
Abstract
Aim:
Sirtuin 1 (Sirt1) is the class III histone/protein deacetylase that interferes with the NF-κB signaling pathway, thereby has anti-inflammatory function. This study was undertaken to investigate whether Sirt1 could protect osteoblasts against TNF-α-induced injury in vitro.
Methods:
Murine osteoblastic cell line, MC3T3-E1, was used. Overexpress of Sirt1 protein in MC3T3-E1 cells was made by transfection the cells with Sirt1-overexpressing adenovirus. The levels of mRNAs and proteins were determined with qRT-PCR and Western blotting, respectively. The activity of NF-κB was examined using NF-κB luciferase assay. The NO concentration was measured using the Griess method.
Results:
Treatment of MC3T3-E1 cells with TNF-α (2.5–10 ng/mL) suppressed Sirt1 protein expression in a concentration-dependent manner. TNF-α (5 ng/mL) resulted in an increase in apoptosis and a reduction in ALP activity in the cells. Overexpression of Sirt1 in the cells significantly attenuated TNF-α-induced injury through suppressing apoptosis, increasing ALP activity, and increasing the expression of Runx2 and osteocalcin mRNAs. Furthermore, overexpression of Sirt1 in the cells significantly suppressed TNF-α-induced NF-κB activation, followed by reducing the expression of iNOS and NO formation. Sirt1 activator resveratrol (10 μmol/L) mimicked the protection of the cells by Sirt1 overexpression against TNF-α-induced injury, which was reversed by the Sirt1 inhibitor EX-527 (5 μmol/L).
Conclusion:
Overexpression of Sirt1 protects MC3T3-E1 osteoblasts aganst TNF-α-induced cell injury in vitro, at least in part, via suppressing NF-κB signaling. Sirt1 may be a novel therapeutic target for treating rheumatoid arthritis-related bone loss.
Similar content being viewed by others

Introduction
Rheumatoid arthritis is characterized by chronic inflammation that results in excessive production of pro-inflammatory cytokines within bone tissue1,2,3,4. One such cytokine, tumor necrosis factor-α (TNF-α), is overproduced within the synovium of rheumatoid arthritis lesions, contributing to both systemic and local bone loss5,6. The pathogenic significance of TNF-α has been established by the clinical effectiveness of blocking TNF-α in the treatment of active rheumatoid arthritis7,8. It has been demonstrated that TNF-α decreases osteoblastic bone formation through the suppression of osteoblast proliferation, the induction of osteoblast apoptosis, the inhibition of osteoblast differentiation and the suppression of matrix proteins, such as osteocalcin, which is produced by mature osteoblasts9,10,11. Therefore, further in vitro studies are needed to identify potential therapeutic targets that protect osteoblasts from TNF-α-induced injury and to elucidate the underlying mechanisms mediating this protection.
Sirtuin 1 (Sirt1), a member of the silent information regulator 2 family in mammals, has recently been found to be involved in age-related diseases, such as cancer, metabolic diseases, cardiovascular disease, neurodegenerative diseases, and osteoporosis. This involvement is primarily mediated through the deacetylation of substrates, which include p53, forkhead box class O, peroxisome proliferator activated receptor γ co-activator 1α, and nuclear factor-κB (NF-κB)12,13. Activation of Sirt1 in mesenchymal stem cells can decrease adipocyte differentiation and increase osteoblast differentiation by regulating p53 and PPARγ12. In addition, the Sirt1 activator, resveratrol, can inhibit the receptor activator of NF-κB ligand (RANKL)-induced osteoclastogenesis in bone-derived cells14. Recently, the inhibition of osteoblast differentiation and bone formation by cytokines, including TNF-α, was demonstrated and correlated with activation of the NF-κB signaling pathway15. Because NF-κB is a direct target of Sirt113, we hypothesize that Sirt1 will protect osteoblasts against TNF-α-induced cytotoxicity by suppressing NF-κB activity. To test this hypothesis, we characterized the in vitro protective effects of Sirt1 overexpression in TNF-α-treated osteoblasts.
In addition, iNOS, a downstream target of NF-κB, plays an important role in the cell injury induced by TNF-α in osteoblasts16,17. Therefore, we also tested the relationship between Sirt1 and iNOS expression in osteoblasts treated with TNF-α.
In this study, we used a murine calvarial osteoblastic cell line (MC3T3-E1) as a model to test the molecular mechanisms of Sirt1 in the protection of osteoblasts against TNF-α-induced cell injury.
Materials and methods
Cell culture
The murine osteoblastic cell line, MC3T3-E1, was cultured at 37 °C in 5% CO2 and 90% humidity in α-MEM medium (Invitrogen, Paisley, UK) supplemented with 10% fetal bovine serum and 100 μg/mL gentamicin (Invitrogen, Life Technologies, Paisley, Scotland). Chemicals, drugs, and reagents were obtained from Sigma Chemical (St Louis, MO, USA) unless otherwise stated.
Experiment 1
MC3T3-E1 cells were seeded in culture dishes and treated with TNF-α (0, 2.5, 5.0, and 10.0 ng/mL) for 24 h. Sirt1 expression was then assessed by Western blot analysis.
Experiment 2
MC3T3-E1 cells were transfected with Ad-Sirt1 or Ad-lacZ for 12 h. After transfection, the cells were washed with PBS and then placed in fresh medium. After culture for 36 h, Sirt1 expression was the assessed by Western blot analysis, and deacetylase activity of Sirt1 was measured using a fluorogenic substrate. NF-κB activity was measured by a luciferase reporter assay, and IκB-α expression was assessed by Western blot analysis.
Both groups of cells (transfected with either Ad-Sirt1 or Ad-lacZ) were treated with TNF-α (50 ng/mL) for 4 h, after which the rate of apoptosis was determined.
Both groups of cells were treated with TNF-α (5.0 ng/mL) for 24 h, and the NOx (NO3–+NO2–) content in the supernatant was then determined using the Griess method. Cellular iNOS expression was determined by Western blot analysis.
Both groups of cells were treated with TNF-α (5.0 ng/mL) for 5 d, after which ALP activity and levels of Runx2 and osteocalcin mRNA were determined.
Experiment 3
MC3T3-E1 cells stimulated with TNF-α (5.0 ng/mL) were treated with BAY-11-7082 (10 μmol/L, an NF-κB inhibitor) or SP600125 (10 μmol/L, a JNK inhibitor) for 24 h. The expression of iNOS was then assayed by Western blot analysis.
Experiment 4
MC3T3-E1 cells were treated with α-MEM alone (control), TNF-α (5.0 ng/mL), TNF-α with resveratrol (10 μmol/L, a Sirt1 activator), or TNF-α with resveratrol and EX-527 (5 μmol/L, a Sirt1 inhibitor) for 1 h. NF-κB activity was then analyzed by luciferase reporter assay.
Cells were treated as described above for 24 h, and the NOx (NO3–+NO2–) content of the supernatant was determined by the Griess method.
Cells were treated as described above for 5 d, after which ALP activity and levels of Runx2 and osteocalcin mRNA were determined.
MC3T3-E1 cells were treated with α-MEM alone (control), TNF-α (50 ng/mL), TNF-α with resveratrol (10 μmol/L, a Sirt1 activator), or TNF-α with resveratrol and EX-527 (5 μmol/L, a Sirt1 inhibitor) for 4 h, after which the rate of cell apoptosis was measured.
Construction of recombinant adenoviruses
A Sirt1-overexpressing adenovirus was constructed according to the method described by Lee et al18. Briefly, mouse Sirt1 cDNA was cloned into KpnI and XhoI sites of the pENTR 2B vector (Invitrogen), and the Sirt1 cDNA insert was then transferred to the pAd/CMV/V5-DEST vector (Invitrogen). The plasmids were linearized with PacI (Promega, Madison, WI) and were transfected into 293A cells using Lipofectamine 2000. As a control, the pAd/CMV/V5-GW/lacZ vector (Invitrogen) was used to produce a lacZ-bearing adenovirus.
Sirt1 deacetylase activity assay
Cells were homogenized in 100 μL of CytoBuster Protein Extraction Buffer (Invitrogen). Enzyme activity was then measured using a fluorescence-based deacetylase activity kit (Millipore, Bedford, MA, USA).
Apoptosis assay
Cells were seeded into a 6-well plate and treated with the indicated reagents for 4 h. After treatment, the cells were washed with PBS and lysed for 30 min at 4 °C with lysis buffer. The expression levels of cleaved and total caspase-3 were determined by Western blot analysis. The ratio of cleaved caspase-3 to total caspase-3 was used as the marker of apoptosis.
Measurement of ALP activity
The induction of ALP is an unequivocal marker of bone cell differentiation. To measure the ALP activity, the cells were seeded into a 12-well plate and treated with the indicated reagents for 5 d. After treatment, cultured osteoblasts were washed with PBS and then lysed with buffer containing 0.9% NaCl, 0.6% Tris, 1 mmol/L EGTA, 1% NP-40, and 0.25% deoxycholate dissolved in RIPA buffer at 4 °C for 30 min. Cell lysates were then sonicated in an ice bath, centrifuged at 1500×g for 5 min, and mixed with buffer containing 0.1 mol/L 2-amino-2-methyl-1-propanol, 1 mmol/L MgCl2, and 8 mmol/L p-nitrophenyl phosphate disodium. After incubation at 37 °C for 20 min, the reaction was halted with 0.1 mol/L NaOH, and the absorbance was read at 405 nm. A standard curve was prepared using p-nitrophenol. Data were normalized to the concentration of total protein.
RNA purification and quantitative RT-PCR analysis
MC3T3-E1 cells were plated at 100 000 cells per well in 6-well plates and treated with the indicated reagents for 5 d. After treatment, total RNA was isolated with the Rneasy RNA purification kit (Qiagen, Solna, Stockholm, Sweden). A total of 1 mg RNA was reverse transcribed to cDNA using Superscript II (Invitrogen, Stockholm, Sweden) according to the manufacturer's protocol. Gene transcription levels of Runx2 (5′-GCCGGGAATGATGAGAACTA-3′; 5′-GGTGAAACTCTTGCCTCGTC-3′) and osteocalcin (5′-GCCATCACCCTGTCTCCTAA-3′; 5′-GCTGTGGAGAAGACACACGA-3′) were analyzed by quantitative PCR using gene-specific primers and a SYBR Green Supermix (Bio-Rad, Hercules, CA). PCR reactions were performed under the following conditions: initial denaturation of one cycle at 95 °C for 10 min, followed by amplification at 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 30 s, for a total of 35 cycles. The housekeeping gene, β-tubulin (5′-CTGCTCATCAGCAAGATCAGAG-3′; 5′-GCATTATAGGGCTCCACCACAG-3′), was used as an endogenous control. Data analysis was performed using Ct values normalized to β-tubulin expression.
Western blot analysis
Osteoblasts were seeded onto 6-well plates at a density of 1×105 cells/well. After incubation with a test reagent, the cells were washed with PBS and lysed for 30 min at 4 °C with lysis buffer. Protein suspensions were loaded and resolved on a 10% SDS-PAGE and were then transferred to a polyvinylidene difluoride (PVDF) membrane (Perkin-Elmer, Norwalk, CT, USA). Sirt1, iNOS, eNOS, IκB-α, total caspase-3, and cleaved caspase-3 were detected using specific antibodies from Santa Cruz Biotechnology (Santa Cruz, CA; sc-74465, sc-650, sc-654, sc-1643, sc-7148, and sc-22171, respectively). Detection of GAPDH using an HRP-conjugated monoclonal antibody (1:8000, Sigma) served as a loading control. Reactivity was visualized using Super Signal West Pico Chemiluminescent Substrate (Pierce, Rockford, IL, USA).
Determination of nitrite and nitrate (NOx) concentrations
Cells were seeded onto a 12-well plate and treated with the indicated reagents for 24 h. After treatment, the supernatants were collected, and the levels of NOx, which are the stable end products of NO, were measured using a Total Nitrite/Nitrate Assay kit (Dojindo, Kumamoto, Japan), which employs the Griess method.
NF-κB luciferase assay
NF-κB activity was measured using an NF-κB luciferase assay. Cells were seeded onto 24-well culture plates at 2×104 cells/well. Cells were incubated for 1 h with plasmids (85 ng of NF-κB-dependent luciferase reporter and 85 ng of pcDNA3-β-gal), 1 μL of Tfx-50 reagent (Promega), and 200 μL of serum-free RPMI-1640. After 1 h, 800 μL of RPMI-1640 containing FBS was added, and the cells were incubated for 24 h. Cells were then treated with the indicated reagents for 1 h. Luciferase activity was measured using a luciferase assay system and normalized against β-galactosidase activity.
MC3T3-E1 cells were transfected with Ad-Sirt1 or Ad-lacZ for 12 h. Cells were then treated as described above to assay for NF-κB activity after treatment with TNF-α (5.0 ng/mL).
Statistical analysis
The in vitro experiments were performed at least three times, and each experiment was performed with replicates. Data are expressed as the mean±SD. The significance level was determined by the Student's t-test. A difference was considered to be statistically significant at P<0.05.
Results
Effect of TNF-α on Sirt1 expression in MC3T3-E1 cells
To investigate the involvement of Sirt1 protein in TNF-α-induced cytotoxicity in osteoblasts, we examined Sirt1 expression levels in response to TNF-α. Western blot analysis showed that incubation with TNF-α (0, 2.5, 5.0, and 10.0 ng/mL) resulted in a reduction of Sirt1 protein expression in osteoblasts (Figure 1).

The effect of TNF-α on Sirt1 expression in MC3T3-E1 cells. MC3T3-E1 cells were cultured with an increasing concentration of TNF-α (2.5, 5.0, and 10 ng/mL) for 24 h. Western blot results and responding quantification of Sirt1 were displayed. GAPDH, glyceraldehyde phosphate dehydrogenase. The group without stimulation of TNF-α served as control. The ratio of Sirt1 to GAPDH of other groups was normalized to the control. bP<0.05 vs control.
Overexpression of Sirt1 protein in MC3T3-E1 cells
To gain insights into the function of Sirt1 in osteoblasts, Sirt1 was overexpressed using a recombinant adenovirus. Transfection of osteoblasts with Ad-Sirt1 markedly increased Sirt1 protein levels (Figure 2A). Sirt1 deacetylase activity was also enhanced in Ad-Sirt1-transfected osteoblasts (Figure 2B).

Sirt1 protein was overexpressed by transfecting MC3T3-E1 cells with Ad-Sirt1 adenovirus. MC3T3-E1 cells were transfected with Ad-Sirt1 or Ad-lacZ for 12 h. The expression of Sirt1 (A) was assayed by Western blotting analysis, and deacetylase activity (B) of overexpressed Sirt1 was measured using fluorogenic substrate. GAPDH, glyceraldehyde phosphate dehydrogenase. bP<0.05 vs Ad-lacZ cells.
Overexpression of Sirt1 attenuated TNF-α-induced cell injury in MC3T3-E1 cells
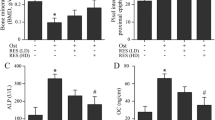
Treatment with TNF-α resulted in an increase in apoptosis and a reduction in ALP activity in osteoblasts. However, overexpression of Sirt1 significantly attenuated TNF-α-induced cell injury through suppressing apoptosis (Figure 3A), increasing ALP activity (Figure 3B), and increasing the expression of Runx2 (Figure 3C) and osteocalcin (Figure 3D) mRNA.

The effect of Sirt1 overexpression on TNF-α-induced cell injury in MC3T3-E1 cells. After successfully transfected with Ad-Sirt1 or Ad-lacZ, two groups of cells were stimulated with TNF-α, the apoptosis (Western blot results and responding quantification of cleaved caspase-3 and caspase-3, A), ALP activity (B), and mRNA levels of Runx2 (C) and osteocalcin (D) were assessed. ALP, alkaline phosphatase; Runx2, runt-related transcription factor 2. bP<0.05 vs Ad-lacZ cells. eP<0.05 vs TNF-α-treated Ad-lacZ cells.
Effect of Sirt1 overexpression on TNF-α-induced NF-κB activation
We investigated the effects of Sirt1 overexpression on NF-κB signaling in MC3T3-E1 cells. Degradation of IκBα induced by TNF-α (5.0 ng/mL) was partially blocked by Sirt1 overexpression (Figure 4A). As demonstrated by the luciferase reporter assay, TNF-α (5.0 ng/mL) increased the activity of NF-κB in MC3T3-E1 cells. This increase was also partially blocked by Sirt1 overexpression (Figure 4B). Without TNF-α treatment, Sirt1 overexpression had no significant effect on NF-κB activity.

The effect of Sirt1 overexpression on TNF-α-induced IkBα degradation (A) and NF-κB activation (B). Control or Sirt1-overexpressing osteoblastic cells were treated with TNF-α (5.0 ng/mL) for 1 h. Activity of NF-κB was analyzed by luciferase reporter assay. bP<0.05 vs Ad-lacZ cells. eP<0.05 vs TNF-α-treated Ad-lacZ cells.
Effect of Sirt1 overexpression on TNF-α-induced NO production and iNOS protein expression
In this study, incubation of osteoblasts with TNF-α (5.0 ng/mL) led to an increase in iNOS expression. To define the link between TNF-α and iNOS expression, we investigated downstream signaling pathways of TNF-α, which primarily activates the JNK and NF-κB pathways19,20. BAY-11-7082, an NF-κB inhibitor, and SP600125, a JNK inhibitor, were used to block activation of NF-κB and JNK, respectively. Data showed that TNF-α-induced iNOS expression was inhibited by BAY-11-7082 (10 μmol/L) and not by SP600125 (10 μmol/L) (Figure 5A), indicating that the TNF-α-induced increase in iNOS expression was mediated by the NF-κB pathway and not the JNK pathway. Because NF-κB activation induced by TNF-α was inhibited by Sirt1 overexpression, we examined the effect of Sirt1 overexpression on TNF-α-induced iNOS expression. We found that Sirt1 overexpression suppressed the iNOS induction by TNF-α (5.0 ng/mL) (Figure 5B). Furthermore, NO production induced by TNF-α (5.0 ng/mL) was also reduced by Sirt1 overexpression (Figure 5C). In untreated osteoblasts, Sirt1 overexpression increased the formation of NO. Because iNOS is expressed at low levels in a steady state20, we examined the effect of Sirt1 overexpression on the expression of eNOS in basal conditions and found it to also be enhanced by Sirt1 overexpression (Figure 5D).

Effect of Sirt1 overexpression on TNF-α-induced NO production and iNOS protein expression. Cells were treated with BAY-11-7082 (10 μmol/L, an NF-κB inhibitor) or SP600125 (10 μmol/L, a JNK inhibitor) in the absence or presence of TNF-α (5.0 ng/mL) for 24 h. The iNOS expression was assayed by Western blotting analysis (A). Control or Sirt1-overexpressing osteoblastic cells were cultured in the absence or presence of TNF-α (5.0 ng/mL) for 24 h. Then, the iNOS expression was assayed by Western blotting analysis (B) and NO formation was detected by the Griess method (C). Control or Sirt1-overexpressing osteoblastic cells were cultured and eNOS expression was assayed by Western blotting analysis (D). NOx, nitrite and nitrate; iNOS, inducible nitric oxide synthase; eNOS, endothelial nitric oxide synthase; GAPDH, glyceraldehyde phosphate dehydrogenase. bP<0.05 vs Ad-lacZ cells. eP<0.05 vs TNF-α-treated Ad-lacZ cells.
Activation of Sirt1 by resveratrol attenuated TNF-α-induced cell injury in MC3T3-E1 cells
Resveratrol (10 μmol/L), a Sirt1 activator, reversed the TNF-α-induced inhibition of Sirt1 deacetylase activity (Figure 6A) and activation of NF-κB activity (Figure 6B). We further assessed the protective effects of resveratrol (10 μmol/L) against cell injury induced by TNF-α in osteoblasts to complement the results obtained by using the overexpression approach. Treatment with resveratrol (10 μmol/L) decreased NO formation (Figure 6C), restored ALP activity (Figure 6D), increased the expression of both Runx2 (Figure 6E) and osteocalcin mRNA (Figure 6F), and suppressed apoptosis (Figure 6G) after treatment with TNF-α. Incubation of osteoblasts with EX-527 (5 μmol/L), a Sirt1 inhibitor, reversed the effect of resveratrol on TNF-α-induced cell injury, affirming the role of Sirt1 in TNF-α-induced cell injury.

The effect of resveratrol on TNF-α-induced cell injury in MC3T3-E1 cells. Cells were treated with α-MEM (control), TNF-α (5 ng/mL), TNF-α+Resveratrol (10 μmol/L, a Sirt1 activator), TNF-α+Resveratrol+EX-527 (5 μmol/L, a Sirt1 inhibitor). Sirt1 deacetylase activity (A), Luciferase activity (B), supernatant NOx content (C), ALP activity (D), and expressions of Runx2 (E) and osteocalcin (F), apoptosis (Western blot results and responding quantification of cleaved caspase-3 and caspase-3, G), were assessed. NOx, nitrite and nitrate; ALP, alkaline phosphatase; TNF-α, tumor necrosis factor alpha; Res, Resveratrol. bP<0.05 vs control. eP<0.05 vs only TNF-α-treated cells. hP<0.05 vs TNF-α+Resveratrol-treated cells.
Discussion
Sirt1, a class III histone/protein deacetylase, interferes with the NF-κB signaling pathway and thereby has an anti-inflammatory function21. Because of the central role of NF-κB in cytokine-mediated injury in osteoblasts, we hypothesized that Sirt1 might play a role in osteoblast models of cytokine-induced cell damage. Our data provide multiple lines of evidence that Sirt1 has a protective effect against TNF-α-induced cell injury in osteoblast cells. First, treatment of osteoblast cells with TNF-α decreased Sirt1 protein levels. Second, Sirt1 overexpression attenuated TNF-α-induced cell injury in osteoblasts. Third, Sirt1 overexpression suppressed the TNF-α-induced NF-κB activation, reduced iNOS expression, and reduced NO formation in the osteoblast cells. Fourth, the Sirt1 activator, resveratrol, mimicked the protective effects of Sirt1 overexpression against TNF-α-induced cell injury in osteoblasts. These data suggest that Sirt1 plays a cytoprotective role against TNF-α in osteoblast cells, at least in part, by suppressing NF-κB activity.
We first showed that Sirt1 protein levels were decreased in osteoblast cells by treatment with TNF-α. A recent report by Takayama et al22 showed that Sirt1 protein levels were downregulated by various stresses, including nutritional stress, catabolic stress, and mechanical shear stress in human chondrocytes. Together with our findings, these data suggest that Sirt1 might be involved in the modulation of osteoporosis induced by inflammation or other factors. However, TNF-α induced elevated expression of Sirt1 in vascular smooth muscle cells23. Thus, the underlying mechanisms of these contradictory observations in different cell types require further investigation.
Several cytokines can regulate inflammatory responses in osteoblast cells by modulating the NF-κB signaling pathway. TNF-α, a pro-inflammatory cytokine, has been implicated in the early events related to osteoblast cell destruction. Suppression of TNF-α production or blockade of its interaction with its cellular receptors significantly inhibits deleterious effects in related osteoporotic diseases24,25,26,27. TNF-α activates several intracellular signaling pathways, including the AP-1 pathway, the MAPK pathway, and the JNK pathway in osteoblastic cells28,29,30. However, to our knowledge, TNF-α exerts deleterious effects primarily through the NF-κB signaling pathway in osteoblastic cells31,32,33. Because NF-κB is a molecular target of Sirt134,35, we investigated the influence of the overexpression of Sirt1 on NF-κB signaling pathways in TNF-α-treated osteoblasts. As expected, Sirt1 overexpression suppressed the NF-κB activity induced by TNF-α. In addition, treatment with resveratrol, a Sirt1 activator, also attenuated TNF-α-induced cell injury, which was, at least in part, due to activation of Sirt1 deacetylase and subsequent inhibition of NF-κB activity.
Sirt1 overexpression also blocked the TNF-α-induced expression of iNOS and formation of NO. Activation of the iNOS system has been shown to partly mediate inflammation-induced osteoporosis16. High concentrations of NO, such as those observed after stimulation with pro-inflammatory cytokines, not only mediate cytokine-induced apoptosis17 but also have potent inhibitory effects on osteoblast growth and differentiation20,36,37. Therefore, Sirt1 exerted its protective role against TNF-α, at least in part, through suppressing iNOS protein expression downstream of NF-κB.
In conclusion, overexpression of Sirt1 protects osteoblasts against TNF-α-induced cell injury, at least in part, by repressing NF-κB activity and genes downstream of NF-κB, including iNOS. Our results suggest that Sirt1 is a novel therapeutic target for treating inflammation-related bone loss.
Author contribution
This research was designed by Wei Huang. The experiments were performed by Wei HUANG, Wei-lin SHANG, Hua-dong WANG, Wen-wen WU, and Shu-xun HOU. The manuscript was written by Wei HUANG, Wei-lin SHANG, and Hua-dong WANG.
References
Pacifici R . Estrogen, cytokines, and pathogenesis of postmenopausal osteoporosis. J Bone Miner Res 1996; 11: 1043–51.
Romas E, Martin TJ . Cytokines in the pathogenesis of osteoporosis. Osteoporos Int 1997; 7: S47–53.
Angeli A, Dovio A, Sartori ML, Masera RG, Ceoloni B, Prolo P, et al. Interactions between glucocorticoids and cytokines in the bone microenvironment. Ann NY Acad Sci 2002; 996: 97–107.
Nowell MA, Richards PJ, Fielding CA, Ognjanovic S, Topley N, Williams AS, et al. Regulation of pre-B cell colony-enhancing factor by STAT-3-dependent interleukin-6 trans-signaling: implications in the pathogenesis of rheumatoid arthritis. Arthritis Rheum 2006; 54: 2084–95.
Feldmann M, Brennan FM, Maini RN . Role of cytokines in rheumatoid arthritis. Annu Rev Immunol 1996; 14: 397–440.
Pfeilschifter J, Köditz R, Pfohl M, Schatz H . Changes in proinflammatory cytokine activity after menopause. Endocr Rev 2002; 23: 90–119.
Scott DL, Kingsley GH . Tumor necrosis factor inhibitors for rheumatoid arthritis. New Engl J Med 2006; 355: 704–12.
Feldmann M, Maini RN . Anti-TNF alpha therapy of rheumatoid arthritis: what have we learned? Annu Rev Immunol 2001; 19: 163–96.
Kuno H, Kurian SM, Hendy GN, White J, deLuca HF, Evans CO, et al. Inhibition of 1,25-dihydroxyvitamin D3 stimulated osteocalcin gene transcription by tumor necrosis factor-alpha: structural determinants within the vitamin D response element. Endocrinology 1994; 134: 2524–31.
Kitajima I, Soejima Y, Takasaki I, Beppu H, Tokioka T, Maruyama I . Ceramide-induced nuclear translocation of NF-kappa B is a potential mediator of the apoptotic response to TNF-alpha in murine clonal osteoblasts. Bone 1996; 19: 263–70.
Gilbert L, He X, Farmer P, Boden S, Kozlowski M, Rubin J, et al. Inhibition of osteoblast differentiation by tumor necrosis factor-alpha. Endocrinology 2000; 141: 3956–64.
Bäckesjö CM, Li Y, Lindgren U, Haldosén LA . Activation of Sirt1 decreases adipocyte formation during osteoblast differentiation of mesenchymal stem cells. J Bone Miner Res 2006; 21: 993–1002.
Haigis MC, Sinclair DA . Mammalian sirtuins: Biological insights and disease relevance. Annu Rev Pathol 2010; 5: 253–95.
Shakibaei M, Buhrmann C, Mobasheri A . Resveratrol-mediated SIRT-1 interactions with p300 modulate receptor activator of NF-κB ligand (RANKL) activation of NF-κB signaling and inhibit osteoclastogenesis in bone-derived cells. J Biol Chem 2011; 286: 11492–505.
Chang J, Wang Z, Tang E, Fan Z, McCauley L, Franceschi R, et al. Inhibition of osteoblastic bone formation by nuclear factor-κB. Nat Med 2009; 15: 682–9.
Armour KE, van't Hof RJ, Grabowski PS, Reid DM, Ralston SH . Evidence for a pathogenic role of nitric oxide in inflammation-induced osteoporosis. J Bone Miner Res 1999; 14: 2137–42.
Chen RM, Chen TL, Chiu WT, Chang CC . Molecular mechanism of nitric oxide-induced osteoblast apoptosis. J Orthop Res 2005; 23: 462–8.
Lee JH, Song MY, Song EY, Kim EK, Moon WS, Han MK, et al. Overexpression of sirt1 protects pancreatic β-cells against cytokine toxicity by suppressing the nuclear factor-κB signaling pathway. Diabetes 2009; 58: 344–51.
Moncada S, Higgs A . The L-arginine nitric oxide pathway. N Engl J Med 1993; 329: 2002–12.
MacPherson H, Noble BS, Ralston SH . Expression and functional role of nitric oxide synthase isoforms in human osteoblast-like cells. Bone 1999; 24: 179–85.
Yang SR, Wright J, Bauter M, Seweryniak K, Kode A, Rahman I . Sirtuin regulates cigarette smoke-induced proinflammatory mediator release via RelA/p65 NF-κB in macrophages in vitro and in rat lungs in vivo: implications for chronic inflammation and aging. Am J Physiol Lung Cell Mol Physiol 2007; 292: L567–6.
Takayama K, Ishida K, Matsushita T, Fujita N, Hayashi S, Sasaki K, et al. Sirt1 regulation of apoptosis of human chondrocytes. Arthritis Rheum 2009; 60: 2731–40.
Zhang HN, Li L, Gao P, Chen HZ, Zhang R, Wei YS, et al. Involvement of the p65/RelA subunit of NF-kappaB in TNF-alpha-induced Sirt1 expression in vascular smooth muscle cells. Biochem Biophys Res Commun 2010; 397: 569–75.
Ammann P, Rizzoli R, Bonjour JP, Bourrin S, Meyer JM, Vassalli P, et al. Transgenic mice expressing soluble tumor necrosis factor-receptor are protected against bone loss caused by estrogen deficiency. J Clin Invest 1997; 99: 1699–703.
Kimble RB, Bain S, Pacifici R . The functional block of TNF but not of IL-6 prevents bone loss in ovariectomized mice. J Bone Miner Res 1997; 12: 935–41.
Brennan FM, Chantry D, Jackson A, Maini R, Feldmann M . Inhibitory effect of TNF alpha antibodies on synovial cell interleukin-1 production in rheumatoid arthritis. Lancet 1989; 2: 244–7.
Redlich K, Hayer S, Maier A, Dunstan CR, Tohidast-Akrad M, Lang S, et al. Tumor necrosis factor alpha-mediated joint destruction is inhibited by targeting osteoclasts with osteoprotegerin. Arthritis Rheum 2002; 46: 785–92.
Aggarwal BB . Tumor necrosis factors receptor associated signaling molecules and their role in activation of apoptosis, JNK and NF-kappaB. Ann Rheum Dis 2000; 59: 6–16.
Chen G, Goeddel DV . TNF-R1 signaling: a beautiful pathway. Science 2002; 296: 1634–5.
Mukai T, Otsuka F, Otani H, Yamashita M, Takasugi K, Inagaki K, et al. TNF-alpha inhibits BMP-induced osteoblast differentiation through activating SAPK/JNK signaling. Biochem Biophys Res Commun 2007; 356: 1004–10.
Wajant H, Pfizenmaier K, Scheurich P . Tumor necrosis factor signaling. Cell Death Differ 2003; 10: 45–65.
Kurokouchi K, Kambe F, Yasukawa K, Izumi R, Ishiguro N, Iwata H, et al. TNF-α increases expression of IL-6 and ICAM-1 genes through activation of NF-κB in osteoblast-like ROS17/2.8 cells. J Bone Miner Res 1998; 13: 1290–9.
Lee HL, Yi T, Woo KM, Ryoo HM, Kim GS, Baek JH . Msx2 mediates the inhibitory action of TNF-α on osteoblast differentiation. Exp Mol Med 2010; 42: 437–45.
Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, et al. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J 2004; 23: 2369–80.
Salminen A, Kauppinen A, Suuronen T, Kaarniranta K . SIRT1 longevity factor suppresses NF-kappaB-driven immune responses: regulation of aging via NF-kappaB acetylation? Bioessays 2008; 30: 939–42.
Damoulis PD, Hauschka PV . Cytokines induce nitric oxide production in mouse osteoblasts. Biochem Biophys Res Commun 1994; 201: 924–31.
Saura M, Tarin C, Zaragoza C . Recent insights into the implication of nitric oxide in osteoblast differentiation and proliferation during bone development. Sci World J 2010; 10: 624–32.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Huang, W., Shang, Wl., Wang, Hd. et al. Sirt1 overexpression protects murine osteoblasts against TNF-α-induced injury in vitro by suppressing the NF-κB signaling pathway. Acta Pharmacol Sin 33, 668–674 (2012). https://doi.org/10.1038/aps.2011.189
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2011.189
Keywords
This article is cited by
-
Sirtuins and Metabolism Biomarkers in Relapsing-Remitting and Secondary Progressive Multiple Sclerosis: a Correlation Study with Clinical Outcomes and Cognitive Impairments
Molecular Neurobiology (2023)
-
Glycyrrhizic Acid Protects Glomerular Podocytes Induced by High Glucose by Modulating SNARK/AMPK Signaling Pathway
Current Medical Science (2023)
-
Pinolenic acid exhibits anti-inflammatory and anti-atherogenic effects in peripheral blood-derived monocytes from patients with rheumatoid arthritis
Scientific Reports (2022)
-
Association of Serum Levels of Silent Information Regulator 1 with Persistent Organ Failure in Acute Pancreatitis
Digestive Diseases and Sciences (2019)
-
Chikusetsusaponin V Inhibits LPS-Activated Inflammatory Responses via SIRT1/NF-κB Signaling Pathway in RAW264.7 Cells
Inflammation (2018)
