


Introduction
Multiple sclerosis (MS) is a chronic demyelinating disease commonly ascribed to abnormal activation of autoaggressive T cells that cross the blood-brain barrier into the central nervous system (CNS) and result in demyelination and axonal loss. However, there is a growing recognition of more widespread immune dysregulation and tissue damage throughout the CNS, underscoring the need for early, more effective intervention to slow the progression of physical and cognitive deficits.
In addition to the focal demyelinating lesions noted by Charcot,Reference Charcot 1 imaging studies have demonstrated widespread inflammation and tissue damage throughout the normal-appearing white (NAWM) and gray matter (NAGM).Reference Simon, Schmidt and Lukas 2 , Reference Gilmore, Donaldson, Bö, Owens, Lowe and Evangelou 3 Histopathological changes can occur in gray matter (GM) in the absence of focal white matter (WM) lesions, suggesting that GM and WM pathology may be independent.Reference Kutzelnigg, Faber-Rod and Bauer 4 Moreover, diffuse inflammation associated with astrocytic and microglial activation is evident both in NAWM and NAGM.Reference Mayo, Trauger and Blain 5 , Reference van Horssen, Singh and van der Pol 6 It remains unclear if activation of innate immune processes is a primary event or is secondary to T-cell activation. Diffuse inflammation is more pronounced in progressive forms of MS,Reference Rissanen, Tuisku and Rokka 7 , Reference Stadelmann and Brück 8 which may indicate secondary activation of the innate immune response and herald the onset of disability progression,Reference Leray, Yaouanq and Le Page 9 in which neurodegeneration resulting from inflammation predominates over focal inflammatory processes. In support of this is the finding that diffuse axonal loss occurs independently of demyelination and is poorly correlated with focal WM lesion burden.Reference Kutzelnigg, Lucchinetti and Stadelmann 10 As such, both diffuse axonal injury and tissue loss in demyelinated plaques appear to contribute to global brain atrophy.Reference Kutzelnigg and Lassmann 11
An accelerated rate of brain atrophy on MRI is already evident early in the disease course, even in clinically isolated syndrome (CIS) and the early relapsing-remitting phases of MS.Reference Henry, Shieh, Okuda, Evangelista, Gorno-Tempini and Pelletier 12 - Reference Audoin, Davies, Rashid, Fisniku, Thompson and Miller 14 Of particular importance to the progression of physical and cognitive disability is the rate of GM atrophy, which has been reported to be about threefold higher in CIS patients converting to relapsing-remitting MS (RRMS), and 14-fold higher in secondary-progressive MS (SPMS) compared with healthy controls.Reference Fisher, Lee, Nakamura and Rudick 15
Cognitive impairment can be detected at the earliest disease phases, affecting patients with CIS and radiologically isolated syndrome, defined as incidental WM pathology without clinical evidence of demyelination.Reference Lebrun, Blanc, Brassat, Zephir and de Seze 16 - Reference Okuda, Mowry and Beheshtian 19 The prevalence of cognitive impairment in MS has been estimated to be as high as 68% in early MS, increasing to 81% later.Reference Fischer, Kunkel and Bublak 20 The earliest and most commonly affected domain is information processing speed, with additional deficits in memory, visual-spatial processing, and language.Reference Van Schependom, D’hooghe and Cleynhens 21 Cognitive impairment has a profound impact on patients’ quality of life,Reference Chruzander, Ytterberg, Gottberg, Einarsson, Widén Holmqvist and Johansson 22 participation in activities of daily living,Reference Ben Ari Shevil, Johansson, Ytterberg, Bergström and von Koch 23 and employment.Reference Krause, Kern, Horntrich and Ziemssen 24 Deterioration in cognitive function has been correlated with neuropathological findings such as brain atrophy,Reference Benedict, Weinstock-Guttman, Fishman, Sharma, Tjoa and Bakshi 25 lesion volume,Reference Randolph, Wishart and Saykin 26 and cortical lesions.Reference Calabrese, Poretto and Favaretto 27 GM atrophy is reportedly the most important predictor of long-term disability and cognitive impairment.Reference Filippi, Preziosa and Copetti 28 In support of this, a recent analysis of early MS patients found significantly lower scores on magnetization transfer imaging in cognitively impaired subjects compared with those without cognitive impairment.Reference Faiss, Dähne and Baum 29 There were no differences in lesion load between the two groups, suggesting that early cognitive impairment may be due primarily to diffuse loss of brain tissue integrity rather than WM lesions.
Clinical Course of MS
MS is diagnosed in accordance with the International Panel’s revised 2010 criteria.Reference Polman, Reingold and Banwell 30 Three phenotypes have been characterized, based on clinical and radiological findings, history, and disease course: CIS, RRMS, and progressive disease with accumulation of disability from onset (primary-progressive MS; PPMS) or after an initial relapsing course (SPMS).Reference Lublin, Reingold and Cohen 31 CIS, RRMS, and progressive disease (PPMS and SPMS) may be further described as active or not active. Disability is evaluated with the Expanded Disability Status Scale (EDSS) on a scale of 0 (no disability) to 10 (death).Reference Kurtzke 32 It is important to note that, because EDSS is heavily weighted to ambulation, cognitive impairment may be underrepresented with this disability measure.
Natural history studies indicate that the median time from onset of RRMS to an EDSS score of 4 (moderate disability but fully ambulatory), 6 (requiring the use of a cane), and 7 (requiring a wheelchair) is approximately 8, 20, and 30 years, respectively.Reference Confavreux, Vukusic and Adeleine 33 , Reference Scalfari, Neuhaus and Degenhardt 34 In pediatric-onset MS, the median time to reach these milestones is longer (approximately 20, 29, and 37 years), but typically occurs when the patient is about 10 years younger compared than those with adult-onset MS.Reference Renoux, Vukusic and Mikaeloff 35
The development of SPMS occurs in adults about 20 years after onsetReference Koch, Kingwell, Rieckmann and Tremlett 36 and is diagnosed retrospectively based on accumulating disability and a declining relapse frequency. Although worsening disability occurs at an earlier stage in PPMS, it is often diagnosed when individuals are older, so that EDSS milestones are typically reached at approximately the same age in PPMS and SPMS.Reference Harding, Wardle and Moore 37 , Reference Kremenchutzky, Rice, Baskerville, Wingerchuk and Ebers 38 With the onset of the progressive phase, neurological decline occurs at the same rate in patients, independent of their initial presentation.Reference Confavreux, Vukusic and Adeleine 33
MS is associated with significant morbidity and an estimated 1.7 increased risk of all-cause mortality compared to age-matched controls.Reference Jick, Li, Falcone, Vassilev and Wallander 39 An analysis of patients registered at MS clinics in British Columbia reported a median survival of 78.6 years for women and 74.3 years for men (standardized mortality ratio, 2.89), or about 6 fewer years compared with the general population.Reference Kingwell, van der Kop, Zhao, Shirani, Zhu, Oger and Tremlett 40 The main causes of early mortality are conditions associated with impaired mobility and neurological dysfunction, such as infections and pulmonary and cardiovascular disease.Reference Lalmohamed, Bazelier and Van Staa 41 , Reference Goodin, Corwin and Kaufman 42
Time Window for Intervention
MS has historically been viewed as a focal inflammatory disease primarily resulting from T-cell activation, and the disease-modifying therapies (DMTs) used in RRMS primarily affect different aspects of immune dysregulation. The therapeutic goals focused on reducing disease activity, as demonstrated by a reduction in relapse frequency and accrual of new lesions on MRI, and slowing the worsening of short-term disability as assessed by EDSS. Because DMTs primarily target the inflammatory component of the disease, there is an urgency to initiate and optimize therapy within the first few years of diagnosis to obtain the most benefit from treatment.
Early in the disease course, worsening disability is largely attributable to relapse-related residual neurological deficits.Reference Lublin, Baier and Cutter 43 , Reference Hirst, Ingram, Pearson, Pickersgill, Scolding and Robertson 44 As a result, the number of relapses in untreated patients in the first 2 years postdiagnosis provides an indication of disability risk, but has limited prognostic value thereafter because of the reduction in relapse frequency that occurs as part of the natural history.Reference Langer-Gould, Popat and Huang 45 , Reference Binquet, Quantin, Le Teuff, Pagliano, Abrahamowicz and Moreau 46 One observation is that any relapse in the first 5 years postdiagnosis increases the risk of reaching EDSS 6 over the short term (hazard ratio, 1.48).Reference Tremlett, Yousefi, Devonshire, Rieckmann and Zhao 47 Also important is worsening disability in the first 5 years after diagnosis, which is predictive of later disability.Reference Degenhardt, Ramagopalan, Scalfari and Ebers 48 An important goal of treatment is the prevention of short-term surrogates of progression, such as accumulation of disability confirmed at 3 or 6 months. This “EDSS progression” is better characterized as sustained accumulation of disability confirmed after a specified number of months, to distinguish it from “progression,” a term now reserved for the progressive phase of MS.Reference Lublin, Reingold and Cohen 31
A limitation of natural history database analyses is that prognostic factors may not be predictive of progression in a treated population. Consequently, efforts have been made to identify clinical and radiological predictors of an inadequate treatment response to enable clinicians to manage suboptimally responding patients more promptly.
Analyses of patients treated with interferon-β have shown that ongoing MRI activity, as evidenced by new gadolinium-enhancing T1-weighted or new/enlarging T2-weighted lesions, is predictive of worsening disability.Reference Rudick, Lee, Simon, Ransohoff and Fisher 49 The presence of one new T2 lesion in the year after initiating an interferon-β agent has been associated with a 10-fold increase in the risk of sustained EDSS worsening (>1 point confirmed at 6 months) during the 4.8-year follow-up.Reference Prosperini, Gallo, Petsas, Borriello and Pozzilli 50
MRI and relapse criteria have been combined to calculate a modified Rio score for predicting an inadequate response to interferon-β.Reference Sormani, Rio and Tintorè 51 Patients are scored from 0 to 3 according to the number of new T2 lesions (>four lesions =1 point) and relapses (one relapse =1, two relapses =2). Sormani et al calculated that at 4 years, the risk of worsening by ≥1 EDSS point was 32%, 42%, and 50% with scores of 0, 1, and 2 or 3, respectively. Applied to the dataset of patients treated with subcutaneous interferon-β-1a in the Prevention of Relapses and Disability by Interferon beta-1a Subcutaneously in Multiple Sclerosis (PRISMS) trial, 52 59.5% of treated patients had >42% risk of worsening disability after 1 year of treatment,Reference Sormani, Rio and Tintorè 51 suggesting that many interferon-treated patients will experience a suboptimal therapeutic response. These observations were extended in a larger meta-analysis of 40 trials of CIS, RRMS, and SPMS, which found a significant association between disease activity (annualized relapse rate, T2 lesion count and volume) during treatment and worsening disability.Reference Fahrbach, Huelin, Martin, Kim, Dastani, Rao and Malhotra 53
A further impetus to early, effective treatment is the high rate of cognitive impairment in CIS and MS, which may occur before the onset of symptomatic disease.Reference Achiron, Chapman and Magalashvili 54 Data from a recent long-term study suggested that there may be a 5-year window after diagnosis, during which reducing disease activity and limiting neurological disability with treatment may decrease cognitive impairment.Reference Achiron, Chapman and Magalashvili 54
Evolution in the Standard of Care
Two decades of DMT use have demonstrated that first-generation injectable therapies have a modest effect on short-term measures of disease activity in RRMS (recently reviewed by Michel et alReference Michel, Larochelle and Prat 55 ). Of greater importance to morbidity and patient quality of life is the impact of treatment on long-term disability progression, but here the findings are mixed. Long-term follow-ups of pivotal trials for interferon-β and glatiramer acetate (GA) have generally reported lower rates of progression to SPMS and less disability, but interpretation of these data is limited by the high proportion of dropouts and that these are uncontrolled observational data.
In the 15-year follow-up of the US GA trial, in the subgroup of 100 patients receiving ongoing therapy (mean drug exposure, 13.6 years), EDSS was stable or improved in 57%, but 39% reached EDSS ≥4 and 35% transitioned to SPMS.Reference Ford, Goodman and Johnson 56 When the modified intention-to-treat cohort (n=232) was stratified by baseline EDSS score, 61% of patients with greater disability at onset reached EDSS ≥4 after a mean of 8.6 years. A meta-analysis found no significant effect of GA on disability.Reference La Mantia, Munari and Lovati 57 In accordance with this lack of effect, GA is not clinically indicated for the reduction of disability progression. 58
In the 8-year follow-up of the PRISMS trial of subcutaneous interferon-β-1a, there appeared to be some benefit with respect to EDSS progression with earlier versus delayed treatment.Reference Kappos, Traboulsee and Constantinescu 59 However, only 49% of the original cohort remained on therapy. Similarly, in a single-center study of patients treated with intramuscular interferon-β-1a for 15 years, ongoing treatment also appeared to be associated with less disability, but the analysis was based on only one-third of the original cohort.Reference Bermel, Weinstock-Guttman, Bourdette, Foulds, You and Rudick 60
The 16-year follow-up of the pivotal trial of interferon-β-1b was able to obtain data for a majority of the original study population.Reference Ebers, Traboulsee, Li and Langdon 61 Patients who started treatment earlier with the higher dose of interferon-β-1b obtained no added benefit with respect to MRI or disability outcomes compared with those initially randomized to placebo. As in the GA trial, long-term disability outcomes were highly dependent on disease severity at baseline.Reference Goodin, Traboulsee and Knappertz 62 A separate analysis at 21 years of follow-up reported a survival advantage with long-term interferon-β-1b exposure.Reference Goodin, Reder and Ebers 63 In contrast, a retrospective analysis of patients in the British Columbia database of MS patients found that interferon-β-1b use was not associated with a reduction in progression of disability compared to untreated contemporary or historical controls.Reference Shirani, Zhao and Karim 64
As noted previously, it is hypothesized that T-cell activation in the periphery and reactivation in the CNS are the primary immunologic events, which then lead to innate immune activation in the CNS, diffuse inflammation throughout the NAWM and NAGM, neurodegeneration, and progressive tissue loss. This is the rationale for treatments targeting different aspects of this process of T-cell activation and migration into the CNS. Accordingly, it may be inferred that the potential long-term benefits of this approach cannot be fully achieved with the limited efficacy of first-generation DMTs and that more aggressive strategies need to be considered. Moreover, because genetic and environmental factors are known to influence the etiopathology of MS, the availability of a broader range of therapies could enable a more individualized approach to patient care, and the selection of an agent that will achieve a better degree of disease control.
Some support for a more aggressive approach can be obtained from studies of rapidly evolving RRMS or progressive MS treated with lymphoablation and stem cell transplantation (SCT). SCT has been shown to produce a rapid and sustained reduction in CNS inflammatory activity, and stabilization or improvement in disability scores.Reference Curro’ D, Vuolo, Gualandi, Bacigalupo, Roccatagliata and Capello 65 - Reference Burt, Loh, Cohen, Stefoski, Balabanov and Katsamakis 67 A recent study reported structural and functional improvements in the optic nerve post-SCT in SPMS patients.Reference Connick, Kolappan, Crawley, Webber, Patani and Michell 68 However, some patients will experience slowly worsening disability despite profound suppression of inflammatory disease activity,Reference Mancardi, Sormani, Di Gioia, Vuolo, Gualandi and Amato 69 , Reference Fassas, Kimiskidis, Sakellari, Kapinas, Anagnostopoulos and Tsimourtou 70 which may be attributed to neurodegenerative mechanisms that may be somewhat independent of inflammation, a differing pathobiology in some subsets of patients, genetic and/or environmental modulators of disease activity, or other factors.
With the development of a broader range of DMTs, there is an opportunity for clinicians to individualize therapy to achieve a more robust response. This in turn has led to a need for more stringent criteria for therapeutic response, as reflected in the emerging treatment goal of “no evidence of disease activity.” This is generally defined with the three metrics of no relapses, no gadolinium-enhancing T1 lesions or new/enlarging T2 lesions, and no EDSS progression confirmed at 3 or 6 months (no evidence of disease activity [NEDA]-3). NEDA-3 was first used in a post-hoc analysis of the Natalizumab Safety and Efficacy in Relapsing-Remitting Multiple Sclerosis (AFFIRM) dataset, which determined that 37% of patients treated with natalizumab achieved NEDA-3 compared with 7% with placebo at 2 years.Reference Havrdova, Galetta and Hutchinson 71 Subsequent analyses have reported that about one-third of patients can also achieve NEDA-3 at 2 years with agents such as fingolimod and alemtuzumab (Table 1).Reference Kappos, Radue and O’Connor 72 - Reference Coles, Twyman and Arnold 74 Even higher NEDA-3 rates may be achievable in younger patients, as suggested by a recent analysis of fingolimod phase III trials.Reference Chitnis, Karlsson, Häring, Ghezzi, Pohl and Putzki 78
Table 1 Rates of NEDA-3Footnote * at 2 years with newer disease-modifying agents

* NEDA, no evidence of disease activity, Formerly called disease activity-free.
† 3-month confirmed disability progression (except for CARE-MS studies).
‡ No relapses + no EDSS progression.
¶ No relapses, no EDSS progression, no Gd+T1, and no new/enlarged T2 lesions.
AFFIRM, Natalizumab (Antegren) Safety and Efficacy in Relapsing Remitting Multiple Sclerosis; BID, twice daily; CARE-MS, Comparison of Alemtuzumab and Rebif Efficacy in Multiple Sclerosis; CONFIRM, Comparator and an Oral Fumarate in Relapsing–Remitting Multiple Sclerosis; DEFINE, The Determination of the Efficacy and Safety of Oral Fumarate in Relapsing-Remitting MS; FREEDOMS, FTY720 Research Evaluating Effects of Daily Oral Therapy in Multiple Sclerosis; GA, glatiramer acetate; TEMSO, Teriflunomide Multiple Sclerosis Oral; Teri, teriflunomide, TID, three times daily.
Note: comparisons cannot be made across trials because of differences in study design, patient characteristics, and different criteria used to define NEDA-3.
NEDA-3 provides an indication of the efficacy of treatments in suppressing focal inflammatory disease activity, but does not capture the full impact of therapy on the neurodegenerative component of MS. Individual measures need to be standardized (e.g. worsening disability confirmed at 3 or 6 months), and extensions of various clinical studies will help to determine the predictive value of NEDA-3 for long-term disability outcomes. A further limitation is that early in the disease course, NEDA-3 results largely reflect an absence of radiologic activity, and traditional MRI markers of disease activity (gadolinium-enhancing T1 or new/enlarging T2 lesions) are known to be only weakly correlated with long-term disability.Reference Bevan and Cree 79
The addition of measures that reflect ongoing tissue loss, such as brain atrophy rates, new hypointense T1 lesions (“black holes”), and magnetization transfer ratio, may provide a more robust evaluation of treatment response and help to refine the predictive value of NEDA for physical and cognitive outcomes. For example, a longitudinal MRI study found that the proportion of patients with cognitive impairment doubled over the 2-year observation period, from 26.4% at baseline to 52.8% at 24 months.Reference Zivadinov, Sepcic and Nasuelli 80 By the end of the study, there was a significant correlation between brain parenchymal volume and cognitive impairment as well as brain atrophy and EDSS change. This loss of brain volume cannot be regained, underscoring the importance of early, effective treatment to minimize cognitive deterioration.
In healthy controls, the rate of brain volume loss is estimated at−0.1% to −0.3% per annum,Reference De Stefano, Sprenger and Freedman 81 so a cutoff of −0.4%/year has been proposed for the metric of NEDA-3 + atrophy (NEDA-4).Reference De Stefano, Sprenger and Freedman 81 Using this value, Kappos et al reported a >fourfold higher likelihood of achieving NEDA-4 with fingolimod vs placebo at 2 years (odds ratio =4.41, p<0.0001).Reference Kappos, Radue and Freedman 82 NEDA-4 analyses have not yet been reported for other DMTs.
Evidence of ongoing disease activity during treatment, as demonstrated by the failure to achieve NEDA-3, should prompt a review of the current regimen with the view to selecting a treatment that may provide improved efficacy or tolerability. The addition of brain volume change in NEDA-4 further refines the efficacy evaluation by incorporating a measure of the underlying pathophysiology of MS that is predictive of physical and cognitive outcomes. Brain atrophy measures are now routinely obtained at some specialized MS centers, and their use may become part of the standard of care in the next few years. However, the optimal method of MRI acquisition (e.g. whole-brain, GM atrophy) and atrophy quantification (e.g. brain parenchymal fraction, Structural Image Evaluation, using Normalisation, of Atrophy [SIENA]), with correction for various confounders such as hydration status, drug/alcohol use, and comorbidities in individual patients, has not been fully determined. Emerging technologies, such as optical coherence tomography of retinal nerve fiber layer thickness as a measure of axonal degeneration,Reference Balk, Steenwijk, Tewarie, Daams, Killestein and Wattjes 83 may also be useful in assessing the extent and progression of WM and GM atrophy.
Clinical Management
An aggressive treatment strategy is not merely a matter of adopting a “zero tolerance” approach to ongoing disease activity and escalating treatment accordingly. Rather, aggressive management also requires more frequent clinical and radiological assessments to allow for early detection of suboptimal response, and identification of factors, such as poor tolerability and adherence, that may contribute to treatment failure.
Close monitoring is playing an increasingly important role in patient management because of the risk of serious adverse events. A 6-hour observation period at first dose is required with fingolimod because of its potential effects on heart rate, blood pressure, and cardiac conduction. The frequency of second-degree atrioventricular block or higher is estimated to be about 1%, and about 4% to 6% in patients with a preexisting cardiac condition.Reference Gold, Comi, Palace, Siever, Gottschalk and Bijarnia 84 With ongoing treatment, the estimated risk of macular edema with fingolimod 0.5 mg is 0.4%.Reference Zarbin, Jampol, Jager, Reder, Francis and Collins 85 Patients should also be monitored for symptoms of infections. There have been isolated case reports of cryptococcal meningitis. 86 Patients should be assessed for prior exposure to varicella zoster virus (VZV) before initiating fingolimod due to the risk of infection; vaccination is recommended in patients who are VZV antibody-negative. Two patients treated with fingolimod without prior natalizumab exposure developed progressive multifocal leukoencephalopathy (PML). Fingolimod should be used with caution in patients with severe respiratory disease. Pulmonary function testing should be performed during treatment if clinically indicated. Liver enzymes should be evaluated every 3 months for the first year and periodically thereafter.
The principal concern with natalizumab is the risk of PML, which is estimated to be 1.3% in anti–John Cunningham (JC) virus antibody-positive patients with prior exposure to immunosuppressants receiving >24 natalizumab infusions. 87 If PML is diagnosed, rapid natalizumab withdrawal can result in a potentially fatal immune reconstitution inflammatory syndrome.Reference Kleinschmidt-DeMasters, Miravalle, Schowinsky, Corboy and Vollmer 88 One case of JC virus granule cell neuronopathy has been reported.Reference Schippling, Kempf, Büchele, Jelcic, Bozinov and Bont 89 Other serious adverse effects may include hepatic injury, infections, anaphylaxis, suicidal ideation, and cholelithiasis. 90 Fatal opportunistic infections have occurred, and there are rare reports of encephalitis and meningitis deaths associated with herpesvirus infection. 90 There is one case of cryptococcal meningitis reported with natalizumab.Reference Valenzuela, Pula, Garwacki, Cotter and Kattah 91
With alemtuzumab, most patients will experience infusion-related reactions and precautionary treatment with corticosteroids is advised; antihistamines and antipyretics may also be used. Patients without a history of prior VZV exposure should undergo VZV antibody testing and immunization should be considered for VZV antibody-negative patients. A 1-month course of antiviral prophylaxis is recommended after each treatment course because of the risk of VZV infection or reactivation. Other potential safety issues include secondary autoimmunity, including thyroid disorders (cumulative incidence, 34.7% at 4 years), immune thrombocytopenic purpura (2.5% at 4 years), and antiglomerular basement membrane disease (<1%). 92 , Reference Havrdova, Giovannoni, Arnold, Coles, Fox and Hartung 93 Thyroid function testing should be performed before treatment initiation and every 3 months thereafter. Patients should be evaluated for active or latent tuberculosis before treatment initiation. Cervical human papillomavirus infection, including cervical dysplasia, has been reported (2%), 92 and annual screening for human papillomavirus is recommended for female patients. Serum creatinine levels and urinalysis should be obtained before treatment initiation and monthly thereafter until 48 months after the last infusion.
Treatment Initiation and Escalation
In accordance with recent recommendations, treatment should be considered for all patients with CIS at high risk of developing clinically definite MS, such as those with extensive MRI burden of disease or positive oligoclonal banding.Reference Freedman, Selchen and Arnold 94 At present, only first-generation injectables and teriflunomide have demonstrated efficacy in this patient population.Reference Jacobs, Beck and Simon 95 - Reference Miller, Wolinsky and Kappos 99 The benefits of early treatment with interferon-β or GA on longer term disability are uncertain.Reference Kinkel, Dontchew and Kollman 100 , Reference Kappos, Freedman and Polman 101 CIS patients who subsequently meet diagnostic criteria for MS may be viewed as having experienced breakthrough disease on treatment, and consideration should be given to employing a more potent therapy to achieve more optimal disease control.
Ideally, all RRMS patients should be started on a DMT, with the initial choice of therapy determined by the clinical presentation. Recent experience indicates that oral therapies are preferred because their efficacy appears to be either comparable, or superior, in cases where head-to-head studies have been conducted, to that of the injectables.Reference Vermersch, Czlonkowska and Grimaldi 102 - Reference Cohen, Barkhof and Comi 104 The oral route of administration may also be preferred by patients and may promote better adherence, as a number of authors have recently suggested.Reference Fox 105 , Reference Kim, Zandoná, Kim and Kim 106 Safety considerations with oral teriflunomide include a risk of elevated liver enzymes (14%) and peripheral neuropathy (2%);Reference O’Connor, Wolinsky, Confavreux, Comi, Kappos and Olsson 107 active elimination is required before pregnancy because of the teratogenic potential. 108 With oral dimethyl fumarate (DMF), a limitation is poor tolerability because of the high incidence of flushing (38%), diarrhea (15%), and nausea (13%).Reference Gold, Kappos, Arnold, Bar-Or, Giovannoni and Selmaj 109 So far, two cases of PML associated with the branded formulation of DMF have been reported in MS patients with lymphopenia,Reference Rosenkranz, Novas and Terborg 110 and one case in a patient with psoriasis and suspected MS treated with a compounded formulation of DMF.Reference van Oosten, Killestein, Barkhof, Polman and Wattjes 111 Although periodic monitoring for severe lymphopenia may mitigate the risk,Reference Sweetser, Dawson and Bozic 112 a case of PML has recently been reported in a patient with psoriasis without severe lymphopenia treated with compounded DMF.Reference Nieuwkamp, Murk, van Oosten, Cremers, Killestein and Viveen 113
A baseline cognitive assessment is recommended in all patients, with routine follow-up evaluations obtained annually. More frequent assessments are advised if cognitive dysfunction is suspected by the patient, family member, clinician, or MS nurse. The Symbol Digit Modalities Test (SDMT) is recommended to evaluate information processing speed and working memory.Reference Smith 114 Additional testing may be required for other cognitive domains. Relapses have been shown to be associated with worsening cognitive function as evaluated by SDMT.Reference Benedict, Morrow and Rodgers 115 A recent meta-analysis also found a moderate-to-strong correlation between T2 lesion volume and information processing speed as evaluated by SDMT.Reference Rao, Martin and Huelin 116 Therefore, a frank decline in cognitive function during therapy may indicate treatment failure and consideration should be given to employing a more potent agent.
As treatment effects may be delayed for several months after initiation, a brain MRI is advised 6 months after starting therapy. This scan will serve as a new baseline for comparison with subsequent MRIs. The therapeutic response should be evaluated 6 to 12 months after the on-treatment reference scan, in accordance with Canadian treatment optimization recommendations.Reference Freedman, Selchen and Arnold 94 Clear evidence of ongoing clinical or radiological disease activity in the first year of treatment should prompt a reexamination of the initial treatment choice and consideration should be given to employing a more potent agent.
Close monitoring is required throughout the disease course to evaluate ongoing treatment response and adverse effects, and semiannual clinic visits and annual brain MRI assessments are recommended.
The most obvious concern with a more aggressive treatment approach is the risk of serious adverse effects. These concerns should not be minimized, and require a full discussion of the side effect profile with the patient. It is important to add that any benefit-risk assessment must be made within the context of the known risks of MS itself. For patients to make an informed choice, they must be aware of the need to adequately control the disease process to minimize the long-term physical and cognitive impairments associated with MS.
Is Escalation Effective?
The core principles of treatment optimization are the prompt recognition of an inadequate response and the selection of a new agent based on its likelihood of achieving better disease control and improved long-term outcomes.Reference Freedman, Selchen and Arnold 94 One strategy for suboptimal responders is to switch to an injectable DMT, teriflunomide, or DMF. An alternative approach would be to switch to an agent with a different mechanism of action and greater potency, such as fingolimod, natalizumab, alemtuzumab, or additional novel therapies as they become available.
Data from randomized controlled phase III studies have suggested that switching treatment can achieve improved disease control (Table 2). In the Comparator and an Oral Fumarate in Relapsing–Remitting Multiple Sclerosis (CONFIRM) extension, the annualized relapse rate (ARR) was initially unchanged in the GA group in the first year after switching to DMF, but declined 24.7% and 35.2% at 2 and 3 years, respectively, after switching to DMF.Reference Gold, Phillips and Bar-Or 117 Because there is a limited time window in which to optimize therapy, escalation to a more potent agent may be more likely to influence the disease course. In the Trial Assessing Injectable Interferon versus FTY720 Oral in Relapsing–Remitting Multiple Sclerosis (TRANSFORMS) extension, there was a 29% reduction in ARR in the interferon-β-1a group in the first 12 months after switching to fingolimod.Reference Khatri, Barkhof and Comi 119 For the Comparison of Alemtuzumab and Rebif Efficacy in Multiple Sclerosis (CARE-MS) extension studies, there was a 69% to 71% reduction in ARR in the interferon-β-1a groups in the 24 months after switching to alemtuzumab.Reference Coles, Arnold and Cohen 120 , Reference Hartung, Arnold, Cohen, Coles, Fox and Giovannoni 121 No phase III studies have examined the benefit of a switch to natalizumab.
Table 2 Phase III extension data on the impact of switching from an injectable disease-modifying therapy to an oral or infusion disease-modifying therapy
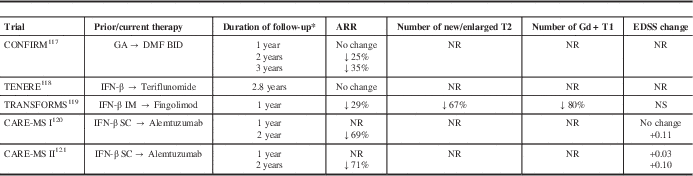
* after switch
CARE-MS, Comparison of Alemtuzumab and Rebif Efficacy in Multiple Sclerosis; CONFIRM, Comparator and an Oral Fumarate in Relapsing–Remitting Multiple Sclerosis; IFN-β, interferon-β-1a; IM, intramuscular; SC, subcutaneous; NR, not reported; NS, not significant; TENERE, Teriflunomide and Rebif®; TRANSFORMS, Trial Assessing Injectable Interferon versus FTY720 Oral in Relapsing-Remitting Multiple Sclerosis.
An earlier switch may also slow the rate of brain volume loss. In the TRANSFORMS trial, brain volume loss was 31% lower with fingolimod versus interferon-β-1a in the first year of treatment.Reference Khatri, Barkhof and Comi 119 In the year after switching to fingolimod, patients in the interferon-β-1a group demonstrated a 51% reduction in the amount of brain volume loss. In the AFFIRM trial, brain volume loss was greater in the first year of natalizumab because of pseudoatrophy, but there was a reduction in the atrophy rate in the second year of treatment.Reference Miller, Soon, Fernando, MacManus, Barker and Yousry 122 Similar results were obtained in the CARE-MS studies. The mean change from baseline in brain parenchymal fraction was similar with alemtuzumab and interferon-β-1a in year 1. However, in year 2, mean brain parenchymal fraction change was 32% lower with alemtuzumab in previously untreated patients with highly active diseaseReference Krieger, Lubetzki and Arnold 123 and 37% lower in previously treated patients receiving alemtuzumab versus interferon-β-1a.Reference Fisher, Barkhof and Cohen 124
An implication of these findings is that agents with more direct CNS effects, such as fingolimod, DMF, or teriflunomide may serve as better platform therapies in the future. Fingolimod crosses the blood-brain barrier and has been shown in laboratory studies and animal models to interact with astrocytes and oligodendrocytes, which may promote remyelination.Reference Wu, Leong and Moore 125 - Reference Slowik, Schmidt, Beyer, Amor, Clarner and Kipp 127 Preclinical data have shown that DMF may have protective effects on axonal and neuronal integrity through its interaction with the nuclear factor (erythroid-derived 2)-like 2 antioxidant pathway,Reference Fox, Kita and Cohan 128 and that teriflunomide may promote oligodendrocyte survival.Reference Petty, Lee and Ying 129
At present, there are limited data on the impact of more potent therapies on cognitive outcomes. The Natalizumab for the Relief of MS Associated Fatigue (ENER-G) study reported that cognitive function was stable or improved with natalizumab.Reference Wilken, Kane and Sullivan 130 Although this may be attributed in part to a reduction in fatigue, a 1-year observational study did find improvements in memory and executive function with natalizumab.Reference Mattioli, Stampatori and Capra 131 An analysis of TRANSFORMS data reported significantly less deterioration in cognition at 1 year, as assessed by the three-second Paced Auditory Serial Addition Test, with fingolimod compared with intramuscular interferon-β.Reference Pelletier, Karlsson and Li 132 A separate 1-year study reported that global cognition scores were stable with fingolimod.Reference Barak, Magalashvili, Paz and Achiron 133 Additional research is needed to confirm these findings.
For patients with rapidly evolving RRMS, the most aggressive approach would be induction with an immunosuppressant followed by maintenance with a DMT. Vollmer and colleagues reported an 70% greater reduction in gadolinium-enhancing lesions at 1 year with mitoxantrone followed by GA compared with GA alone.Reference Vollmer, Panitch, Bar-Or, Dunn, Freedman and Gazda 134 A separate study reported a 65% reduction in disability worsening with mitoxantrone induction before interferon-β-1b.Reference Edan, Comi, Le Page, Leray, Rocca and Filippi 135 Cyclophosphamide induction with pulse maintenance therapy has also been shown to produce benefits in PPMS (with adrenocorticotropic hormone) and in aggressive pediatric MS.Reference Weiner, Mackin, Orav, Hafler, Dawson and LaPierre 136 , Reference Makhani, Gorman, Branson, Stazzone, Banwell and Chitnis 137 Although long-term maintenance with immunosuppressants is limited by toxicities, a form of induction may be achieved with alemtuzumab, with a maintenance regimen of annual courses of alemtuzumab, or ongoing treatment with another DMT. This approach has not yet been examined with alemtuzumab, and more data on the long-term efficacy and safety of induction strategies are required.
Conclusions
The past 2 decades have seen significant advances in our understanding of the MS disease process and how therapeutic interventions need to be evaluated. Some newer, high-efficacy DMTs have been shown to be more effective than first-generation agents in reducing inflammation and tissue damage, as assessed by clinical and radiological endpoints, with about one-third of patients achieving NEDA. The caveat is that these potential benefits must be weighed against the risk of possible adverse effects. Natalizumab, fingolimod, and alemtuzumab produce rapid and profound reductions in inflammatory disease activity, and fingolimod has been shown to have an early, consistent effect on the rate of brain volume loss. Thus, some of the newer-generation DMTs may be the first agents to modify the MS disease process. A more complete understanding of the risks associated with these agents will become evident over time.
Even with the most potent agents available, there is a limited time window for effective intervention. Time is brain, and the tissue loss that occurs during the years of switching from one first-line agent to another cannot be regained. There is sufficient evidence to show that NEDA-3 can be achieved in a substantial proportion of RRMS patients, and NEDA-4 data demonstrate that effective therapy can slow the development of early brain atrophy. In the near future, it can be expected that more aggressive treatment strategies will be routinely employed in patients with evidence of an accelerated rate of brain volume loss.
NEDA-3 and NEDA-4 are redefining the standard for treatment efficacy. They provide the rationale for an early, more aggressive treatment strategy in RRMS patients, with the goal of more effectively reducing the extensive tissue damage and minimizing the physical and cognitive impairments that occur during the clinical course of MS.
Disclosures
The authors take full responsibility for the content of the paper and wish to thank Steven Manners (Communications Lansdowne) for manuscript drafting and editorial assistance under their guidance. Editorial help was funded by Novartis Pharmaceuticals Canada Inc.
PD has received honoraria for speaking, consulting, and advisory board participation; support to attend meetings from Bayer HealthCare, Biogen Idec, EMD Serono, Genzyme, Novartis, Roche and Teva Neuroscience; research support from Biogen Idec, EMD Serono, and Genzyme; participated in clinical phrase 2 to phrase 4 trials, investigator-initiated research, and CME activities for Novartis, EMD Serono, Biogen-Idec, Genzyme, and TEVA. He has acted as a local principal investigator for clinical trials financed by Bayer HealthCare, Biogen Idec, Elan, EMD Serono, Novartis, Sanofi-Aventis, and Teva Neuroscience. He has also received funding from the Canadian Institutes of Health Research and the Multiple Sclerosis Society of Canada.
PG has received honoraria for speaking, consulting, and advisory board participation from Allergan, Bayer HealthCare, Biogen Idec, EMD Serono, Genzyme, Novartis, Roche, and Teva Neuroscience. He has received research support from Biogen Idec and Teva Neuroscience; has been a consultant for NeuroRx Research, an imaging contract research organization; and has acted as a principal investigator or subinvestigator for clinical trials for Alexion, Bayer HealthCare, Biogen Idec, Elan, EMD Serono, GlaxoSmithKline, Novartis, Ono, Roche-Genentech, Sanofi-Aventis, and Teva Neuroscience.
VB has received honoraria for speaking, consulting, and advisory board participation from Bayer HealthCare, Biogen Idec, EMD Serono, Genzyme, Novartis, Roche, and Teva Neuroscience. He has acted as a principal investigator for clinical trials for Biogen Idec, Elan, EMD Serono, Novartis, Roche-Genentech, Sanofi-Aventis, and Teva Neuroscience.
MH has received honoraria for speaking, consulting, and/or advisory board participation from Bayer HealthCare, Biogen Idec, EMD Serono, Genzyme, Novartis, Roche, and Teva Neuroscience.
RS is an employee of Novartis Pharmaceuticals Canada Inc.



 Open access
Open access