, long-term exposure to inhalable nickel, arsenic, and iron was associated with increased histone 3 lysine 4 dimethylation (H3K4Me2) and histone 3 lysine 9 acetylation (H3K9Ac) on histones from blood leukocytes [56]. In an animal study, neonatal exposure to B[a]P decreased the acetylation of H3K14 in the steroidogenic acute regulatory protein (StAR), an important protein in testosterone production [57]. B[a]P damaged testosterone production and sperm counts in these rats, plausibly as the result of a direct effect on epigenetic regulation of StAR. In HeLa cells challenged with B[a]P, histone H3 trimethylated at lysine 4 (H3K4Me3) and H3K9Ac were enriched, whereas the association of DNA methyltransferase-1 (DNMT1) with the L1 promoter was reduced [24]. Cytosine methylation within the L1 promoter CpG island was also reduced. Pharmacological inhibition altered these BaP-mediated histone epigenetic modifications [24]. Lastly, experiments using knockout mice demonstrated that Sirt1, a member of class III histone deacetylase, controls lung inflammation and coagulation after PM exposure. PM exposure aggravated lung vascular leakage and inflammation in Sirt1 knockout mice, and these effects were correlated with increased NF-kappaB acetylation and activation [58].
Conclusion
The last several years have produced an emerging set literature on the hypothesis that epigenetic modifications are susceptible to air pollution exposure, both during pregnancy and in childhood or adulthood. For histone modifications and miRNAs, the data are still much too sparse to make any meaningful conclusions. The same is true for many of the specific air pollutants, namely NO2, O3, and SO4. However, it is becoming clear that PAHs and PM2.5 may have modest effects on methylation levels of certain CpG sites within candidate genes of interest in cardiovascular and respiratory disease as well as cancer. The application of epigenome-wide approaches, such as DNA methylation array platforms and bisulfite-sequencing, as they become more widely used and the results publicly available will undoubtedly shed additional light on these associations and discover new ones. Moreover, the extent to which data from large-scale methylation studies are shared for common exposures such as PAH and PM2.5 holds the promise of maximizing the potential inferences.
In all likelihood, many of the candidate gene associations observed today are some of the more robust associations, plausible biomarkers of exposure. They may not be as sensitive to the cell type used or timing of exposure, and thus they have been observed despite potential confounding by cell type in mixed blood. As an example, the consistent associations observed for PM2.5 and iNOS have been found using buccal cell DNA in children as well as whole blood in adults. As the field evolves and we improve sensitivity of laboratory assays, develop advanced statistical methods specific for analyzing epigenetic data, and integrate information across genotype, epigenotype, and exposure, we will undoubtedly gain further insights, although likely raising as many questions as are answered.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Franchini M, Mannucci PM. Air pollution and cardiovascular disease. Thromb Res. 2012;129:230–4.
Kelly FJ, Fussell JC. Air pollution and airway disease. Clin Exp Allergy. 2011;41:1059–71.
Takizawa H. Impact of air pollution on allergic diseases. Korean J Intern Med. 2011;26:262–73.
Stanek LW, Brown JS, Stanek J, Gift J, Costa DL. Air pollution toxicology–a brief review of the role of the science in shaping the current understanding of air pollution health risks. Toxicol Sci. 2011;120 Suppl 1:S8–S27.
Minelli C, Wei I, Sagoo G, Jarvis D, Shaheen S, Burney P. Interactive effects of antioxidant genes and air pollution on respiratory function and airway disease: a HuGE review. Am J Epidemiol. 2011;173:603–20.
Webster AL, Yan MS, Marsden PA. Epigenetics and cardiovascular disease. Can J Cardiol. 2013;29:46–57.
Clifford RL, Singer CA, John AE. Epigenetics and miRNA emerge as key regulators of smooth muscle cell phenotype and function. Pulm Pharmacol Ther. 2013;26:75–85.
Jenke AC, Zilbauer M. Epigenetics in inflammatory bowel disease. Curr Opin Gastroenterol. 2012;28:577–84.
Alagiakrishnan K, Gill SS, Fagarasanu A. Genetics and epigenetics of Alzheimer's disease. Postgrad Med J. 2012;88:522–9.
Salam MT, Zhang Y, Begum K. Epigenetics and childhood asthma: current evidence and future research directions. Epigenomics. 2012;4:415–29.
Waldmann T, Schneider R. Targeting histone modifications–epigenetics in cancer. Curr Opin Cell Biol. 2013;25:184–9.
Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27.
Inbar-Feigenberg M, Choufani S, Butcher DT, Roifman M, Weksberg R. Basic concepts of epigenetics. Fertil Steril. 2013;99:607–15.
Bollati V, Baccarelli A. Environmental epigenetics. Heredity (Edinb). 2010;105:105–12.
Durham AL, Wiegman C, Adcock IM. Epigenetics of asthma. Biochim Biophys Acta. 1810;2011:1103–9.
Edwards TM, Myers JP. Environmental exposures and gene regulation in disease etiology. Environ Health Perspect. 2007;115:1264–70.
Jardim MJ. microRNAs: implications for air pollution research. Mutat Res. 2011;717:38–45.
Herbstman JB, Tang D, Zhu D, et al. Prenatal exposure to polycyclic aromatic hydrocarbons, benzo[a]pyrene-DNA adducts, and genomic DNA methylation in cord blood. Environ Health Perspect. 2012;120:733–8.
Lukashevich OV, Baskunov VB, Darii MV, Kolbanovskiy A, Baykov AA, Gromova ES. Dnmt3a-CD is less susceptible to bulky benzo[a]pyrene diol epoxide-derived DNA lesions than prokaryotic DNA methyltransferases. Biochemistry. 2011;50:875–81.
Verma N, Pink M, Rettenmeier AW, Schmitz-Spanke S. Review on proteomic analyses of benzo[a]pyrene toxicity. Proteomics. 2012;12:1731–55.
Duan H, He Z, Ma J, et al. Global and MGMT promoter hypomethylation independently associated with genomic instability of lymphocytes in subjects exposed to high-dose polycyclic aromatic hydrocarbon. Arch Toxicol. 2013;87(11):2013–22.
Pavanello S, Pesatori AC, Dioni L, et al. Shorter telomere length in peripheral blood lymphocytes of workers exposed to polycyclic aromatic hydrocarbons. Carcinogenesis. 2010;31:216–21.
Madrigano J, Baccarelli A, Mittleman MA, et al. Air pollution and DNA methylation: interaction by psychological factors in the VA Normative Aging Study. Am J Epidemiol. 2012;176:224–32.
Teneng I, Montoya-Durango DE, Quertermous JL, Lacy ME, Ramos KS. Reactivation of L1 retrotransposon by benzo(a)pyrene involves complex genetic and epigenetic regulation. Epigenetics. 2011;6:355–67.
Madrigano J, Baccarelli A, Wright RO, et al. Air pollution, obesity, genes and cellular adhesion molecules. Occup Environ Med. 2010;67:312–7.
Madrigano J, Baccarelli A, Mittleman MA, et al. Prolonged exposure to particulate pollution, genes associated with glutathione pathways, and DNA methylation in a cohort of older men. Environ Health Perspect. 2011;119:977–82.
Peluso M, Bollati V, Munnia A, et al. DNA methylation differences in exposed workers and nearby residents of the Ma Ta Phut industrial estate, Rayong, Thailand. Int J Epidemiol. 2012;41:1753–60. discussion 61-3.
Bellavia A, Urch B, Speck M, et al. DNA hypomethylation, ambient particulate matter, and increased blood pressure: findings from controlled human exposure experiments. J Am Heart Assoc. 2013;2:e000212.
Bind MA, Baccarelli A, Zanobetti A, et al. Air pollution and markers of coagulation, inflammation, and endothelial function: associations and epigene-environment interactions in an elderly cohort. Epidemiology. 2012;23:332–40.
Ouyang B, Baxter CS, Lam HM, et al. Hypomethylation of dual specificity phosphatase 22 promoter correlates with duration of service in firefighters and is inducible by low-dose benzo[a]pyrene. J Occup Environ Med. 2012;54:774–80.
Tang SC, Sheu GT, Wong RH, et al. Expression of glutathione S-transferase M2 in stage I/II non-small cell lung cancer and alleviation of DNA damage exposure to benzo[a]pyrene. Toxicol Lett. 2010;192:316–23.
Rossnerova A, Tulupova E, Tabashidze N, et al. Factors affecting the 27K DNA methylation pattern in asthmatic and healthy children from locations with various environments. Mutat Res. 2013;741–742:18–26.
Ye F, Xu XC. Benzo[a]pyrene diol epoxide suppresses retinoic acid receptor-beta2 expression by recruiting DNA (cytosine-5-)-methyltransferase 3A. Mol Cancer. 2010;9:93.
Janssen BG, Godderis L, Pieters N, et al. Placental DNA hypomethylation in association with particulate air pollution in early life. Part Fibre Toxicol. 2013;10:22.
Fang X, Corrales J, Thornton C, Scheffler BE, Willett KL. Global and gene specific DNA methylation changes during zebrafish development. Comp Biochem Physiol B Biochem Mol Biol. 2013;166:99–108.
Fang X, Thornton C, Scheffler BE, Willett KL. Benzo[a]pyrene decreases global and gene specific DNA methylation during zebrafish development. Environ Toxicol Pharmacol. 2013;36:40–50.
Kile ML, Fang S, Baccarelli AA, Tarantini L, Cavallari J, Christiani DC. A panel study of occupational exposure to fine particulate matter and changes in DNA methylation over a single workday and years worked in boilermaker welders. Environ Health. 2013;12:47.
Breton CV, Salam MT, Wang X, Byun HM, Siegmund KD, Gilliland FD. Particulate matter, DNA methylation in nitric oxide synthase, and childhood respiratory disease. Environ Health Perspect. 2012;120:1320–6.
Salam MT, Byun HM, Lurmann F, et al. Genetic and epigenetic variations in inducible nitric oxide synthase promoter, particulate pollution, and exhaled nitric oxide levels in children. J Allergy Clin Immunol. 2012;129:232–9 e231-7. This paper was the first to illustrate complex interactions between PM 2.5 , genetic variation in iNOS and iNOS methylation on the gene product.
Tarantini L, Bonzini M, Apostoli P, et al. Effects of particulate matter on genomic DNA methylation content and iNOS promoter methylation. Environ Health Perspect. 2009;117:217–22.
Sofer T, Baccarelli A, Cantone L, et al. Exposure to airborne particulate matter is associated with methylation pattern in the asthma pathway. Epigenomics. 2013;5:147–54. This paper applied an interesting pathway approach to identify disease pathways particularly affected by air pollutant exposures.
Nadeau K, McDonald-Hyman C, Noth EM, et al. Ambient air pollution impairs regulatory T-cell function in asthma. J Allergy Clin Immunol. 2010;126:845–52 e810. This paper focused on addressing effects of pollution on a candidate gene in a very specific cell type motivated by a clear hypothesis, and has found consistently interesting results.
Kohli A, Garcia MA, Miller RL, et al. Secondhand smoke in combination with ambient air pollution exposure is associated with increasedx CpG methylation and decreased expression of IFN-gamma in T effector cells and Foxp3 in T regulatory cells in children. Clin Epigenetics. 2012;4:17.
Soberanes S, Gonzalez A, Urich D, et al. Particulate matter air pollution induces hypermethylation of the p16 promoter via a mitochondrial ROS-JNK-DNMT1 pathway. Sci Rep. 2012;2:275.
Dioni L, Hoxha M, Nordio F, et al. Effects of short-term exposure to inhalable particulate matter on telomere length, telomerase expression, and telomerase methylation in steel workers. Environ Health Perspect. 2011;119:622–7.
Tarantini L, Bonzini M, Tripodi A, et al. Blood hypomethylation of inflammatory genes mediates the effects of metal-rich airborne pollutants on blood coagulation. Occup Environ Med. 2013;70:418–25.
Fu A, Leaderer BP, Gent JF, Leaderer D, Zhu Y. An environmental epigenetic study of ADRB2 5'-UTR methylation and childhood asthma severity. Clin Exp Allergy. 2012;42:1575–81.
Tomankova T, Petrek M, Gallo J, Kriegova E. MicroRNAs: emerging regulators of immune-mediated diseases. Scand J Immunol. 2011. doi:10.1111/j.1365-3083.2011.02650.x.
Jardim MJ, Fry RC, Jaspers I, Dailey L, Diaz-Sanchez D. Disruption of microRNA expression in human airway cells by diesel exhaust particles is linked to tumorigenesis-associated pathways. Environ Health Perspect. 2009;117:1745–51. The author of this paper was the first to investigate air pollution effects on miRNAs using an array, and has since provided a nice review of miRNAs in the context of pollution.
Bollati V, Marinelli B, Apostoli P, et al. Exposure to metal-rich particulate matter modifies the expression of candidate microRNAs in peripheral blood leukocytes. Environ Health Perspect. 2010;118:763–8.
Wilker EH, Alexeeff SE, Suh H, Vokonas PS, Baccarelli A, Schwartz J. Ambient pollutants, polymorphisms associated with microRNA processing and adhesion molecules: the Normative Aging Study. Environ Health. 2011;10:45.
Li Y, Li M, Liu Y, Song G, Liu N. A microarray for microRNA profiling in spermatozoa from adult men living in an environmentally polluted site. Bull Environ Contam Toxicol. 2012;89:1111–4.
Krishnan S, Horowitz S, Trievel RC. Structure and function of histone H3 lysine 9 methyltransferases and demethylases. Chembiochem. 2011;12:254–63.
Rando OJ. Combinatorial complexity in chromatin structure and function: revisiting the histone code. Curr Opin Genet Dev. 2012;22:148–55.
Wang G, Chen Z, Bartell T, Wang X. Early life origins of metabolic syndrome: the role of environmental toxicants. Curr Environ Health Rep. 2014;1: in press.
Cantone L, Nordio F, Hou L, et al. Inhalable metal-rich air particles and histone H3K4 dimethylation and H3K9 acetylation in a cross-sectional study of steel workers. Environ Health Perspect. 2011;119:964–9.
Liang J, Zhu H, Li C, Ding Y, Zhou Z, Wu Q. Neonatal exposure to benzo[a]pyrene decreases the levels of serum testosterone and histone H3K14 acetylation of the StAR promoter in the testes of SD rats. Toxicology. 2012;302:285–91.
Wu Z, Liu MC, Liang M, Fu J. Sirt1 protects against thrombomodulin down-regulation and lung coagulation after particulate matter exposure. Blood. 2012;119:2422–9.
Acknowledgments
Carrie V. Breton is supported by the following grants: NIEHS grants K01ES017801, R01ES022216 and 5P30ES007048
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
Carrie V. Breton and Amy N. Marutani declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Breton, C.V., Marutani, A.N. Air Pollution and Epigenetics: Recent Findings. Curr Envir Health Rpt 1, 35–45 (2014). https://doi.org/10.1007/s40572-013-0001-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40572-013-0001-9