
Abstract
Amyloid beta peptide (Aβ) is produced through the proteolytic processing of a transmembrane protein, amyloid precursor protein (APP), by β- and γ-secretases. Aβ accumulation in the brain is proposed to be an early toxic event in the pathogenesis of Alzheimer's disease, which is the most common form of dementia associated with plaques and tangles in the brain. Currently, it is unclear what the physiological and pathological forms of Aβ are and by what mechanism Aβ causes dementia. Moreover, there are no efficient drugs to stop or reverse the progression of Alzheimer's disease. In this paper, we review the structures, biological functions, and neurotoxicity role of Aβ. We also discuss the potential receptors that interact with Aβ and mediate Aβ intake, clearance, and metabolism. Additionally, we summarize the therapeutic developments and recent advances of different strategies for treating Alzheimer's disease. Finally, we will report on the progress in searching for novel, potentially effective agents as well as selected promising strategies for the treatment of Alzheimer's disease. These prospects include agents acting on Aβ, its receptors and tau protein, such as small molecules, vaccines and antibodies against Aβ; inhibitors or modulators of β- and γ-secretase; Aβ-degrading proteases; tau protein inhibitors and vaccines; amyloid dyes and microRNAs.
Similar content being viewed by others

Introduction
Alzheimer's disease is the most common type of dementia. It affects tens of millions of people worldwide, and this number is rising dramatically. The social and economic burden of Alzheimer's disease is high. The amyloid hypothesis1,2,3 proposes β-amyloid (Aβ) as the main cause of the disease and suggests that misfolding of the extracellular Aβ protein accumulated in senile plaques4 and the intracellular deposition of misfolded tau protein in neurofibrillary tangles cause memory loss and confusion and result in personality and cognitive decline over time. Accumulated Aβ peptide is the main component of senile plaques and derives from the proteolytic cleavage of a larger glycoprotein named amyloid precursor protein (APP). APP is a type 1 membrane glycoprotein that plays an important role in a range of biological activities, including neuronal development, signaling, intracellular transport, and other aspects of neuronal homeostasis. Several APP cleavage products may be major contributors to Alzheimer's disease, causing neuronal dysfunction. Deposits of Aβ peptides are mainly observed in the region of the hippocampus and the neocortex as well as in the cerebrovasculature (CAA)5.
As Aβ peptides are the main components of senile plaques, understanding the structures and biochemical properties of Aβ will advance our understanding of Alzheimer's disease at the molecular level. Aβ monomers aggregate into different forms of oligomers, which can then form regular fibrils. The peptides share a common structural motif and aggregation pathway, providing a powerful conceptual framework for understanding the pathogenic mechanism and disease-specific factors. Here, we review the structure and biology of Aβ, which may constitute a core pathway for the growing number of neurodegenerative diseases, including Alzheimer's, Parkinson's, and Huntington's diseases, as well as structure-based drug discovery, which may contribute to the development of novel treatment strategies against different degenerative diseases.
Structure of the amyloid beta peptide
Molecular architecture of APP and its proteolysis in the amyloidogenic pathway
The Aβ peptides are cleaved from the much larger precursor APP. APP is an integral membrane protein expressed in many tissues, especially in the synapses of neurons, which plays a central role in Alzheimer's disease (AD) pathogenesis. APP consists of a single membrane-spanning domain, a large extracellular glycosylated N-terminus and a shorter cytoplasmic C-terminus. It is one of three members of a larger gene family in humans. The other two family members are the APP-related proteins (APLPs) APLP1 and APLP26. APP has been implicated as a regulator of synaptic formation and repair 7, anterograde neuronal transport8 and iron export9. It is produced as several different isoforms, ranging in size from 695 to 770 amino acids. The most abundant form in the brain (APP695) is produced mainly by neurons and differs from longer forms of APP in that it lacks a Kunitz-type protease inhibitor sequence in its ectodomain10 . APP isoform 695 is mainly expressed in neurons, whereas APP751 and APP770, which contain the Kunitz-type serine protease inhibitory domain KPI, are mainly expressed on peripheral cells and platelets11,12 (Figure 1).

Molecular architecture of APP. Schematic representation of human APP isoforms and the APP-like proteins (APLP), APLP1 and APLP2. APP isoforms range in size from 695 to 770 amino acids. The most abundant form in brain is APP695, which lacks a Kunitz type protease inhibitor sequence in its ectodomain. APP751 and APP770 contain the Kunitz type serine protease inhibitory domain (KPI) are mainly expressed on the surface of peripheral cells and platelets.
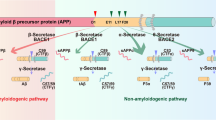
APP is best known as the precursor molecule cut by β-secretases and γ-secretases to produce a 37 to 49 amino acid residue peptide, Aβ13, that lies at the heart of the amyloid cascade hypothesis and whose amyloid fibrillar form is the primary component of amyloid plaques found in the brains of Alzheimer's disease patients. Human APP can be processed via two alternative pathways: amyloidogenic and nonamyloidogenic. APP is first cleaved by α-secretase (nonamyloidogenic pathway) or β-secretase (amyloidogenic pathway), generating membrane-tethered α- or β-C terminal fragments (CTFs). The cleavage of APP by α-secretase releases sAPPα from the cell surface and leaves an 83-amino-acid C-terminal APP fragment (C83). The production of sAPPα increases in response to electrical activity and the activation of muscarinic acetylcholine receptors, suggesting that neuronal activity increases the α-secretase cleavage of APP 14. Further processing involves the intramembrane cleavage of α- and β-CTFs by γ-secretase, which liberates the P3 (3 kDa) and Aβ (4 kDa) peptides, respectively15,16. The amyloidogenic processing of APP thus involves sequential cleavages by β- and γ-secretase at the N and C termini of Aβ, respectively (Figure 2)17 . The 99-amino-acid C-terminal fragment of APP (C99) generated by β-secretase cleavage can be internalized and further processed by γ-secretase at multiple sites to produce cleavage fragments of 43, 45, 46, 48, 49 and 51 amino acids that are further cleaved to the main final Aβ forms, the 40-amino-acid Aβ40 and the 42-amino-acid Aβ42, in endocytic compartments18,19. The cleavage of C99 by γ-secretase liberates an APP intracellular domain (AICD) that can translocate to the nucleus, where it may regulate gene expression, including the induction of apoptotic genes. The cleavage of APP/C99 by caspases produces a neurotoxic peptide (C31)20. The β-site APP cleaving enzyme is abundant in neurons, which may accelerate the amyloidogenic processing pathway in the brain and impair neuronal survival. The three-dimensional structure of human γ-secretase was determined by single-particle cryo-electron microscopy in 201421. The γ-secretase complex comprises a horseshoe-shaped transmembrane domain, which contains 19 transmembrane segments (TMs), and a large extracellular domain (ECD) from the nicastrin subunit, which localizes immediately above the hollow space formed by the TM horseshoe. This structure serves as an important basis for understanding the mechanisms of γ-secretase function. The γ-secretase complex consists of four different proteins, presenilin, nicastrin, presenilin enhancer 2 and anterior pharynx-defective 1. Presenilin is activated by auto-processing to generate N- and C-terminal cleavage products that both contain aspartyl protease sites that together are required for the activity of the mature γ-secretase. Nicastrin, presenilin enhancer 2 and anterior pharynx-defective 1 are critical components of γ-secretase and may modulate enzyme activity in response to physiological stimuli22,23,24. This unique cleavage process of APP provides essential targets for AD therapeutics25.

Human APP proteolytic pathways. Human APP proteolysis in the non-amyloidogenic pathway and amyloidogenic pathway. Non-amyloidogenic processing of APP refers to the sequential processing of APP by membrane bound α-secretases, which cleave within the Aβ domain to generate the membrane-tethered α-C terminal fragment CTFα (C83) and the N-terminal fragment sAPPα. CTFα is then cleaved by γ-secretases to generate extracellular P3 and the APP intracellular domain (AICD). Amyloidogenic processing of APP is carried out by the sequential action of membrane bound β- and γ-secretases. β-Secretase cleaves APP into the membrane-tethered C-terminal fragments β (CTFβ or C99) and N-terminal sAPPβ. CTFβ is subsequently cleaved by γ-secretases into the extracellular Aβ and APP intracellular domain (AICD).
Aβ monomer
Aβ monomers aggregate into various types of assemblies, including oligomers, protofibrils and amyloid fibrils. Amyloid fibrils are larger and insoluble, and they can further assemble into amyloid plaques, while amyloid oligomers are soluble and may spread throughout the brain. The primary amino acid sequence of Aβ was first discovered from extracellular deposits and amyloid plaques in 19842. The primary amino acid sequence of the 42-amino-acid Aβ isoform Aβ42 is shown here (Figure 3A). Aβ encompasses a group of peptides ranging in size from 37 to 49 residues. Amyloid plaques with Aβ as the main component are most commonly found in the neocortex in the brain of Alzheimer's disease patients26.

Structures of Aβ monomer, fibril and oligomers. (A) The primary amino acid sequence of the 42 amino acid Aβ isoform Aβ42. Aβ encompasses a group of peptides ranging in size from 37–49 residues. (B) The structure of amyloid beta peptide (1–28), which forms a predominately alpha-helical structure that can be converted to a beta-sheet structure in membrane-like media (PDB code: 1AMC, 1AMB), it's the major proteinaceous component of amyloid deposits in Alzheimer's disease. The side chains of histidine-13 and lysine-16 residing on the same face of the helix are close. (C) Solution structure of amyloid beta peptide (1–40), in which the C-terminal two-thirds of the peptide form an alpha-helix conformation between residues 15 and 36 with a kink or hinge at 25–27 in aqueous sodium dodecyl sulfate (SDS) micelles with a bend centered at residue 12, while the peptide is unstructured between residues 1 and 14 which are mainly polar and likely solvated by water (PDB code: 1BA4, 1BA6) . It collapsed into a compact series of loops, strands, and turns with no alpha-helical or beta-sheet structure. The van der Waals and electrostatic forces maintain its conformational stabilization. Approximately 25% of the surface is uninterrupted hydrophobic, and the compact coil structure is meta-stabled, which may lead to a global conformational rearrangement and formation of intermolecular beta-sheet secondary structure caused by fibrillization. (D) Amyloid beta peptide (10–35) forms a collapsed coil structure (PDB code: 1HZ3). It collapsed into a compact series of loops, strands, and turns with no alpha-helical or beta-sheet structure. The van der Waals and electrostatic forces maintain its conformational stabilization. Approximately 25% of the surface is uninterrupted hydrophobic, and the compact coil structure is meta-stabled, which may lead to a global conformational rearrangement and formation of intermolecular beta-sheet secondary structure caused by fibrillization. (E) Proposed pathway for the conversion of amyloid beta monomers to higher order oligomers, protofibrils and fibrils. Aβ monomers can form higher order assemblies ranging from low molecular weight oligomers, including dimers, trimers, tetramers, and pentamers, to mid-range molecular weight oligomers including hexamers, nonamers and dodecamers to protofibrils and fibrils.
Aβ is commonly thought to be intrinsically unstructured and hence cannot be crystallized by common methods . Many studies therefore focus on optimizing conditions that can stabilize Aβ peptides. The three-dimensional solution structure of different fragments of the Aβ peptide was determined using nuclear magnetic resonance (NMR) spectroscopy, molecular dynamic (MD) techniques and X-ray crystallography. Most structural knowledge about Aβ comes from NMR and molecular dynamics.
Early NMR-derived models of the solution structure of Aβ peptide (1-28) indicated that it folds into a predominately α-helical structure with β-sheet conversion in membrane-like media that may also occur during the early stages of amyloid formation in Alzheimer's disease27 (Figure 3B). It is the major proteinaceous component of amyloid deposits in Alzheimer's disease, where the side chains of histidine-13 and lysine-16 residing on the same face of the helix are in close proximity. The solution structure of Aβ peptide (1-40) suggests that the C-terminus of the peptide has an α-helix conformation between residues 15 and 36 with a kink or hinge at 25-27 in aqueous sodium dodecyl sulfate (SDS) micelles, while the peptide is unstructured between residues 1 and 14, which are mainly polar and likely solvated by water . The deprotonation of two acidic amino acids in the helix promotes a helix-to-coil conformational transition that precedes the aggregation of Aβ1-4028 (Figure 3C). Solid-state NMR spectroscopy-derived models of the solution structure of Aβ peptide (10-35) show that in water29 (Figure 3D), the peptide collapses into a compact series of loops, strands, and turns without alpha-helical or beta-sheet structure. The van der Waals and electrostatic forces maintain its conformational stabilization. Approximately 25% of the surface is uninterruptedly hydrophobic, and the compact coil structure is meta-stable, which may lead to a global conformational rearrangement and the formation of an intermolecular beta-sheet secondary structure caused by fibrillization . The 3D NMR structures of Aβ peptide (8-25) and Aβ peptide (28-38) show two helical regions connected by a regular type I β-turn. Aβ peptide (25-35) is a highly toxic synthetic derivative of Aβ peptides. Researchers have used NMR and CD investigation of Aβ peptide (25-35) and fluoro-alcohols to scan its conformational properties. The peptide behaves as a typical transmembrane helix in a lipidic environment, forming fibrillar aggregates, which suggests a direct mechanism of neurotoxicity30,31.
NMR-guided simulations of Aβ peptides 1-40 (Aβ40) and 1-42 (Aβ42) also suggested very different conformational states32, with the C-terminus of Aβ42 being more structured and residues 31-34 and 38-41 forming a β-hairpin that reduces the C-terminal flexibility, which may be responsible for the greater propensity of Aβ42 than Aβ40 to form amyloids. Replica exchange molecular dynamics studies suggested that Aβ40 and Aβ42 can indeed populate multiple discrete conformations, comprising α-helix or β-sheet conformers, and the structural states transition rapidly33. More recent studies identified a multiplicity of discrete conformational clusters by statistical analysis34. However, the most recent NMR structure of Aβ40 shows significant secondary and tertiary structure35. The hydrophobic C-terminal of the Aβ is critical in triggering the transformation from α-helical to β-sheet structure and plays a key role in determining the state of protein aggregation in Alzheimer's disease36.
Aggregation of Aβ into fibrils
Early proposals supported the so-called “amyloid cascade hypothesis,” which proposes that Aβ aggregation into plaques leads to neurotoxicity and dementia37 through common cytopathic effects that contribute to the pathogenesis of Alzheimer disease and other amyloidosis. While the Aβ peptide can rapidly aggregate to form fibrils that deposit into the amyloid plaques, which are found to be linked with Alzheimer's disease, later studies demonstrated that there is no direct correlation between amyloid plaques38 and the loss of synapses and neurons in brains with Alzheimer's disease39,40. Many pathways may lead to the peptide aggregation. Early studies indicated that the amyloid polypeptide is organized in a characteristic “cross β” pattern in a regular manner, in which adjacent chain segments are folded in an anti-parallel manner within the fiber lattice41. Later research revealed that the peptide chains of β-strand segments run perpendicular to the long fibril, and the intermolecular hydrogen bonds of β-strands run parallel to the axis in a “cross β” structural pattern42.
Solid-state NMR measurements have shown that amyloid fibril “cross β” structures exist in two patterns: parallel and antiparallel. The crosslinking of Aβ peptides by tissue transglutaminase (tTg) indicated that the Aβ fibril is a hydrogen-bonded, parallel β-sheet that defines the long axis of the Aβ fibril propagation43. Specific amino acid contacts have implications for the overall fibril formation of the extended Aβ (10-35) and its stability, morphology and parallel organization44. Multiple quantum (MQ) 13C NMR data indicate an in-register, parallel organization45. These measurements, known as experimental EM, STEM, and solid-state NMR, suggest that the supramolecular structures of Aβ peptide (1-40) fibrils, Aβ peptide (10-35), and Aβ peptide (1-42) fibrils are organized as β-sheets46,47. As Aβ peptide aggregation pathways are determined by the primary amino acid sequence and the intermolecular interactions, later studies in the structural organization of disease-related amyloid fibrils have led to the identification of the exact register motif. In addition to the parallel pattern, several short peptide segments of Aβ can adopt an antiparallel pattern48. Solid-state NMR spectroscopy indicates amyloid fibrils with a simple and intriguing structural motif49. Site-directed spin labeling and electron paramagnetic resonance (SDSLEPR) spectroscopy for amyloid fibrils confirmed that this parallel, exact register structural motif is highly conserved50,51,52. Progress has been made by disulfide cross-linking within preformed fibrils, with results indicating that they are located proximally inside the hairpin turn. Residues 17 and 34 could be efficiently cross-linked by a disulfide bond, while residues 17/35 and 17/36 were not efficiently cross-linked in fibrils. Purified double mutant proteins consisted of disulfide-bonded monomers that were able to assemble into amyloid fibrils53. The 17/35 residues on the C-terminal strand would need to flip 180 degrees to provide the structural flexibility allowing Aβ to assemble into at least two slightly different forms54,55,56. These results are inconsistent with the hairpin model based on electrostatic interactions, with the exception of the side chains of Glu22 and Lys2857. It is unclear whether small differences in the fibril structure are pathologically significant; however, the slight two-residue difference in Aβ40 and Aβ42 leads to great differences in their biophysical, biological, and clinical behaviors. The 3D structure of residues 15-42 of Aβ42 adopts a double-horseshoe-like cross-β-sheet entity with maximally buried hydrophobic side chains, in which residues 1-14 are partially ordered and in a β-strand conformation, which is the more neurotoxic species, aggregates much faster, and dominates in senile plaque in Alzheimer's disease patients58. Further studies reported that cognitive deficits appeared before plaque deposition or the detection of insoluble amyloid fibrils59,60. In contrast, the amount of oligomeric Aβ61,62 is increased in Alzheimer's disease brain extracts63, which is the basis for the Aβ oligomer hypothesis64,65,66, which posits that soluble Aβ oligomers rather than insoluble fibrils or plaques trigger synapse failure and memory impairment67, resulting in impaired brain function in the final stages of the disease.
Aβ oligomers
While amyloid fibrils are larger, insoluble, and assemble into amyloid plaques forming histological lesions that are characteristic of Alzheimer's disease, Aβ oligomers are soluble and may spread throughout the brain. The size distribution of Aβ oligomers is heterogeneous. There is a broad consensus for the preferential accumulation of a soluble high-molecular-weight species of approximately 100–200 kDa under relatively physiological conditions in vitro68,69,70,71,72. Aβ monomers can form higher-order assemblies ranging from low-molecular-weight oligomers, including dimers, trimers, tetramers, and pentamers, to midrange molecular weight oligomers, including hexamers, nonamers and dodecamers, to protofibrils and fibrils (Figure 3E). In contrast to the fibril structure, relatively little is known about the structure of amyloid oligomers. Soluble oligomers prepared in the presence of detergents seem to feature substantial beta sheet content with mixed parallel and antiparallel character73. The structural characterization of oligomers is complicated because their oligomeric states are more transient than fibrils, and preparing homogeneous populations of oligomers is difficult74. They can be stabilized by detergents, which may help to alleviate this problem75. There was little structural information on the oligomeric state of amyloid beta until 2010, when low temperature and low salt conditions made it possible to isolate pentameric disc-shaped oligomers devoid of beta structure76. Circular dichroism and infrared spectroscopy indicate that Aβ oligomers are extended coil or beta sheet structures63. Hydrogen deuterium exchange analysis also indicates that they have a stable core, which is consistent with substantial beta sheet character, as 40% of the total backbone hydrogen bonds are resistant to exchange in the oligomeric conformation with a stable beta sheet secondary structure77. In contrast, fifty percent of the backbone hydrogen bonds are resistant to exchange in the mature amyloid fibril, indicating that a small increase in main chain hydrogen bonding accompanies the transition to the fibrillar conformation78. Computational studies suggest that Aβ oligomers form an antiparallel beta-turn-beta motif79. The solution conformation of Aβ is of significant importance during self-assembly in water environments. The soluble peptide has no alpha-helical or beta-sheet character but adopts a collapsed coil structure80. A particular conformation that forms ring-shaped pentamers and hexamers is stable by microsecond all-atom MD simulations81.
The relationship between oligomers and fibrils remains to be established. There seem to be some similar structural elements, as they both appear to be extended or beta sheet structures and both display similar amounts of main chain hydrogen bonding that is resistant to exchange. On the other hand, amyloid oligomers and fibrils appear to contain mutually exclusive and non-overlapping conformations recognized as generic antibody epitopes that are common to amyloids of different sequences74,82. Oligomers are a kinetic intermediate waxing at early times during the development of fibrils83. Different types of soluble amyloid oligomers have a common structure and share a common mechanism of toxicity63. It is also unknown whether the oligomer structures represent basic units of amyloid protein that then assemble into fibrils or are just in equilibrium with monomers, which directly form fibrils without intermediate oligomeric structure. Oligomers appear as spherical aggregates at early times and then elongate by the coalescence of spherical subunits with a “bead” appearance, forming the precursor of protofibrils on the pathway to mature fibers. The parallelism between Aβ monomers represents a key organizing principle for amyloid oligomers and may also serve as a common structural motif for amyloid fibrils71,84. Other studies suggest that the spherical oligomers simply dilute the Aβ monomer concentration and may be off-pathway intermediates85 or that both on-pathway and off-pathway concurrence is possible under special conditions69.
The structure of Aβ aggregate forms and the aggregation pathways remain challenging research issues, though considerable progress has been made recently. The interactions of Aβ with transition metals have revealed potential pathogenic interactions and structural consequences. Oligomers that may normally be embedded in the membrane bind to transition metals such as Cu, Zn and Fe86,87. Constitutively metal-bound senile plaques play a role in accelerating the aggregation of amyloid beta peptide88, and the expression of Aβ oligomers may, in turn, regulate metal transition homeostasis89,90,91. NMR data have provided information on the structure of the Aβ-(1-16)-Zn2+ complex in aqueous solution. The residues His(6), His (13), and His (14) and the Glu (11) carboxylate were identified as ligands that tetrahedrally coordinate the Zn(II) cation92.
All these different structures have been generated in different environments and determined by different techniques. The special form of Aβ structures may be not stable or may be stabilized only in a unique solution; they may be similar but not the same as each other; one structure may depict one representative form of Aβ, and all the forms of Aβ may co-exist in vivo. Aβ forms a myriad of structures in the monomeric and oligomeric states, all of which result in similar fibril structures. Amyloid fibrils of Aβ form a parallel, in-register cross β-sheet structure. The accumulation of Aβ into long, unbranched fibrils is a hallmark of the disease, as is the loss of neurons due to cell death in parallel with the Aβ aggregation process. These new insights into the structures and aggregation pathways may help to uncover the mechanisms of amyloid pathogenesis in degenerative diseases, ultimately leading to new therapeutic strategies to prevent the formation of toxic aggregates (Table 1).
Biological function of amyloid beta
Aβ production
Alzheimer's disease is characterized by abnormal accumulation of the Aβ protein, which is important for memory and cognition, in the brain regions. Aβ is a normal product of the cellular metabolism derived from the amyloid precursor protein (APP). APP is synthesized in the endoplasmic reticulum (ER) and then transported to the Golgi complex, where it completes maturation and is finally transported to the plasma membrane. Mature APP at the plasma membrane is cleaved by the successive action of the β-secretase and γ-secretase to generate Aβ (Figure 2)93. The newly generated Aβ either is released to the extracellular space or remains associated with the plasma membrane and lipid raft structures. The binding of Aβ to ganglioside GM1 in the lipid rafts strongly favors Aβ aggregation94 . The binding of ApoE to Aβ taken up by the cells through receptor-mediated endocytosis mediated by LRP (LDL receptor-related protein), and LDLR regulates aggregation but also the cellular uptake of Aβ95 . Endocytosed Aβ also has access to other subcellular compartments through the vesicular transport system. Earlier studies pointed to Aβ fibrils as the neurotoxic agent leading to cellular death, memory loss, and other AD characteristics. Over the last two decades, further investigation has suggested that oligomeric or prefibrillar species of the Aβ peptide are the most damaging to neuronal cells. Soluble Aβ can bind to numerous molecules in the extracellular space, including cell surface receptors, metals and cellular membranes.
Aβ binding receptors
The extracellular accumulation of Aβ in neuritic plaques and the binding of Aβ to a variety of receptors appear to be the characteristic hallmarks of Alzheimer's disease. The binding of Aβ to a variety of receptors has been proposed as a cause for the neuronal toxicity: Aβ oligomers were proposed to induce mitochondrial dysfunction and oxidative stress in AD neurons, resulting in a massive calcium influx and toxicity in neurons96. Furthermore, soluble oligomeric Aβ was proposed to be toxic through binding to a variety of receptors, including lipids, proteoglycans, and proteins, such as the Aβ-binding p75 neurotrophin receptor (P75NRT), the low-density lipoprotein receptor-related protein (LRP), cellular prion protein (PrPc), metabotropic glutamate receptors (mGluR5), α subunit containing nicotinic acetylcholine receptor (α7nAChR), N-methyl-D-aspartic acid receptor (NMDAR), β-adrenergic receptor (β-AR), erythropoietin-producing hepatoma cell line receptor (EphR), and paired immunoglobulin-like receptor B (PirB)97. The Aβ/Aβ receptor interactions are proposed to generate and transduce neurotoxic signals into neurons, causing cellular defects such as mitochondrial dysfunction and the ER stress response. In addition, some Aβ receptors are most likely to internalize Aβ into neurons to display distinct cellular defects (Figure 4).

Biological functions of Aβ. Aβ monomers can form higher order assemblies ranging from low molecular weight oligomers (including dimers, trimers, tetramers, and pentamers) to midrange-molecular weight oligomers, high molecular weight oligomers, protofibrils fibrils and senile plaques. Soluble Aβ can interact with potential receptors and activate downstream pathways to generate reactive oxygen species, hyperphosphorylate Tau protein, and cause inflammatory responses, which may result in neuronal death and lead to Alzheimer's disease.
NMDAR and α7nAChR
NMDAR and α7nAChR are both ion channel receptors. Several reports suggest that Aβ interacts with NMDARs at postsynaptic terminals, and antibodies raised against the GluN1 or GluN2B subunit of NMDARs markedly block the binding of the Aβ oligomer to neurons98,99, indicating that Aβ oligomers partially co-localize with the GluN2B subunits of NMDARs at the cell surface100. Indeed, Aβ directly or indirectly binds to NMDAR subunits to activate NMDAR, and thus Aβ oligomers induce calcium dysregulation, neuronal death 101, and synaptic dysfunction102,103. Moreover, Aβ oligomers promote the endocytosis of NMDARs, which requires the activation of α7nAChR signaling104. The receptor α7nAChR is another candidate Aβ-binding receptor and binds to soluble Aβ with high affinity105. The α7nAChR-expressing cells are susceptible to Aβ-induced toxicity in vitro106, and it mediates Aβ-induced tau phosphorylation via the ERK and JNK pathways107. In a mouse model of AD, α7nAChR may exacerbate AD pathology in a mouse model, while its deficiency may improve cognitive deficits and synaptic pathology108.
Aβ binding p75 neurotrophin receptor (p75NTR)
The receptor p75NTR is a TNF family low affinity receptor for neurotrophins. Aβ binds to both p75NTR monomers and trimers (Figure 4), which activates their intracellular signaling to induce apoptosis in human neuroblastoma cells. Early studies compared neuroblastoma cell clones that either did not express any of the neurotrophin receptors or had been engineered to express full-length or various truncated forms of the p75NTR109 . These studies showed that p75NTR binds to Aβ via its extracellular domain, which directly signals cell death via its death domain. In fact, this signaling leads to the activation of caspase 8 and caspase 3 and to the production of reactive oxygen species (ROS) and cellular oxidative stress110 . In addition, Aβ can interact synergistically with cytokines TNFα and IL1β, which markedly strengthens the neurotoxic actions of Aβ/p75NTR signaling and potentiates neuronal damage. Aβ-bound p75NTR triggers cell death in the hippocampus of human Alzheimer's disease brains. Taken together, these findings indicated that p75NTR-expressing neurons endowed with receptors for proinflammatory cytokines might be the reason for the target selectivity of Aβ cytotoxic actions in Alzheimer's disease111,112.
Low-density lipoprotein receptor-related protein (LRP)
The low-density lipoprotein receptor-related protein (LRP), also known as alpha-2-macroglobulin receptor (A2MR), apolipoprotein E receptor (APOER) or cluster of differentiation 91 (CD91), is a protein receptor found in the plasma membrane of cells involved in receptor-mediated endocytosis. LRP1 is involved in various biological processes such as lipoprotein metabolism and cell motility, and pathologically in neurodegenerative diseases, atherosclerosis and cancer113.
LRP is a multifunctional cell surface receptor of more than 600 kDa in size with a single transmembrane-spanning domain. LRP has more than 20 identified ligands, many of which are localized to the central nervous system. The broad categories of these ligands include apolipoprotein E (apoE) and lipid-related ligands as well as protease and protease inhibitor complexes such as APP containing Kunitz proteinase inhibitor, α2M, tissue plasminogen activator and plasminogen activator inhibitor 1 complexes, and others such as lactoferrin. Cholesterol is imported into neurons by apoE via LRP1 receptors. Starving neurons of cholesterol and malfunction of the neuronal cholesterol metabolism is thought to be a causal factor in Alzheimer's disease114. In addition, over-accumulation of copper in the brain is associated with reduced LRP1-mediated clearance of Aβ across the blood brain barrier. This defective clearance may contribute to the buildup of neurotoxic Aβ115. Together, these studies support a critical role of the multifunctional receptor LRP in Aβ metabolism and Alzheimer's disease.
LRP also interacts with the amyloid precursor protein itself. LRP regulates APP trafficking and processing by different mechanisms. SorLA (also called SORL1, SORLA1, or LR11) is a neuronal apolipoprotein E receptor that can regulate the intracellular transport and processing of the APP in neurons. It alters the localization of APP to discrete intracellular compartments, resulting in a decrease of extracellular Aβ levels116. LRP and LRP1B expression and endocytosis are thought to play opposing roles in APP endocytosis, resulting in increased APP processing to Aβ levels in the presence of LRP for a rapid fast endocytosis rate and decreased Aβ production in the presence of LRP1B for a slower endocytosis rate117.
PrPc and mGlu5 receptors in astrocyte upregulation by Aβ
PrPc is a glycosylphosphatidylinositol (GPI)-anchored membrane protein that can undergo a conformational change to an infectious, pathological state called scrapie prion protein (PrPSc), which is linked to transmittable spongiform encephalopathies and causes terminal neurodegenerative disorders 118. PrPc-binding ligands include the laminin γ1-chain, Cu2+ ions and Aβ42 oligomers119,120, the latter of which binds PrPc with high affinity121.
Metabotropic glutamate receptors (mGluR5) are members of the G-protein coupled receptor superfamily. mGluR5 has been specifically implicated in neurodegenerative diseases such as Alzheimer's disease, Parkinson's disease and Huntington's disease122,123,124. The activation of mGluR5 has been shown to decrease the fragile X mental retardation protein (FMRP)-mediated translation repression of APP and to stimulate sAPPα secretion, which induces the β-secretase pathway and Aβ production. FMRP can stimulate neural pruning and synaptic plasticity, which results in neuroprotection under normal physiological conditions125. More recently, mGluR5 has been suggested to be the primary co-receptor for both PrPc and Aβ oligomers126 (Figure 4). The extracellular domain of mGluR5 interacts with both PrPc and Aβ42, which results in the activation of Ca2+ release from intracellular stores, thus promoting PKC translocation and ERK1/2 phosphorylation. Aβ42 and PrPc also activate mGluR5 to stimulate Fyn kinase-mediated APP protein translation.
Immune globin receptors FccRIIb and PirB
Two immune globin receptors, FccRIIb and PirB, originally believed to function exclusively in the immune system, were recently shown to play neuropathic roles as Aβ receptors in Alzheimer's disease brains97,127,128. These two proteins show similarity in their structures and in the high binding affinity for Aβ oligomers. Both have immunoglobulin (Ig) domains in their extracellular domains and immunoreceptor tyrosine-based inhibitory (ITI) motifs in their intracellular domains. FccRIIb has two Ig domains and one ITI motif, whereas PirB has six Ig domains and four ITI motifs. FccRIIb interacts with low-molecular-weight oligomers via its second Ig domain, and PirB binds to high-molecular weight-oligomers via its first two Ig domains.
Other receptors
Other receptors, such as microglia receptors, are also involved in the amyloid cascade. Microglial membrane receptors bind Aβ and contribute to microglial activation and Aβ phagocytosis and clearance. These receptors can be categorized into several groups. The scavenger receptors (SRs) include scavenger receptor A-1 (SCARA-1), MARCO, scavenger receptor B-1 (SCARB-1), CD36 and the receptor for advanced glycation end product (RAGE)129. The G-protein coupled receptor (GPCR) group includes formyl peptide receptor 2 (FPR2) and chemokine-like receptor 1 (CMKLR1)130, and the toll-like receptor (TLR) group includes TLR2, TLR4, and the co-receptor CD14131. Functionally, SCARA-1 and CMKLR1 participate the uptake of Aβ, and RAGE is responsible for the activation of microglia and production of proinflammatory mediators in response to Aβ binding. CD36, CD36/CD47/α6β1-integrin, CD14/TLR2/TLR4, and FPR2 display functions in both Aβ binding and microglia activation. In addition, MARCO and SCARB-1 exhibit the ability to bind Aβ and may be involved in the progression of Alzheimer's disease132.
A variety of microglia receptors are involved in Aβ clearance and in triggering an inflammatory response. Some receptors, including RAGE and NLRP3, are mainly implicated in the generation of an inflammatory response by triggering a signaling cascade that results in the production of proinflammatory mediators133. Other receptors, such as SR-AI and TREM2, participate the clearance of Aβ by inducing internalization of Aβ fibrils. Complement receptors, Fc receptors, FPRL1/FPR2, CD36, and TLRs are involved in both Aβ clearance and the generation of inflammatory responses, while the microglia receptor CD33 seems to accelerate Aβ accumulation.
Aβ degradation
The production of Aβ is normally counterbalanced by several processes, including proteolytic degradation, cell-mediated clearance (which may itself involve proteolytic degradation), active transport out of the brain, and deposition into insoluble aggregates. A growing body of evidence suggests that proteolytic degradation is a particularly important determinant of cerebral Aβ levels and, by extension, of Aβ-associated pathology134. The individual Aβ-degrading proteases neprilysin, endothelin-converting enzymes, insulin-degrading enzyme, plasmin and other Aβ-degrading proteases play important roles in Aβ degradation and Alzheimer's disease, although their relative importance remains to be established (Figure 5).

Aβ homeostasis involves production, aggregation, transport, degradation, and clearance. Aβ is produced in peripheral tissues and the CNS, where it can aggregate and form insoluble fibrils. Soluble Aβ can be transported across the BBB from blood to brain via RAGE, and from brain to blood via LRP. Aβ can also bind to transport proteins, eg., apoE, apoJ, α2-macroglobulin (α2M), which may influence Aβ sequestration as well as the form of its accumulation in brain. Aβ can be proteolytically degraded by the proteases Neprilysin (Nep), endothelin converting enzymes (ECE), insulin degrading enzyme (IDE), plasmin and other Aβ-degrading proteases (MMP, Cathepsin D), as well as by microglia-mediated degradation.
Neprilysin (NEP) is a 93-kDa zinc metallo-endopeptidase implicated in the degradation of a wide array of bioactive peptides135 and is the most efficient Aβ peptidase. It is a type 2 membrane glycoprotein with its active site located in the intraluminal/extracellular space, into which Aβ peptides are normally secreted136. NEP is also localized to the early Golgi and endoplasmic reticulum and other subcellular compartments. Synthetic Aβ was first demonstrated to undergo proteolysis by NEP, and Aβ degradation was most strongly inhibited by a potent and selective inhibitor of NEP, thiorphan137. The overexpression of NEP resulted in a lack of amyloid accumulation in APP transgenic mice, while the absence of NEP expression resulted in amyloid aggregation in APP transgenic mice138,139. These experimental and clinical observations, therefore, support the hypothesis that Aβ degradation and the development of idiopathic Alzheimer's disease may be greatly affected by the regulation of an aging-induced reduction of NEP activity.
Endothelin converting enzymes 1 and 2 (ECE1 and ECE2) are membrane-bound zinc metalloproteinases belonging to the same family as NEP (M13 family). While other members of the M13 family are capable of degrading Aβ in cell culture or in vitro140,141, the addition of phosphoramidon, a known inhibitor of ECEs, can significantly elevate the secretion of Aβ into the medium of cultured cells. The overexpression of ECEs in cultured cells stably expressing APP led to a reduction of more than ninety percent in the level of secreted Aβ, and this effect was reversed by treatment with phosphoramidon. Taken together, the involvement of ECEs may play a causal role in the degradation of Aβ142.
Insulin-degrading enzyme (IDE) is a 110-kDa zinc metallo-endopeptidase that degrades a broad range of substrates, including insulin, glucagon, and amylin, along with a range of other bioactive peptides143, as well as the intracellular domain of APP144. Early studies identified IDE as the first protease to degrade Aβ in vitro within crude brain homogenates145, and IDE was later identified independently as the major Aβ degrading component secreted into the medium by a range of cultured cells146. Moreover, IDE might indirectly affect Aβ levels via its effects on AICD levels, which has recently been implicated in the transcriptional regulation of APP147 and neprilysin expression148,149.
Plasmin is a serine protease that is the ultimate effector in the fibrinolytic cascade. Plasmin can degrade and reduce the toxicity of both monomer and fibril Aβ150,151,152,153.
Other candidate Aβ-degrading proteases remain to be identified. The matrix metalloproteases (MMPs) MMP2 and MMP9154,155 have been shown to degrade Aβ in vitro. Angiotensin-converting enzyme is yet another metalloprotease that may play an important role in the pathogenesis of Alzheimer's disease and that has been shown to degrade Aβ in vitro156. Cathepsin D, an aspartyl protease localized within lysosomes and endosomes, was identified as a major Aβ-degrading enzyme in brain homogenates157, and its expression level in the brain is altered in Alzheimer's disease158.
Aβ transport
In addition to degradation, Aβ released into the extracellular space can be transported between different compartments, such as from the brain to the blood or from the blood to the brain, and can also be cleared by chaperones, such as apoE, which can affect Aβ metabolism after it is released by cells and influence Aβ aggregation, clearance, and transport159.
The carrier- and receptor-mediated transport of Aβ across the blood brain barrier (BBB) regulates brain Aβ levels160,161,162,163,164,165. The concentration of soluble Aβ in the CNS, which is central to the formation of neurotoxic oligomeric Aβ species166 and vascular aggregated forms of Aβ, is critically influenced by Aβ transport exchange across the BBB. This transport process has been reported to be regulated by receptors, such as advanced glycation end products (RAGE)167, or the low-density lipoprotein receptor-related protein 1 (LRP1)168,169. Moreover, other receptors such as glycoprotein 330 (gp330/megalin)170 and P-glycoprotein171 may also contribute to the transport of Aβ across the BBB, and the Aβ-binding proteins α2-macroglobulin, apoE and apoJ appear to influence this process172. The levels and form of Aβ may be greatly determined by not only the vascular clearance and BBB transport of Aβ, proteolytic degradation173,174, oligomerization, and aggregation but also by the production175,176 and clearance of different forms of Aβ (fibrillar vs soluble) by other cells of the neurovascular unit, such as astrocytes177,178,179. These activities may also play a major role in determining the brain accumulation and associated neuronal and vascular toxicity. Plasma Aβ levels may contribute more to Alzheimer's disease toxicity in cognitively normal elderly individuals180,181. Strategies to clear Aβ from the vascular system can reduce the Aβ levels and amyloid load in the CNS. These strategies include the use of an anti-Aβ antibody182,183,184, non-immune approaches with gelsolin, GM1185, sRAGE186 or soluble forms of LRP-1, sLRP-1 fragments187 and insulin-like growth factor I187. Many receptors are involved in inducing Aβ transport and clearance. Among them, RAGE is an influx transport receptor that binds soluble Aβ and mediates pathophysiological cellular responses188,189,190. RAGE also mediates the transport of plasma Aβ across the BBB. LRP-1 functions as a clearance receptor for Aβ at the BBB191,192 (Figure 5).
ApoE can regulate Aβ transport, clearance, and aggregation. ApoE is a 299-amino-acid lipid transport protein expressed as three different isoforms: apoE2, apoE3 and apoE4. E3 is the most common isoform, and E4 is responsible for a genetic predisposition to Alzheimer's disease, increasing the risk of Alzheimer's disease by approximately 3-fold more than the E3 allele, whereas E2 decreases AD risk193. Glial cells such as astrocytes and microglia secrete apoE into the interstitial fluid (ISF) of the brain. When Aβ is secreted into the brain ISF, mostly by neurons, apoE-containing high-density lipoproteins (HDL) interact with Aβ and influence its clearance into cells via the endocytic LDL receptor family member LDLR. The binding of apoE/Aβ complexes to heparin sulfate proteoglycans (HSPG) can increase the retention of Aβ in the extracellular matrix of the brain and arterioles. This process may play a role in the development of cerebral amyloid angiopathy (CAA)194 . ApoE and Aβ have been shown to colocalize in detergent-insoluble glycolipid-rich membrane domains (DIGs), which may promote their interaction. In the ISF, apoE/Aβ interactions likely determine whether and when Aβ will aggregate. ApoE may also play a role in Aβ transport out of the brain via ISF/bulk flow, which can modulate both soluble and fibrillary Aβ clearance as well as transport and fibrillogenesis and, in doing so, plays an important role in Alzheimer's disease and CAA pathogenesis195.
Aβ forms and their toxicity
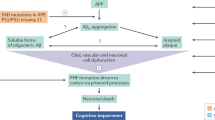
The different forms of Aβ include soluble Aβ, Aβ oligomer and Aβ present in amyloid plaques. In addition, a dynamic compartmentalization of the different types of Aβ may exist between plaques and soluble Aβ196, and the different Aβ forms may contribute to neurodegeneration at different stages of the disease197. Aβ has also been reported to form aggregates in two fundamental types of reactions: non-metal-dependent association and metal-dependent association. Non-metal Aβ aggregates form soluble oligomers and amyloid fibrils, while metal Aβ aggregates form ionically bridged aggregates, covalently crosslinked oligomers, and seeds for non-metal-dependent Aβ fibrillization198. Accumulating Aβ first forms Aβ oligomers and gradually deposits as fibrils and senile plaques. In addition, tau protein becomes hyperphosphorylated in response to kinase/phosphatase activity changes mediated by Aβ aggregation, leading to the formation of neurofibrillary tangles (NFTs), neuronal and eventual synaptic dysfunction, and finally Alzheimer's disease (Figure 4). When the process of self-aggregation occurs on neuron membranes, it generates a toxic aldehyde called 4-hydroxynonenal and leads to lipid peroxidation, which can damage the function of ion-motive ATPases, glucose transporters and glutamate transporters. In turn, Aβ promotes depolarization of the synaptic membrane, excessive calcium influx and mitochondrial damage, which impairs the ability of cells to conduct normal physiological activities79.
Furthermore, the aggregation of Aβ may also produce free radicals as ROS that react rapidly with proteins or lipids, resulting in the formation of “toxic” oxidized proteins and peroxided lipids. Oxidized proteins are harmful to the membrane integrity and may also alter the sensitivity to oxidative modifications of enzymes such as glutamine synthetase (GS) and creatine kinase (CK), which are critical to neuronal function199,200. Peroxidized lipids can generate toxic products such as 4-hydroxy-2-nonenal (HNE) and 2-propenal (acrolein) that migrate to different parts of neurons and cause multiple harmful alterations to cellular activity. The deleterious functions associated with neuronal death include the inhibition of ion-motive ATPases and glial cell Na+-dependent glutamate transport, loss of Ca2+ homoeostasis, and disruption of signaling pathways201,202,203. In addition to proteins and lipids, oxidative stress induced by Aβ aggregation has also been reported to cause DNA oxidation, leading to DNA damage.
Sustained elevation of Aβ levels and continuous aggregation might also promote a chronic response of the innate immune system by activating microglia, which can lead to neuronal loss through direct phagocytosis. The immunological receptors that are activated by Aβ include toll-like receptor 2 (TLR2), TLR4, TLR6, and their co-receptors CD14, CD36, and CD47204,205. In addition, Aβ aggregation also causes inflammatory responses and the release of inflammation-related mediators, such as eicosanoids, chemokines, proinflammatory cytokines and complement factors, which can increase neuronal death and the loss of neuronal synapses and impair the clearance of Aβ and the neuronal debris mediated by microglia. In addition to the microglia driven neuroinflammatory response206, this processes is probably also mediated indirectly by regulating kinase/ phosphatase activity.
Moreover, when the Aβ precursor APP accumulates at the mitochondrial membrane, it blocks the translocation of inner mitochondrial metabolites and proteins, leading to disruption of the electron-transport chain and mitochondrial dysfunction, which may in turn increase excessive Aβ generation and result in greater toxicity in a feed-forward loop207,208. Excessive Aβ levels also activate the mitochondrial fission proteins Fis1 and Drp1, thereby inducing mitochondrial fragmentation209. Aβ localized in the mitochondria can interact with the proapoptotic factors Aβ-binding alcohol dehydrogenase (ABAD) and cyclophilin D (CypD), resulting in increased neuronal cell death210. Therefore, there may be a vicious feedback loop between increased Aβ production and mitochondrial dysfunction.
Extracellular deposits of fibrils or amorphous aggregates of Aβ peptide form plaques and diffuse deposits, while intracellular fibrillar aggregates of hyperphosphorylated and oxidated tau can form neurofibrillar tangles. These plaques and neurofibrillary tangles are deposited mainly in brain regions, such as the hippocampus, amygdala, entorhinal cortex, and basal forebrain, that influence memory and learning and emotional behaviors. Aβ can damage synapses and neurites, and plaque deposits in brain regions reduce the number of synapses. Aβ specifically damages neurons that produce serotonin and norepinephrine or that employ glutamate or acetylcholine as neurotransmitters. After synthetic Aβ fragments were found to kill cultured neurons211, the chemical and cell biological bases for the synaptic dysfunction and death of neurons in Alzheimer's disease were reported by a series of studies. Aβ, particularly in its aggregating forms, can impair synaptic ion and glucose transporters, and electrophysiological studies have shown that Aβ impairs synaptic plasticity. Decreasing sAPPα levels can increase the resistance of neurons to oxidative and metabolic insults, which is consistent with sAPPα contributing to the demise of neurons, which is coincident with the increased production of Aβ in Alzheimer's disease212,213. Memory deficits correlate with the formation of Aβ oligomers, which appear relatively early in the process of Aβ deposition in APP mutant mice214. Remarkably, the immunization of APP mutant mice with human Aβ42 resulted in the removal of Aβ deposits from the brain and the reversal of cognitive deficits, adding to the evidence that Aβ deposition is a pivotal event in Alzheimer's disease215.
Aβ and Alzheimer's disease
The cause of most Alzheimer's cases is still unknown. Although it is characterized mostly by the formation of amyloid plaques in the brain, there are several other competing hypotheses regarding the cause of the disease.
The amyloid hypothesis proposed that the fundamental cause of the Alzheimer's disease is the deposits of extracellular Aβ peptides216. Mutations in the human APP gene cause the development of amyloid plaques and Alzheimer's-like brain pathology, especially in early-onset familial Alzheimer's disease (EOFAD)217,218. Mutations in the human APP gene are close to the γ-secretase site and can increase the Aβ42/Aβ40-ratio. It is reported that mutations that alter residues C-terminal to the Aβ42 site reduce cleavage efficiency and increase the Aβ42/Aβ40 ratio219. AD-causing mutations also occur in the genes PSEN1 and PSEN2. Mutations in the human PSEN1 and PSEN2 genes affect γ-secretase activity and can increase the Aβ42/Aβ40 ratio. Some early-onset families do not show mutations in APP, PSEN1 or PSEN2. Several additional key proteins may be involved in γ-and β-secretase cleavage events, as well as in the hyperphosphorylation of tau and the development of neurofibrillary tangles65,220. Late-onset Alzheimer's disease (LOAD) is characterized by a pattern of interwoven genetic and non-genetic factors. These risk-factor genes each affect one or more of the known pathogenic mechanisms: increased Aβ production and aggregation; decreased Aβ clearance and degradation; increased inflammation; and resistance to γ-secretase activity, and thus lead to neurodegeneration in AD221. Among these risk genes, for APOE, the alleles occurring at the APOE loci ɛ2, ɛ3 and ɛ4 were tested and shown to be associated with increased risk of AD222,223. Other researchers have been led to suspect that non-plaque Aβ oligomers are toxic and might be the main cause of neurodegenerative disorders such as Alzheimer's disease224.
Non-Aβ hypothesis
The cause of most Alzheimer's cases is still unknown. Numerous reports on genetic evidence suggest that Aβ and its aggregation in senile plaques play an important role in the pathogenesis of AD. Aβ cleavage by β-secretase and γ-secretase from APP can result in oligomers that form higher-order fibrils, which then give rise to Aβ plaques. However, genetic causes only explain the small proportion (1% to 5%) of AD cases in which genetic differences have been identified225. The dominant mutations in the genes APP, PSEN1 and PSEN2226, which are implicated in AD pathology, are only present in a very small portion of Alzheimer's cases. It has also been reported that the accumulation of the more insoluble Aβ42 over Aβ40 is an important trigger for AD pathogenesis, while APP can alternatively be cleaved by α-secretase to generate non-plaque-forming extracellular peptides in the non-amyloidogenic processing pathway227. Our previous studies have tested the effect of all 28 FAD-linked C99 mutations and found that most familiar Alzheimer's disease (FAD)-linked APP mutant proteins cause partial resistance to γ-secretase cleavage228. Among them, only mutations that affect residues C-terminal to the Aβ42 cleavage site (Aβ42-53) markedly affect cleavage efficiency and increase the Aβ42 production that leads to AD.
AD pathogenesis includes both Aβ-dependent and Aβ-independent mechanisms. There are still many doubts about the real pathogenesis of AD and the β-amyloid contribution to the onset of the disease. Aβ or Aβ oligomers or plaques are not solely responsible for the onset of the disease. More than 30 mutations responsible for FAD are localized in the APP gene; however, the “type London” APP mutations, causing only a slight increase in β-amyloid production, cause the onset of the pathology earlier than the “type Swedish” mutations, which induce a greater increase of the protein229, suggesting that there are other mechanisms involved in the onset of AD. In fact, the Aβ-independent mechanisms are mediated via APP, intracellular fragments and PS1 via the cellular processes, such as inflammation, oxidative stress and Ca2+ dysregulation, implicated in AD pathogenesis230. Cdk5 may be influenced by or interact with both pathways, and its activation triggers DNA damage, cell cycle activation and neurodegeneration231. Non-Aβ factors such as Tau and ApoE also contribute to AD pathology232. All these pathways can lead to synaptic dysfunction, neurodegeneration and AD.
The senile plaques also do not seem to be an exclusive feature of Alzheimer's disease. They increase with age, even in healthy subjects, and the number of plaques in healthy controls is often comparable with the number found in age-matched affected individuals233. Moreover, β-amyloid is physiologically produced in healthy brains during neuronal activity and is necessary for synaptic plasticity and memory234. Furthermore, in the AD population, there is only a weak correlation between the number of senile plaques and the severity of the pathology. The cleavage of APP by γ-secretase produces some fragments called AICD (APP intracellular domain), which appear to play an important role in the onset of AD. In fact, it is known that transgenic mice for AICD show tau phosphorylation and aggregation and decreased cell proliferation/survival, even in the absence of endogenous APP235. High levels of AICD may also play an important role in the pathology in human brains236. The challenges to the Amyloid Hypothesis of Alzheimer's Disease are sharply formulated237. There are several other competing hypotheses, such as the cholinergic hypothesis, the tau hypothesis, and the hypothesis that some other environmental risk factors, may contribute additional causes of the disease.
The cholinergic hypothesis proposed that AD is caused by cholinergic effects such as reduced synthesis of the neurotransmitter acetylcholine, or the initiation of large-scale aggregation of amyloid and neuroinflammation238,239. Most currently available drug therapies are based on this hypothesis240.
The genetic heritability of Alzheimer's disease reveals that most AD is caused by mutations in one of the genes that encoding APP and presenilins 1 and 2241. Most mutations in these genes increase Aβ42 production. Environmental and genetic risk factors, such as the ɛ4 allele of the apolipoprotein E (APOE)242, increase the risk of the disease by threefold. Mutations in the TREM2 gene make the risk of developing Alzheimer's disease several times higher243. Other genes also appear to affect the risk, including CASS4, CELF1, FERMT2, HLA-DRB5, INPP5D, MEF2C, NME8, PTK2B, SORL1, ZCWPW1, SlC24A4, CLU, PICALM, CR1, BIN1, MS4A, ABCA7, EPHA1, and CD2AP244.
The tau hypothesis postulates that tau protein abnormalities initiate the disease cascade as hyperphosphorylated tau forms neurofibrillary tangles, leading to the disintegration of microtubules in brain cells245, which may result in dysfunction of the biological activity between neurons and later in the death of the cells.
Other hypotheses include such environmental risk factors as smoking and infection, and a neurovascular hypothesis has been proposed, suggesting that the blood-brain barrier is critical for brain Aβ homeostasis and regulates Aβ transport via the LRP receptor and RAGE, as mentioned before246. These findings point to new therapeutic targets for AD.
Aβ and inflammation
Aβ peptides are the major components of the senile plaques present in Alzheimer's disease. Recent studies have shown that the soluble assemblies of Aβ also stimulate neuronal dysfunction and may play a prominent role in stimulating the proinflammatory activation of primary microglia247. In the context of inflammation, compared to fibrillar assemblies, oligomer Aβ preparations induce greater or differential proinflammatory cytokine production by microglia and astrocytes in vitro248. Indeed, studies in primary glia demonstrate that the oligomer Aβ-induced increase in proinflammatory cytokines, such as nitric oxide, NO, TNFα and TNFβ secretion, occurs earlier and is greater than the increase induced by fibrillar assemblies of Aβ249. Thus, for different forms of Aβ, identifying their levels at different stages of AD, the inflammatory response they produce, and their underlying mechanisms (eg, receptor mediated) may provide critical information for therapeutic development.
Other aspects of biology of Aβ
In addition to the key role in the pathology of AD, Aβ generated in the brain and peripheral tissues also function in many other aspects of biology. Aβ has been shown to be a ligand with various receptors, as mentioned in the previous section. It can also be transported between tissues and across the blood-brain barrier by complex trafficking pathways176 to destinations where it can induce and modulate proinflammatory activities in response to a variety of environmental stressors250,251. Aβ also functions similarly to a group of biomolecules collectively known as “antimicrobial peptides” (AMPs) that function in the innate immune system. It inhibits the growth of eight of 12 clinically important pathogens screened252 and acts as an anti-microbial peptide in several infection models including mice, C elegans, and cell culture models253. This new function stands in stark contrast to current models of Aβ-dependent pathology and will play significant roles in the development of future AD treatment strategies.
Therapeutic approaches for the treatment of Alzheimer's disease
Drugs approved by the FDA
To date, only a total of five drugs developed to improve the symptoms of Alzheimer's disease have been approved by the FDA. It is important to note that a new drug, Namzaric (donepezil and memantine)254 was approved in 2014. The five drugs function by two different mechanisms. One is cholinesterase inhibition, which delays Alzheimer's disease by blocking hydrolysis of the critical neurotransmitter acetylcholine. This category of drugs includes donepezil (Aricept)255,256, approved in 1996; rivastigmine (Exelon)255,256, approved in 2000; and galantamine (Razadyne)257, approved in 2001. The other one is memantine (Namenda)258, approved in 2003, a non-competitive N-methyl-D-aspartate (NMDA) channel blocker that reduces the activity of the neurotransmitter glutamate, which plays an important role in learning and memory by binding to the NMDA receptor. Memantine can inhibit the prolonged influx of Ca2+ ions, particularly from extrasynaptic receptors, that forms the basis of neuronal excitotoxicity. It is an option for the management of patients with moderate to severe Alzheimer's disease. Namzaric is a combination of the two drugs to reduce the levels of both acetylcholine and glutamate (Table 2).
Novel therapeutic approaches for Alzheimer's disease
Researchers have identified several novel therapeutic approaches for Alzheimer's disease that focus on the reduction of amyloid oligomer levels. Methods that are currently under development include the inhibition of oligomerization using small molecule inhibitors, the neutralization of oligomeric species using immunotherapy, the overexpression of Aβ-degrading enzymes to control Aβ oligomer levels in the brain, catalytic Aβ antibodies to hydrolyze specific aggregates, β-sheet breakers to break existing β-sheet structures, Aβ blockers to block amyloid channels, and therapies directed against the tau protein to lead to the partial reversal of brain pathologies. All these approaches are in preclinical research stages, and their therapeutic efficiency remains unknown.
Small molecule inhibitors
These molecules (2-amino-4-chlorophenol, 4-aminophenol, 4-aminoanisole, 3,4-dihydroxybenzoic acid, 2-hydroxy-3-ethoxy benzoaldehyde) block Aβ oligomerization or fibrillization259. Fourteen naturally occurring polyphenolic compounds and polyphenol-containing black tea extracts inhibit the assembly of alpha-synuclein into multimeric oligomers, which are cytotoxic and share common structural elements with amyloid oligomers260,261. Polyphenols derived from red wine prevent Aβ oligomerization and attenuate cognitive deterioration. The main phenolic component of olive oil, oleuropein, has been shown to possess antioxidant262, anti-inflammatory263 and hypolipidemic activities264. Small molecules (NQTrp, ClNQTrp, coumarin, furosemide, D737) that inhibit Aβ aggregation265,266 or remodel toxic soluble oligomers of Aβ267 inhibit oligomer formation268,269,270,271,272,273,274 (Table 3).
Secretase inhibitors and modulators
Since β-secretase and γ-secretase are responsible for the release of Aβ from the intracellular domain of APP, compounds that can partially inhibit the activity of either β- or γ-secretase have been extensively explored. β-Secretase inhibitors (eg, MK-8931, CTS21166) can block the first cleavage of APP inside the cell275,276. A novel orally active β-secretase inhibitor, AZD3293, was tested in phase II/III clinical trials by Astra Zeneca and Eli Lilly277. γ-Secretase inhibitors can block the second cleavage of APP in the cell membrane and were expected to stop the subsequent formation of Aβ and its toxic fragments278. One γ-secretase inhibitor, semagacestat, was a candidate drug for a causal therapy against Alzheimer's disease, originally developed by Eli Lilly and Élan, but is unfortunately being stopped as there is no effect in phase III clinical trials279. An alpha-secretase agonist, EHT-0202276, biases APP processing towards the non-amyloidogenic pathway. A new γ-secretase modulator, CHF5074, showed a longer survival time for treated animals280. Selective Aβ42-reducing agents (eg, tarenflurbil) modulate γ-secretase to decrease Aβ42 production in favor of shorter Aβ versions281 (Table 4).
Immunotherapeutic approach
Immunotherapy stimulates the host immune system to recognize and attack Aβ or produces antibodies that enhance the clearance of Aβ oligomers or plaques to prevent plaque deposition. Active or passive Aβ immunization can prevent Aβ oligomerization, which is why antibodies to Aβ can be used to decrease cerebral plaque levels. This decrease is accomplished by promoting microglial clearance and redistributing the peptide from the brain to the systemic circulation. Several epitopes of Aβ are exposed and available for antibody capture of the soluble peptides, while others are available for antibodies to bind with oligomers. One such Aβ vaccine is CAD106, currently in clinical trial265, which induced efficacious Aβ antibody titers of different IgG subclasses mainly recognizing the Aβ3-6 epitope. The 20-amino-acid SDPM1 protein can bind to Aβ40 and Aβ42 tetramers and block subsequent Aβ amyloid accumulation. Aβ42 immunization leads to the clearance of amyloid plaques in patients with Alzheimer's disease but does not prevent progressive neurodegeneration282. A more recent study showed that programmed death 1 (PD-1) inhibitors, which are FDA-approved cancer drugs, may be effective in clearing Aβ plaques and improving cognitive performance in a mouse model of Alzheimer's disease283. Anti-Aβ antibodies (solanezumab, gantenerumab, crenezumab, IVIG), which can bind soluble Aβ and improve cognitive performance, are currently in clinical trials165,284,285. Solanezumab accommodates a large Aβ epitope (960 Å2 buried interface over residues 16 to 26) that forms extensive contacts and hydrogen bonds to the antibody, largely via main-chain Aβ atoms and a deeply buried Phe19-Phe20 dipeptide core Solanezumab and crenezumab both share identity with the Aβ KLVFF epitope286. The human anti-Aβ monoclonal antibody, gantenerumab, binds Aβ plaques and targets the N-terminus and central portion of Aβ287. Intravenous immune globulin (IVIG) derived from human plasma contains IgGs that recognize conformational epitopes of Aβ fibrils and oligomers288. Intravenous immunoglobulin G289 and 2E6290 bind to soluble Aβ and reduce amyloid aggregation (Table 5).
Anti-aggregation agents
Anti-aggregation agents291, such as apomorphine, can prevent Aβ peptides from aggregating or clear aggregates once they are formed292. The hormone melatonin may be effective against amyloid by interacting with dimers of the soluble Aβ peptide and inhibiting their aggregation293,294,295. The cannabinoid HU-210296 has been shown to prevent Aβ-induced inflammation297. The endocannabinoids anandamide and noladin have also been shown to be neuro-protective against Aβ in vitro298. Apomorphine299, melatonin300, and tannic acid301 can prevent Aβ aggregation. A number of small molecules extracted from traditional Chinese herbal medicine have been shown to be capable of inhibiting Aβ aggregation. Among them, LJW0F2 purified from the flowers of Lonicera japonica Thunb could inhibit Aβ42 aggregation and attenuate the cytotoxicity induced by Aβ42 aggregation302. Resveratrol, curcumin, EGb761, isoliquiritigenin, protocatechuic acid, atractylenolide III, chlorogenic acid, euphorbiafactor L3, euphorbiafactor L2, ganoderic acid D, and ganoderic acid DM extracts from Chinese herbal medicine can inhibit Aβ aggregation303,304. A series of substituted bisphenol A derivatives function as Aβ aggregation inhibitors and can inhibit neurotoxicity and increase cell viability305 (Table 6).
Aβ-degrading proteases (AβDPs)
Aβ can be degraded by a number of peptidases and proteinases, collectively known as Aβ-degrading proteases (AβDPs). Aβ-degrading proteases play an important role in Aβ degradation and may be a good target for the treatment of Alzheimer's disease. NEP has been reported to degrade Aβ oligomers that impair neuronal plasticity and cognitive function306. Several close homologues of NEP, NEP2307 and human membrane metalloendopeptidase-like protein (hMMEL)308, are also implicated in the degradation of Aβ308. Members of the M13 family of zinc metalloproteases, endothelin converting enzymes ECE1, ECE2, and ACE, are also known to be endogenous regulators of Aβ levels142,309. The serine proteases plasmin, urokinase type and tissue type plasminogen activators (uPA and tPA, respectively) and acyl peptide hydrolase (APH) have been found to degrade Aβ both directly and indirectly150,310,311,312. Several cysteine proteases, including cathepsin D141, cathepsin B282,313, BACE1283,291, and BACE2 314 are also involved in Aβ degradation (Table 7).
Therapies directed against the tau protein
Neurofibrillary tangles (NFTs) caused by hyperphosphorylated tau are an important pathogenic factor in Alzheimer's disease, and the tau protein is therefore also an important biological target for innovative therapies. The inhibition of tau protein oligomerization and aggregation, tau phosphorylation, microtubule stabilization [epothilone D (BMS-241027), TPI-287]315, and the enhancement of tau degradation as well as tau immunotherapy (ACI-35275) are all potential strategies for Alzheimer's disease therapy. The tau protein hyperphosphorylation inhibitor LMTX can facilitate the clearance of tau from the brain and reduce Aβ aggregation and has reached phase three clinical trials316. The anti-tau AADvac1 vaccine is currently being investigated in phase II trials. AADvac1 has been reported to significantly improve neurobehavioral deficits and reduce neurofibrillary degeneration and mortality317. Moreover, glycogen synthase kinase 3 beta (GSK-3β) inhibitors, such as tideglusib and humulin R, can block the phosphorylation of tau protein and thus are potential drug targets for Alzheimer's disease318 (Table 8).
Other blockers
A drug that is currently under investigation is liraglutide (Victoza), which is typically used as a diabetes drug. Treatment with Victoza improved object recognition and spatial recognition and resulted in cognitive benefits. Other histological benefits include a reduced inflammatory response and an increase in the number of young neurons in the dentate gyrus, where the Aβ level was also found to be significantly reduced319. The β-sheet breakers or blockers that are capable of binding Aβ consist of short synthetic peptides. They destabilize the β-sheet structure and inhibit the formation of Aβ oligomers or amyloids320,321. Aβ oligomers can form calcium channels in membranes. Calcium conductance through these channels can be blocked by compounds MRS2481 and MRS2485, which destabilize the β-sheet structure and decrease Aβ-promoted neuronal toxicity322. Bexarotene might serve as another class of anti-Alzheimer compounds by efficiently preventing the cholesterol-dependent increase in calcium fluxes promoted by Aβ in neural cells323. Voltage-gated calcium channel blockers, such as verapamil, diltiazem, isradipine and nimodipine, protect cultured neurons from Aβ-induced toxicity and thus could be potential candidates for treating Alzheimer's disease324. The 5-HT6 receptor antagonist idalopirdine, in combination with a cholinesterase inhibitor, may also increase cognitive function325. Huperzine A326,327, 2,2′,4′-trihydroxychalcone (TDC)328 and bis(7)-cognitin329 exhibit neuroprotective effects. Agenin330 and clioquinol331 can inhibit Aβ deposition. Other inhibitors, such as the RAGE inhibitor azeliragon, the α7-nAChR inhibitor encenicline and the calcium antagonist nilvaldipine, can improve memory and could be further candidates for Alzheimer's disease therapeutics332,333,334 (Table 9).
Amyloid dyes
The traditional method of identifying amyloid fibrils in tissue sections is by the use of amyloid-staining dyes. The first of these dyes was Congo red335, whose staining is linked to the presence of the cross-β structure of fibrils. Other amyloid dyes include iodine-sulfuric acid336, thioflavin T or S337, crystal violet338, methyl violet339, BTA-1340, chrysamine G341, ANS (1-anilinonaphthalene-8-sulfonic acid)342, bisANS (4,4′-dianilino-1,1′-binaphthyl-5,5′-disulfonic acid)342, Nile red343, K114 ((trans, trans)-1-bromo-2,5-bis (4-hydroxystyryl) benzene)344, FSB345, curcumin346, nanocurcumin346, and others. The thioflavin T staining method is widely used to identify and classify amyloid proteins in tissues. Specific BTA-1 binding to Aβ plaques inhibits Aβ aggregation and cytotoxicity, making it a good drug candidate for Alzheimer's disease340. These amyloid dyes not only indicate the presence of mature amyloids but also function as a tool for dissecting their structure and the mechanism of amyloid formation. They bind selectively to Aβ in the human brain and blood vessels in vitro. They might thus lead to further compounds for the development of tracer agents for the in vivo diagnosis of Alzheimer's disease and of inhibitors of Aβ aggregation as a novel therapy for Alzheimer's disease (Table 10).
MicroRNAs
MicroRNAs (miRNAs) are a class of conserved endogenous small noncoding RNAs known to regulate the expression of complementary messenger RNAs involved in AD development347. MiRNAs in the brain play an important role in Aβ generation, targeting the mRNAs of APP, β-secretase and γ-secretase and altering Aβ expression. MiRNAs may provide a novel therapeutic approach to the treatment of AD while also providing new insights into the etiology of this neurological disorder348. A series of specific miRNAs can regulate APP expression and serve as an ideal target for AD therapeutic drug design. Both miR-107 and miR-29 may potentially target the mRNA of BACE1, which is the key enzyme responsible for generating Aβ protein from APP349.
Conclusions
Aβ is the major component of senile plaques and participates in Alzheimer's disease progression through its neurotoxic effects. Identifying Aβ structures, biology, receptors and Aβ-based therapeutic approaches for the treatment of Alzheimer's disease therefore remains of paramount importance. In this review, we have addressed the different structures involved in Aβ accumulation and have discussed the current understanding of the biological function and neurotoxic role of Aβ and the potential receptors that interact with Aβ and mediate Aβ intake, clearance and metabolism. Identification of the key Aβ receptor under relevant physiological conditions and obtaining crystal structures of full-length Aβ in different states are critical for the development of new therapeutic agents.
Over the last decade, advances have been made in understanding the structures of Aβ peptide forms. Aβ peptide rapidly aggregates to form oligomers, protofibrils and fibrils that lead to the deposition of amyloid plaques. Different structural approaches, such as NMR spectroscopy, distance geometry, molecular dynamic techniques, and X-ray crystallography, have shown that the structural conversion of Aβ oligomers to fibrils involves the association of these loosely aggregated strands into α-helical and parallel β-sheet structures, as well as that the structural states transition quickly. Different signal transduction pathways are involved in Aβ expression, degradation, transport and clearance. The phosphorylation and activation of specific intracellular kinases represent common events in these signaling cascades, and these signaling molecules are potential targets for new Alzheimer's disease drugs.
Several therapeutic approaches for Alzheimer's disease target amyloid oligomers. Methods that are currently under development include the inhibition of Aβ oligomerization using small molecule inhibitors, the neutralization of oligomeric species using immunotherapy, the overexpression of Aβ-degrading enzymes in the brain, catalytic Aβ antibodies for hydrolyzing specific aggregates, β-sheet breakers for destabilizing existing β-sheet structure and Aβ blockers for blocking amyloid channels and thereby leading to the partial reversal of brain pathologies. The therapeutic targeting of microglia receptors implicated in the response to Aβ and their associated signaling pathways could reduce the inflammation associated with Alzheimer's disease. Tau protein inhibitors or vaccines and amyloid dyes that selectively bind Aβ and inhibit Aβ aggregation offer additional novel therapeutic approaches for the treatment of Alzheimer's disease.
We have summarized new progress in developing treatments targeting Aβ and its receptors. Existing Alzheimer's disease drugs only treat the symptoms of Alzheimer's disease; they do not decelerate or cure it. The last drug that was approved by the Food and Drug Administration for therapeutic Alzheimer's disease treatment was namzaric in 2014. In the last decade, several candidate drugs have failed to reach statistical significance in their primary outcomes. The drugs currently undergoing clinical trials are inhibitors of Aβ production and aggregation, Aβ antibodies and vaccines. Identification of the key physiological Aβ receptors and the determination of their crystal structures in complex with Aβ will play a critical role in mitigating Alzheimer's disease progression and symptoms.
Abbreviations
Alzheimer's disease, (Aβ), amyloid precursor protein (APP), Aβ binding p75 neurotrophic receptor (P75NTR), low-density lipoprotein receptor-related protein (LRP), cellular prion protein (PrPc), metabotropic glutamate receptor (mGluR5), α subunit-containing nicotinic acetylcholine receptor (α7nAChR), N-methyl-D-aspartic acid receptor (NMDAR), β-adrenergic receptor (β-AR), erythropoietin-producing hepatocellular receptor (EphR), paired immunoglobulin-like receptor B (PirB).
References
Haass C, Selkoe DJ . Cellular processing of beta-amyloid precursor protein and the genesis of amyloid beta-peptide. Cell 1993; 75: 1039–42.
Glenner GG, Wong CW . Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein (reprinted from Biochemical and Biophysical Research Communications, Vol 120, Pg 885-890, 1984). Biochem Biophys Res Commun 2012; 425: 534–9.
Selkoe DJ, Hardy J . The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med 2016; 8: 595–608.
Bloom GS . Amyloid-beta and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol 2014; 71: 505–8.
Viswanathan A, Greenberg SM . Cerebral amyloid angiopathy in the elderly. Ann Neurol 2011; 70: 871–80.
Coulson EJ, Paliga K, Beyreuther K, Masters CL . What the evolution of the amyloid protein precursor supergene family tells us about its function. Neurochem Int 2000; 36: 175–84.
Priller C, Bauer T, Mitteregger G, Krebs B, Kretzschmar HA, Herms J . Synapse formation and function is modulated by the amyloid precursor protein. J Neurosci 2006; 26: 7212–21.
Turner PR, O'Connor K, Tate WP, Abraham WC . Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory. Prog Neurobiol 2003; 70: 1–32.
Duce JA, Tsatsanis A, Cater MA, James SA, Robb E, Wikhe K, et al. Iron-export ferroxidase activity of beta-amyloid precursor protein is inhibited by Zinc in Alzheimer's disease. Cell 2010; 142: 857–67.
Guerreiro RJ, Gustafson DR, Hardy J . The genetic architecture of Alzheimer's disease: beyond APP, PSENs and APOE. Neurobiol Aging 2012; 33: 437–56.
Vannostrand WE, Rozemuller AJM, Chung R, Cotman CW, Saporitoirwin SM . Amyloid beta-protein precursor in cultured leptomeningeal smooth-muscle cells. Amyloid-Int J Exp Clin Invest 1994; 1: 1–7.
Hardy J . Amyloid, the presenilins and Alzheimer's disease. Trends Neurosci 1997; 20: 154–9.
Nunan J, Small DH . Regulation of APP cleavage by alpha-, beta- and gamma-secretases. FEBS Lett 2000; 483: 6–10.
Haass C, Lemere CA, Capell A, Citron M, Seubert P, Schenk D, et al. The Swedish mutation causes early-onset Alzheimer's disease by beta-secretase cleavage within the secretory pathway. Nat Med 1995; 1: 1291–6.
Kahle PJ, De Strooper B . Attack on amyloid. EMBO Rep 2003; 4: 747–51.
Iwatsubo T . The gamma-secretase complex: machinery for intramembrane proteolysis. Curr Opin Neurobiol 2004; 14: 379–83.
Joshi G, Wang Y . Golgi defects enhance APP amyloidogenic processing in Alzheimer's disease. BioEssays 2015; 37: 240–7.
Olsson F, Schmidt S, Althoff V, Munter LM, Jin S, Rosqvist S, et al. Characterization of intermediate steps in amyloid beta (Abeta) production under near-native conditions. J Biol Chem 2014; 289: 1540–50.
Takami M, Nagashima Y, Sano Y, Ishihara S, Morishima-Kawashima M, Funamoto S, et al. Gamma-secretase: successive tripeptide and tetrapeptide release from the transmembrane domain of beta-carboxyl terminal fragment. J Neurosci 2009; 29: 13042–52.
Lu DC, Rabizadeh S, Chandra S, Shayya RF, Ellerby LM, Ye X, et al. A second cytotoxic proteolytic peptide derived from amyloid beta-protein precursor. Nat Med 2000; 6: 397–404.
Lu P, Bai XC, Ma D, Xie T, Yan C, Sun L, et al. Three-dimensional structure of human gamma-secretase. Nature 2014; 512: 166–70.
Carter DB, Dunn E, Pauley AM, McKinley DD, Fleck TJ, Ellerbrook BR, et al. Changes in gamma-secretase activity and specificity caused by the introduction of consensus aspartyl protease active motif in Presenilin 1. Mol Neurodegener 2008; 3: 6.
Francis R, McGrath G, Zhang J, Ruddy DA, Sym M, Apfeld J, et al. aph-1 and pen-2 are required for Notch pathway signaling, gamma-secretase cleavage of betaAPP, and presenilin protein accumulation. Dev Cell 2002; 3: 85–97.
Takasugi N, Tomita T, Hayashi I, Tsuruoka M, Niimura M, Takahashi Y, et al. The role of presenilin cofactors in the gamma-secretase complex. Nature 2003; 422: 438–41.
Carroll CM, Li YM . Physiological and pathological roles of the gamma-secretase complex. Brain Res Bull 2016; 126: 199–206.
Haass C, Schlossmacher MG, Hung AY, Vigopelfrey C, Mellon A, Ostaszewski BL, et al. Amyloid beta-peptide is produced by cultured-cells during normal metabolism. Nature 1992; 359: 322–5.
Talafous J, Marcinowski KJ, Klopman G, Zagorski MG . Solution structure of residues-1-28 of the Amyloid beta-peptide. Biochemistry 1994; 33: 7788–96.
Coles M, Bicknell W, Watson AA, Fairlie DP, Craik DJ . Solution structure of amyloid beta-peptide(1-40) in a water-micelle environment. Is the membrane-spanning domain where we think it is? Biochemistry 1998; 37: 11064–77.
Zhang S, Iwata K, Lachenmann MJ, Peng JW, Li S, Stimson ER, et al. The Alzheimer's peptide A beta adopts a collapsed coil structure in water. J Struct Biol 2000; 130: 130–41.
Crescenzi O, Tomaselli S, Guerrini R, Salvadori S, D'Ursi AM, Temussi PA, et al. Solution structure of the Alzheimer amyloid beta-peptide (1-42) in an apolar microenvironment–Similarity with a virus fusion domain. Eur J Biochem 2002; 269: 5642–8.
D'Ursi AM, Armenante MR, Guerrini R, Salvadori S, Sorrentino G, Picone D . Solution structure of amyloid beta-peptide (25-35) in different media. J Med Chem 2004; 47: 4231–8.
Sgourakis NG, Yan Y, McCallum SA, Wang C, Garcia AE . The Alzheimer's peptides Abeta40 and 42 adopt distinct conformations in water: a combined MD/NMR study. J Mol Biol 2007; 368: 1448–57.
Yang M, Teplow DB . Amyloid beta-protein monomer folding: free-energy surfaces reveal alloform-specific differences. J Mol Biol 2008; 384: 450–64.
Sgourakis NG, Merced-Serrano M, Boutsidis C, Drineas P, Du ZM, Wang CY, et al. Atomic-level characterization of the ensemble of the A beta(1-42) monomer in water using unbiased molecular dynamics simulations and spectral algorithms. J Mol Biol 2011; 405: 570–83.
Vivekanandan S, Brender JR, Lee SY, Ramamoorthy A . A partially folded structure of amyloid-beta(1-40) in an aqueous environment. Biochem Bioph Res Commun 2011; 411: 312–6.
Mirza Z, Pillai VG, Kamal MA . Protein interactions between the C-terminus of Abeta-peptide and phospholipase A2–a structure biology based approach to identify novel Alzheimer's therapeutics. CNS Neurol Disorders Drug Targets 2014; 13: 1224–31.
Hensley K, Carney JM, Mattson MP, Aksenova M, Harris M, Wu JF, et al. A model for beta-Amyloid aggregation and neurotoxicity based on free-radical generation by the peptide–relevance to Alzheimer-disease. Proc Natl Acad Sci U S A 1994; 91: 3270–4.
Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, et al. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 1991; 30: 572–80.
Masliah E, Terry RD, Alford M, DeTeresa R, Hansen LA . Cortical and subcortical patterns of synaptophysinlike immunoreactivity in Alzheimer's disease. Am J Pathol 1991; 138: 235–46.
Masliah E, Achim CL, Ge N, DeTeresa R, Terry RD, Wiley CA . Spectrum of human immunodeficiency virus-associated neocortical damage. Ann Neurol 1992; 32: 321–9.
Eanes ED, Glenner GG . X-ray diffraction studies on amyloid filaments. J Histochem Cytochem 1968; 16: 673–7.
Sunde M, Blake CC . From the globular to the fibrous state: protein structure and structural conversion in amyloid formation. Quart Rev Biophys 1998; 31: 1–39.
Benzinger TLS, Gregory DM, Burkoth TS, Miller-Auer H, Lynn DG, Botto RE, et al. Propagating structure of Alzheimer's beta-amyloid((10-35)) is parallel beta-sheet with residues in exact register. Proc Natl Acad Sci U S A 1998; 95: 13407–12.
Benzinger TLS, Gregory DM, Burkoth TS, Miller-Auer H, Lynn DG, Botto RE, et al. Two-dimensional structure of beta-amyloid(10-35) fibrils. Biochemistry 2000; 39: 3491–9.
Antzutkin ON, Balbach JJ, Leapman RD, Rizzo NW, Reed J, Tycko R . Multiple quantum solid-state NMR indicates a parallel, not antiparallel, organization of beta-sheets in Alzheimer's beta-amyloid fibrils. Proc Natl Acad Sci U S A 2000; 97: 13045–50.
Balbach JJ, Petkova AT, Oyler NA, Antzutkin ON, Gordon DJ, Meredith SC, et al. Supramolecular structure in full-length Alzheimer's beta-amyloid fibrils: evidence for a parallel beta-sheet organization from solid-state nuclear magnetic resonance. Biophys J 2002; 83: 1205–16.
Antzutkin ON, Leapman RD, Balbach JJ, Tycko R . Supramolecular structural constraints on Alzheimer's beta-amyloid fibrils from electron microscopy and solid-state nuclear magnetic resonance. Biochemistry 2002; 41: 15436–50.
Petkova AT, Buntkowsky G, Dyda F, Leapman RD, Yau WM, Tycko R . Solid state NMR reveals a pH-dependent antiparallel beta-sheet registry in fibrils formed by a beta-amyloid peptide. J Mol Biol 2004; 335: 247–60.
Petkova AT, Ishii Y, Balbach JJ, Antzutkin ON, Leapman RD, Delaglio F, et al. A structural model for Alzheimer's beta-amyloid fibrils based on experimental constraints from solid state NMR. Proc Natl Acad Sci U S A 2002; 99: 16742–7.
Jayasinghe SA, Langen R . Identifying structural features of fibrillar islet amyloid polypeptide using site-directed spin labeling. J Biol Chem 2004; 279: 48420–5.
Margittai M, Langen R . Template-assisted filament growth by parallel stacking of tau. Proc Natl Acad Sci U S A 2004; 101: 10278–83.
Der-Sarkissian A, Jao CC, Chen J, Langen R . Structural organization of alpha-synuclein fibrils studied by site-directed spin labeling. J Biol Chem 2003; 278: 37530–5.
Shivaprasad S, Wetzel R . An intersheet packing interaction in A beta fibrils mapped by disulfide cross-linking. Biochemistry 2004; 43: 15310–7.
Harper JD, Lieber CM, Lansbury PT . Atomic force microscopic imaging of seeded fibril formation and fibril branching by the Alzheimer's disease amyloid-beta protein. Chem Biol 1997; 4: 951–9.
Goldsbury C, Green J . Time-lapse atomic force microscopy in the characterization of amyloid-like fibril assembly and oligomeric intermediates. Methods Mol Biol 2005; 299: 103–28.
Petkova AT, Leapman RD, Guo Z, Yau WM, Mattson MP, Tycko R . Self-propagating, molecular-level polymorphism in Alzheimer's beta-amyloid fibrils. Science 2005; 307: 262–5.
Tycko R, Ishii Y . Constraints on supramolecular structure in amyloid fibrils from two-dimensional solid-state NMR spectroscopy with uniform isotopic labeling. J Am Chem Soc 2003; 125: 6606–7.
Proceedings of the National Academy of Sciences of the United States of America. Annual subject and author indexes. Proc Natl Acad Sci U S A 1990; 87 Suppl: 10069–240.
Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, et al. Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc Natl Acad Sci U S A 1999; 96: 3228–33.
Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, et al. High-level neuronal expression of A beta(1-42) in wild-type human amyloid protein precursor transgenic mice: Synaptotoxicity without plaque formation. J Neurosci 2000; 20: 4050–8.
Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717f beta-amyloid precursor protein. Nature 1995; 373: 523–7.
Podlisny MB, Ostaszewski BL, Squazzo SL, Koo EH, Rydell RE, Teplow DB, et al. Aggregation of secreted amyloid beta-protein into sodium dodecyl sulfate-stable oligomers in cell-culture. J Biol Chem 1995; 270: 9564–70.
Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003; 300: 486–9.
Klein WL, Krafft GA, Finch CE . Targeting small Abeta oligomers: the solution to an Alzheimer's disease conundrum? Trends Neurosci 2001; 24: 219–24.
Hardy J, Selkoe DJ . The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 2002; 297: 353–6.
Walsh DM, Klyubin I, Fadeeva JV, Rowan MJ, Selkoe DJ . Amyloid-beta oligomers: their production, toxicity and therapeutic inhibition. Biochem Soc T 2002; 30: 552–7.
Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002; 416: 535–9.
Goldsbury CS, Wirtz S, Muller SA, Sunderji S, Wicki P, Aebi U, et al. Studies on the in vitro assembly of A beta 1-40: Implications for the search for A beta fibril formation inhibitors. J Struct Biol 2000; 130: 217–31.
Nichols MR, Moss MA, Reed DK, Lin WL, Mukhopadhyay R, Hoh JH, et al. Growth of beta-amyloid(1-40) protofibrils by monomer elongation and lateral association. Characterization of distinct products by light scattering and atomic force microscopy. Biochemistry 2002; 41: 6115–27.
Lashuel HA, Hartley DM, Petre BM, Wall JS, Simon MN, Walz T, et al. Mixtures of wild-type and a pathogenic (E22G) form of Abeta40 in vitro accumulate protofibrils, including amyloid pores. J Mol Biol 2003; 332: 795–808.
Walsh DM, Lomakin A, Benedek GB, Condron MM, Teplow DB . Amyloid beta-protein fibrillogenesis. Detection of a protofibrillar intermediate. J Biol Chem 1997; 272: 22364–72.
Soreghan B, Kosmoski J, Glabe C . Surfactant properties of Alzheimer's A beta peptides and the mechanism of amyloid aggregation. J Biol Chem 1994; 269: 28551–4.
Yu L, Edalji R, Harlan JE, Holzman TF, Lopez AP, Labkovsky B, et al. Structural characterization of a soluble amyloid beta-peptide oligomer. Biochemistry 2009; 48: 1870–7.
Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, et al. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 2003; 39: 409–21.
LeVine H . Alzheimer's beta-peptide oligomer formation at physiologic concentrations. Anal Biochem 2004; 335: 81–90.
Ahmed M, Davis J, Aucoin D, Sato T, Ahuja S, Aimoto S, et al. Structural conversion of neurotoxic amyloid-beta(1-42) oligomers to fibrils. Nat Struct Mol Biol 2010; 17: 561–7.
Kheterpal I, Wetzel R, Cook KD . Enhanced correction methods for hydrogen exchange-mass spectrometric studies of amyloid fibrils. Protein Sci 2003; 12: 635–43.
Kheterpal I, Zhou S, Cook KD, Wetzel R . Abeta amyloid fibrils possess a core structure highly resistant to hydrogen exchange. Proc Natl Acad Sci U S A 2000; 97: 13597–601.
Mattson MP . Pathways towards and away from Alzheimer's disease. Nature 2004; 430: 631–9.
Zhang S, Iwata K, Lachenmann MJ, Peng JW, Li S, Stimson ER, et al. The Alzheimer's peptide a beta adopts a collapsed coil structure in water. J Struct Biol 2000; 130: 130–41.
Tran L, Basdevant N, Prevost C, Ha-Duong T . Structure of ring-shaped Abeta(42) oligomers determined by conformational selection. Sci Rep 2016; 6: 21429.
O'Nuallain B, Wetzel R . Conformational Abs recognizing a generic amyloid fibril epitope. Proc Natl Acad Sci U S A 2002; 99: 1485–90.
Harper JD, Wong SS, Lieber CM, Lansbury PT . Observation of metastable Abeta amyloid protofibrils by atomic force microscopy. Chem Biol 1997; 4: 119–25.
Blackley HK, Sanders GH, Davies MC, Roberts CJ, Tendler SJ, Wilkinson MJ . In-situ atomic force microscopy study of beta-amyloid fibrillization. J Mol Biol 2000; 298: 833–40.
Goldsbury C, Frey P, Olivieri V, Aebi U, Muller SA . Multiple assembly pathways underlie amyloid-beta fibril polymorphisms. J Mol Biol 2005; 352: 282–98.
Curtain CC, Ali F, Volitakis I, Cherny RA, Norton RS, Beyreuther K, et al. Alzheimer's disease amyloid-beta binds copper and zinc to generate an allosterically ordered membrane-penetrating structure containing superoxide dismutase-like subunits. J Biol Chem 2001; 276: 20466–73.
Curtain CC, Ali FE, Smith DG, Bush AI, Masters CL, Barnham KJ . Metal ions, pH, and cholesterol regulate the interactions of Alzheimer's disease amyloid-beta peptide with membrane lipid. J Biol Chem 2003; 278: 2977–82.
Lovell MA, Robertson JD, Teesdale WJ, Campbell JL, Markesbery WR . Copper, iron and zinc in Alzheimer's disease senile plaques. J Neurol Sci 1998; 158: 47–52.
Maynard CJ, Cappai R, Volitakis I, Cherny RA, White AR, Beyreuther K, et al. Overexpression of Alzheimer's disease amyloid-beta opposes the age-dependent elevations of brain copper and iron. J Biol Chem 2002; 277: 44670–6.
Bayer TA, Schafer S, Simons A, Kemmling A, Kamer T, Tepest R, et al. Dietary Cu stabilizes brain superoxide dismutase 1 activity and reduces amyloid Abeta production in APP23 transgenic mice. Proc Natl Acad Sci U S A 2003; 100: 14187–92.
Phinney AL, Drisaldi B, Schmidt SD, Lugowski S, Coronado V, Liang Y, et al. In vivo reduction of amyloid-beta by a mutant copper transporter. Proc Natl Acad Sci U S A 2003; 100: 14193–8.
Zirah S, Kozin SA, Mazur AK, Blond A, Cheminant M, Segalas-Milazzo I, et al. Structural changes of region 1-16 of the Alzheimer disease amyloid beta-peptide upon zinc binding and in vitro aging. J Biol Chem 2006; 281: 2151–61.
Haass C, Koo EH, Mellon A, Hung AY, Selkoe DJ . Targeting of cell-surface beta-amyloid precursor protein to lysosomes–alternative processing into amyloid-bearing fragments. Nature 1992; 357: 500–3.
Amaro M, Sachl R, Aydogan G, Mikhalyov, II, Vacha R, Hof M . GM1 ganglioside inhibits beta-amyloid oligomerization induced by sphingomyelin. Angew Chem Int Ed Engl 2016; 55: 9411–5.
Ruzali WA, Kehoe PG, Love S . Influence of LRP-1 and apolipoprotein E on amyloid-beta uptake and toxicity to cerebrovascular smooth muscle cells. J Alzheimer's <0x000A>Dis 2013; 33: 95–110.
Canevari L, Abramov AY, Duchen MR . Toxicity of amyloid beta peptide: tales of calcium, mitochondria, and oxidative stress. Neurochem Res 2004; 29: 637–50.
Kim T, Vidal GS, Djurisic M, William CM, Birnbaum ME, Garcia KC, et al. Human LilrB2 is a beta-amyloid receptor and its murine homolog PirB regulates synaptic plasticity in an Alzheimer's model. Science 2013; 341: 1399–404.
Costa RO, Lacor PN, Ferreira IL, Resende R, Auberson YP, Klein WL, et al. Endoplasmic reticulum stress occurs downstream of GluN2B subunit of N-methyl-D-aspartate receptor in mature hippocampal cultures treated with amyloid-beta oligomers. Aging Cell 2012; 11: 823–33.
De Felice FG, Velasco PT, Lambert MP, Viola K, Fernandez SJ, Ferreira ST, et al. A beta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J Biol Chem 2007; 282: 11590–601.
Deshpande A, Kawai H, Metherate R, Glabe CG, Busciglio J . A role for synaptic Zinc in activity-dependent A beta oligomer formation and accumulation at excitatory synapses. J Neurosci 2009; 29: 4004–15.
Alberdi E, Victoria Sanchez-Gomez M, Cavaliere F, Perez-Samartin A, Luis Zugaza J, Trullas R, et al. Amyloid beta oligomers induce Ca2+ dysregulation and neuronal death through activation of ionotropic glutamate receptors. Cell Calcium 2010; 47: 264–72.
Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL . Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci 2007; 27: 2866–75.
Roenicke R, Mikhaylova M, Roenicke S, Meinhardt J, Schroeder UH, Faendrich M, et al. Early neuronal dysfunction by amyloid beta oligomers depends on activation of NR2B-containing NMDA receptors. Neurobiol Aging 2011; 32: 2219–28.
Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, et al. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci 2005; 8: 1051–8.
Wang HY, Lee DH, Davis CB, Shank RP . Amyloid peptide Abeta(1-42) binds selectively and with picomolar affinity to alpha7 nicotinic acetylcholine receptors. J Neurochem 2000; 75: 1155–61.
Nagele RG, D'Andrea MR, Anderson WJ, Wang HY . Intracellular accumulation of beta-amyloid(1-42) in neurons is facilitated by the alpha 7 nicotinic acetylcholine receptor in Alzheimer's disease. Neuroscience 2002; 110: 199–211.
Wang HY, Li W, Benedetti NJ, Lee DH . Alpha 7 nicotinic acetylcholine receptors mediate beta-amyloid peptide-induced tau protein phosphorylation. J Biol Chem 2003; 278: 31547–53.
Hernandez CM, Kayed R, Zheng H, Sweatt JD, Dineley KT . Loss of alpha7 nicotinic receptors enhances beta-amyloid oligomer accumulation, exacerbating early-stage cognitive decline and septohippocampal pathology in a mouse model of Alzheimer's disease. J Neurosci 2010; 30: 2442–53.
Naveilhan P, Neveu I, Baudet C, Funakoshi H, Wion D, Brachet P, et al. 1,25-Dihydroxyvitamin D3 regulates the expression of the low-affinity neurotrophin receptor. Brain Res Mol Brain Res 1996; 41: 259–68.
Hashimoto Y, Kaneko Y, Tsukamoto E, Frankowski H, Kouyama K, Kita Y, et al. Molecular characterization of neurohybrid cell death induced by Alzheimer's amyloid-beta peptides via p75NTR/PLAIDD. J Neurochem 2004; 90: 549–58.
Perini G, Della-Bianca V, Politi V, Della Valle G, Dal-Pra I, Rossi F, et al. Role of p75 neurotrophin receptor in the neurotoxicity by beta-amyloid peptides and synergistic effect of inflammatory cytokines. J Exp Med 2002; 195: 907–18.
Chiarini A, Dal Pra I, Whitfield JF, Armato U . The killing of neurons by beta-amyloid peptides, prions, and pro-inflammatory cytokines. Ital J Anat Embryol 2006; 111: 221–46.
Etique N, Verzeaux L, Dedieu S, Emonard H . LRP-1: a checkpoint for the extracellular matrix proteolysis. Biomed Res Int 2013; 2013: 152163.
Wijnberg MJ, Quax PHA, Nieuwenbroek NME, Verheijen JH . The migration of human smooth muscle cells in vitro is mediated by plasminogen activation and can be inhibited by alpha(2)-macroglobulin receptor associated protein. Thromb Haemost 1997; 78: 880–6.
Yepes M, Sandkvist M, Moore EG, Bugge TH, Strickland DK, Lawrence DA . Tissue-type plasminogen activator induces opening of the blood-brain barrier via the LDL receptor-related protein. J Clin Invest 2003; 112: 1533–40.
Marzolo MP, Bu G . Lipoprotein receptors and cholesterol in APP trafficking and proteolytic processing, implications for Alzheimer's disease. Semin Cell Dev Biol 2009; 20: 191–200.
Lillis AP, Mikhailenko I, Strickland DK . Beyond endocytosis: LRP function in cell migration, proliferation and vascular permeability. J Thromb Haemost 2005; 3: 1884–93.
Linden R, Martins VR, Prado MAM, Cammarota M, Izquierdo I, Brentani RR . Physiology of the prion protein. Physiol Rev 2008; 88: 673–728.
Beraldo FH, Arantes CP, Santos TG, Machado CF, Roffe M, Hajj GN, et al. Metabotropic glutamate receptors transduce signals for neurite outgrowth after binding of the prion protein to laminin gamma 1 chain. FASEB J 2011; 25: 265–79.
Kessels HW, Nguyen LN, Nabavi S, Malinow R . The prion protein as a receptor for amyloid-beta. Nature 2010; 466: E3–E4.
Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM . Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 2009; 457: 1128–U84.
Ribeiro FM, Paquet M, Cregan SP, Ferguson SS . Group I metabotropic glutamate receptor signalling and its implication in neurological disease. CNS Neurol Disord Drug Targets 2010; 9: 574–95.
Bordi F, Ugolini A . Group I metabotropic glutamate receptors: implications for brain diseases. Prog Neurobiol 1999; 59: 55–79.
Bruno V, Battaglia G, Copani A, Cespedes VM, Galindo MF, Cena V, et al. An activity-dependent switch from facilitation to inhibition in the control of excitotoxicity by group I metabotropic glutamate receptors. Eur J Neurosci 2001; 13: 1469–78.
Sokol DK, Maloney B, Long JM, Ray B, Lahiri DK . Autism, Alzheimer disease, and fragile X APP, FMRP, and mGluR5 are molecular links. Neurology 2011; 76: 1344–52.
Selkoe DJ . Toward a comprehensive theory for Alzheimer's disease - Hypothesis: Alzheimer's disease is caused by the cerebral accumulation and cytotoxicity of amyloid beta-protein. Ann Ny Acad Sci 2000; 924: 17–25.
Fernandez-Vizarra P, Lopez-Franco O, Mallavia B, Higuera-Matas A, Lopez-Parra V, Ortiz-Munoz G, et al. Immunoglobulin G Fc receptor deficiency prevents Alzheimer-like pathology and cognitive impairment in mice. Brain 2012; 135: 2826–37.
Kam TI, Song S, Gwon Y, Park H, Yan JJ, Im I, et al. FcgammaRIIb mediates amyloid-beta neurotoxicity and memory impairment in Alzheimer's disease. J Clin Invest 2013; 123: 2791–802.
Choi B-R, Cho W-H, Kim J, Lee HJ, Chung C, Jeon WK, et al. Increased expression of the receptor for advanced glycation end products in neurons and astrocytes in a triple transgenic mouse model of Alzheimer's disease. Exp Mol Med 2014; 46: Article No.: e75.
Peng L, Yu Y, Liu J, Li SQ, He HQ, Cheng N, et al. The chemerin Receptor CMKLR1 is a functional receptor for amyloid-beta peptide. J Alzheimers Dis 2015; 43: 227–42.
Tahara K, Kim HD, Jin JJ, Maxwell JA, Li L, Fukuchi K . Role of toll-like receptor signalling in Abeta uptake and clearance. Brain 2006; 129: 3006–19.
Yu Y, Ye RD . Microglial Abeta receptors in Alzheimer's disease. Cell Mol Neurobiol 2015; 35: 71–83.
Rozemuller A, Awan T, Ghetti B, Puoti G, Eikelenboom P, Tagliavini F . Microglia and complement in the cortex of Creutzfeldt-Jakob disease and comparison with Alzheimer's disease. Brain Pathol 2000; 10: 658.
Iwata N, Tsubuki S, Takaki Y, Watanabe K, Sekiguchi M, Hosoki E, et al. Identification of the major Abeta1-42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat Med 2000; 6: 143–50.
Turner AJ, Tanzawa K . Mammalian membrane metallopeptidases: NEP, ECE, KELL, and PEX. FASEB J 1997; 11: 355–64.
Turner RT, Koelsch G, Hong L, Castenheira P, Ghosh A, Tang J . Subsite specificity of memapsin 2 (beta-secretase): implications for inhibitor design. Biochemistry 2001; 40: 10001–6.
Angus RM, Millar EA, Chalmers GW, Thomson NC . Effect of inhaled thiorphan, a neutral endopeptidase inhibitor, on the bronchodilator response to inhaled atrial natriuretic peptide (ANP). Thorax 1996; 51: 71–4.
Shi J, Zhang S, Tang M, Ma C, Zhao J, Li T, et al. Mutation screening and association study of the neprilysin gene in sporadic Alzheimer's disease in Chinese persons. J Gerontol A Biol Sci Med Sci 2005; 60: 301–6.
Iwata N, Higuchi M, Saido TC . Metabolism of amyloid-beta peptide and Alzheimer's disease. Pharmacol Ther 2005; 108: 129–48.
Takaki Y, Iwata N, Tsubuki S, Taniguchi S, Toyoshima S, Lu B, et al. Biochemical identification of the neutral endopeptidase family member responsible for the catabolism of amyloid beta peptide in the brain. J Biochem (Tokyo) 2000; 128: 897–902.
Shirotani K, Tsubuki S, Iwata N, Takaki Y, Harigaya W, Maruyama K, et al. Neprilysin degrades both amyloid beta peptides 1-40 and 1-42 most rapidly and efficiently among thiorphan- and phosphoramidon-sensitive endopeptidases. J Biol Chem 2001; 276: 21895–901.
Eckman EA, Reed DK, Eckman CB . Degradation of the Alzheimer's amyloid beta peptide by endothelin-converting enzyme. J Biol Chem 2001; 276: 24540–8.
Duckworth WC, Bennett RG, Hamel FG . Insulin acts intracellularly on proteasomes through insulin-degrading enzyme. Biochem Biophys Res Commun 1998; 244: 390–4.
Edbauer D, Willem M, Lammich S, Steiner H, Haass C . Insulin-degrading enzyme rapidly removes the beta-amyloid precursor protein intracellular domain (AICD). J Biol Chem 2002; 277: 13389–93.
Kurochkin IV, Goto S . Alzheimer's beta-amyloid peptide specifically interacts with and is degraded by insulin degrading enzyme. FEBS Lett 1994; 345: 33–7.
Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, et al. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci U S A 2003; 100: 4162–67.
von Rotz RC, Kohli BM, Bosset J, Meier M, Suzuki T, Nitsch RM, et al. The APP intracellular domain forms nuclear multiprotein complexes and regulates the transcription of its own precursor. J Cell Sci 2004; 117: 4435–48.
Pardossi-Piquard R, Petit A, Kawarai T, Sunyach C, Alves da Costa C, Vincent B, et al. Presenilin-dependent transcriptional control of the Abeta-degrading enzyme neprilysin by intracellular domains of betaAPP and APLP. Neuron 2005; 46: 541–54.
Miller BC, Eckman EA, Sambamurti K, Dobbs N, Chow KM, Eckman CB, et al. Amyloid-beta peptide levels in brain are inversely correlated with insulysin activity levels in vivo. Proc Natl Acad Sci U S A 2003; 100: 6221–6.
Van Nostrand WE, Porter M . Plasmin cleavage of the amyloid beta-protein: alteration of secondary structure and stimulation of tissue plasminogen activator activity. Biochemistry 1999; 38: 11570–6.
Tucker HM, Kihiko M, Caldwell JN, Wright S, Kawarabayashi T, Price D, et al. The plasmin system is induced by and degrades amyloid-beta aggregates. J Neurosci 2000; 20: 3937–46.
Bertram L, Blacker D, Mullin K, Keeney D, Jones J, Basu S, et al. Evidence for genetic linkage of Alzheimer's disease to chromosome 10q. Science 2000; 290: 2302–3.
Ertekin-Taner N, Graff-Radford N, Younkin LH, Eckman C, Baker M, Adamson J, et al. Linkage of plasma Abeta42 to a quantitative locus on chromosome 10 in late-onset Alzheimer's disease pedigrees. Science 2000; 290: 2303–4.
Roher AE, Kasunic TC, Woods AS, Cotter RJ, Ball MJ, Fridman R . Proteolysis of A-beta-peptide from Alzheimer-disease brain by gelatinase-A. Biochem Biophys Res Commun 1994; 205: 1755–61.
Backstrom JR, Lim GP, Cullen MJ, Tokes ZA . Matrix metallo-proteinase-9 (MMP-9) is synthesized in neurons of the human hippocampus and is capable of degrading the amyloid-beta peptide (1-40). J Neurosci 1996; 16: 7910–9.
Hu JG, Igarashi A, Kamata M, Nakagawa H . Angiotensin-converting enzyme degrades Alzheimer amyloid beta-peptide (A beta); Retards A beta aggregation, deposition, fibril formation, and inhibits cytotoxicity. J Biol Chem 2001; 276: 47863–8.
McDermott JR, Gibson AM . Degradation of Alzheimer's beta-amyloid protein by human cathepsin D. Neuroreport 1996; 7: 2163–6.
Cataldo AM, Hamilton DJ, Barnett JL, Paskevich PA, Nixon RA . Properties of the endosomal-lysosomal system in the human central nervous system: Disturbances mark most neurons in populations at risk to degenerate in Alzheimer's disease. J Neurosci 1996; 16: 186–99.
Strittmatter WJ, Saunders AM, Schmechel D, Pericakvance M, Enghild J, Salvesen GS, et al. Apolipoprotein-E - high-avidity binding to beta-amyloid and increased frequency of type-4 allele in late-onset familial Alzheimer-disease. Proc Natl Acad Sci U S A 1993; 90: 1977–81.
Zlokovic BV, Martel CL, Mackic JB, Matsubara E, Wisniewski T, McComb JG, et al. Brain uptake of circulating apolipoproteins J and E complexed to Alzheimer's amyloid beta. Biochem Biophys Res Commun 1994; 205: 1431–7.
Maness LM, Banks WA, Podlisny MB, Selkoe DJ, Kastin AJ . Passage of human amyloid beta-protein-1-40 across the murine blood-brain-barrier. Life Sci 1994; 55: 1643–50.
GhersiEgea JF, Gorevic PD, Ghiso J, Frangione B, Patlak CS, Fenstermacher JD . Fate of cerebrospinal fluid-borne amyloid beta-peptide: Rapid clearance into blood and appreciable accumulation by cerebral arteries. J Neurochem 1996; 67: 880–3.
Martel CL, Mackic JB, Matsubara E, Governale S, Miguel C, Miao W, et al. Isoform-specific effects of apolipoproteins E2, E3, and E4 on cerebral capillary sequestration and blood-brain barrier transport of circulating Alzheimer's amyloid beta. J Neurochem 1997; 69: 1995–2004.
Zlokovic BV, Yamada S, Holtzman D, Ghiso J, Frangione B . Clearance of amyloid beta-peptide from brain: transport or metabolism? Nature Med 2000; 6: 718–18.
DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM . Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A 2001; 98: 8850–5.
Hardy J, Selkoe DJ . Medicine–the amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 2002; 297: 353–6.
Deane R, Yan SD, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med 2003; 9: 907–13.
Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, et al. Clearance of Alzheimer's amyloid-beta(1-40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest 2000; 106: 1489–99.
Deane R, Wu Z, Sagare A, Davis J, Du Yan S, Hamm K, et al. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron 2004; 43: 333–44.
Zlokovic BV . Cerebrovascular transport of Alzheimer's amyloid beta and apolipoproteins J and E: possible anti-amyloidogenic role of the blood-brain barrier. Life Sci 1996; 59: 1483–97.
Cirrito JR, Deane R, Fagan AM, Spinner ML, Parsadanian M, Finn MB, et al. P-glycoprotein deficiency at the blood-brain barrier increases amyloid-beta deposition in an Alzheimer disease mouse model. J Clin Invest 2005; 115: 3285–90.
Zlokovic BV, Deane R, Sallstrom J, Chow N, Miano JM . Neurovascular pathways and Alzheimer amyloid beta-peptide. Brain Pathol 2005; 15: 78–83.
Iwata N, Tsubuki S, Takaki Y, Shirotani K, Lu B, Gerard NP, et al. Metabolic regulation of brain Abeta by neprilysin. Science 2001; 292: 1550–2.
Selkoe DJ . Clearing the brain's amyloid cobwebs. Neuron 2001; 32: 177–80.
Hardy J, Selkoe DJ . The amyloid hypothesis of Alzheimer's disease: Progress and problems on the road to therapeutics. Science 2002; 297: 353–6.
Tanzi RE, Moir RD, Wagner SL . Clearance of Alzheimer's Abeta peptide: the many roads to perdition. Neuron 2004; 43: 605–8.
Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, et al. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat Med 2003; 9: 453–7.
Koistinaho M, Lin S, Wu X, Esterman M, Koger D, Hanson J, et al. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat Med 2004; 10: 719–26.
Zlokovic BV, Deane R, Sallstrom J, Chow N, Miano JM . Neurovascular pathways and Alzheimer amyloid beta-peptide. Brain Pathol 2005; 15: 78–83.
Mayeux R, Tang MX, Jacobs DM, Manly J, Bell K, Merchant C, et al. Plasma amyloid beta-peptide 1-42 and incipient Alzheimer's disease. Ann Neurol 1999; 46: 412–6.
Mayeux R, Honig LS, Tang MX, Manly J, Stern Y, Schupf N, et al. Plasma A[beta]40 and A[beta]42 and Alzheimer's disease: relation to age, mortality, and risk. Neurology 2003; 61: 1185–90.
DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM . Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A 2001; 98: 8850–5.
Sigurdsson EM, Scholtzova H, Mehta PD, Frangione B, Wisniewski T . Immunization with a nontoxic/nonfibrillar amyloid-beta homologous peptide reduces Alzheimer's disease-associated pathology in transgenic mice. Am J Pathol 2001; 159: 439–47.
Bales KR, Dodart JC, DeMattos RB, Holtzman DM, Paul SM . Apolipoprotein E, amyloid, and Alzheimer disease. Mol Interv 2002; 2: 363–75, 39.
Burns M, Gaynor K, Olm V, Mercken M, LaFrancois J, Wang L, et al. Presenilin redistribution associated with aberrant cholesterol transport enhances beta-amyloid production in vivo. J Neurosci 2003; 23: 5645–9.
Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med 2003; 9: 907–13.
Deane R, Wu ZH, Zlokovic BV . RAGE (Yin) versus LRP (Yang) balance regulates Alzheimer amyloid beta-peptide clearance through transport across the blood-brain barrier. Stroke 2004; 35: 2628–31.
Mackic JB, Stins M, McComb JG, Calero M, Ghiso J, Kim KS, et al. Human blood-brain barrier receptors for Alzheimer's amyloid-beta 1-40–Asymmetrical binding, endocytosis, and transcytosis at the apical side of brain microvascular endothelial cell monolayer. J Clin Invest 1998; 102: 734–43.
Stern DM, Du Yan S, Yan SF, Schmidt AM . Receptor for advanced glycation endproducts (RAGE) and the complications of diabetes. Ageing Res Rev 2002; 1: 1–15.
LaRue B, Hogg E, Sagare A, Jovanovic S, Maness L, Maurer C, et al. Method for measurement of the blood-brain barrier permeability in the perfused mouse brain: application to amyloid-beta peptide in wild type and Alzheimer's Tg2576 mice. J Neurosci Meth 2004; 138: 233–42.
Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, et al. Clearance of Alzheimer's amyloid-beta1-40 peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest 2000; 106: 1489–99.
Deane R, Wu Z, Sagare A, Davis J, Yan SD, Hamm K, et al. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron 2004; 43: 333–44.
Lowe LC, Gaser C, Franke K, Alzheimer's Disease Neuroimaging I . The effect of the APOE genotype on individual brain AGE in normal aging, mild cognitive impairment, and Alzheimer's disease. PLoS One 2016; 11: e0157514.
Holtzman DM . Role of ApoE/Abeta interactions in the pathogenesis of Alzheimer's disease and cerebral amyloid angiopathy. J Mol Neurosci 2001; 17: 147–55.
Fagan AM, Watson M, Parsadanian M, Bales KR, Paul SM, Holtzman DM . Human and murine ApoE markedly alters A beta metabolism before and after plaque formation in a mouse model of Alzheimer's disease. Neurobiol Dis 2002; 9: 305–18.
Hudry E, Dashkoff J, Roe AD, Takeda S, Koffie RM, Hashimoto T, et al. Gene transfer of human ApoE isoforms results in differential modulation of amyloid deposition and neurotoxicity in mouse brain. Sci Transl Med 2013; 5: 212ra161.
Lesne SE, Sherman MA, Grant M, Kuskowski M, Schneider JA, Bennett DA, et al. Brain amyloid-beta oligomers in ageing and Alzheimer's disease. Brain 2013; 136: 1383–98.
Atwood CS, Scarpa RC, Huang X, Moir RD, Jones WD, Fairlie DP, et al. Characterization of copper interactions with alzheimer amyloid beta peptides: identification of an attomolar-affinity copper binding site on amyloid beta1-42. J Neurochem 2000; 75: 1219–33.
Aksenov MY, Aksenova MV, Harris ME, Hensley K, Butterfield DA, Carney JM . Enhancement of beta-amyloid peptide A-beta(1-40)-mediated neurotoxicity by glutamine-synthetase. J Neurochem 1995; 65: 1899–902.
Yatin SM, Yatin M, Aulick T, Ain KB, Butterfield DA . Alzheimer's amyloid beta-peptide associated free radicals increase rat embryonic neuronal polyamine uptake and ornithine decarboxylase activity: protective effect of vitamin E. Neurosci Lett 1999; 263: 17–20.
Mark RJ, Hensley K, Butterfield DA, Mattson MP . Amyloid beta-peptide impairs ion-motive ATPase activities–evidence for a role in loss of neuronal Ca2+ homeostasis and cell-death. J Neurosci 1995; 15: 6239–49.
Varadarajan S, Yatin S, Aksenova M, Butterfield DA . Review: Alzheimer's amyloid beta-peptide-associated free radical oxidative stress and neurotoxicity. J Struct Biol 2000; 130: 184–208.
Ezeani M, Omabe M . A new perspective of lysosomal cation channel-dependent homeostasis in Alzheimer's disease. Mol Neurobiol 2016; 53: 1672–8.
Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, et al. A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature 2001; 414: 212–6.
Neniskyte U, Neher JJ, Brown GC . Neuronal death induced by nanomolar Amyloid beta is mediated by primary phagocytosis of neurons by microglia. J Biol Chem 2011; 286: 39904–13.
Heneka MT, Golenbock DT, Latz E . Innate immunity in Alzheimer's disease. Nat Immunol 2015; 16: 229–36.
Anandatheerthavarada HK, Biswas G, Robin MA, Avadhani NG . Mitochondrial targeting and a novel transmembrane arrest of Alzheimer's amyloid precursor protein impairs mitochondrial function in neuronal cells. J Cell Biol 2003; 161: 41–54.
Devi L, Prabhu BM, Galati DF, Avadhani NG, Anandatheerthavarada HK . Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer's disease brain is associated with mitochondrial dysfunction. J Neurosci 2006; 26: 9057–68.
Barsoum MJ, Yuan H, Gerencser AA, Liot G, Kushnareva YE, Graber S, et al. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J 2006; 25: 3900–11.
Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, et al. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science 2004; 304: 448–52.
Yankner BA, Duffy LK, Kirschner DA . Neurotrophic and neurotoxic effects of amyloid beta protein: reversal by tachykinin neuropeptides. Science 1990; 250: 279–82.
Mattson MP . Cellular actions of beta-amyloid precursor protein and its soluble and fibrillogenic derivatives. Physiol Rev 1997; 77: 1081–132.
Chapman PF, White GL, Jones MW, Cooper-Blacketer D, Marshall VJ, Irizarry M, et al. Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat Neurosci 1999; 2: 271–6.
Koistinaho M, Ort M, Cimadevilla JM, Vondrous R, Cordell B, Koistinaho J, et al. Specific spatial learning deficits become severe with age in beta-amyloid precursor protein transgenic mice that harbor diffuse beta-amyloid deposits but do not form plaques. Proc Natl Acad Sci U S A 2001; 98: 14675–80.
Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, et al. Abeta peptide vaccination prevents memory loss in an animal model of Alzheimer's disease. Nature 2000; 408: 982–5.
Hardy J, Allsop D . Amyloid deposition as the central event in the etiology of Alzheimer's disease. Trend Pharmacol Sci 1991; 12: 383–8.
Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature 1995; 373: 523–7.
Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, et al. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 1996; 274: 99–102.
Suarez-Calvet M, Belbin O, Pera M, Badiola N, Magrane J, Guardia-Laguarta C, et al. Autosomal-dominant Alzheimer's disease mutations at the same codon of amyloid precursor protein differentially alter A beta production. J Neurochem 2014; 128: 330–9.
Tanzi RE, Bertram L . Twenty years of the Alzheimer's disease amyloid hypothesis: a genetic perspective. Cell 2005; 120: 545–55.
Bertram L, Tanzi RE . Alzheimer's disease: one disorder, too many genes? Human Mol Genet 2004; 13 Spec No 1: R135–41.
Martinoli MG, Trojanowski JQ, Schmidt ML, Arnold SE, Fujiwara TM, Lee VM, et al. Association of apolipoprotein epsilon 4 allele and neuropathologic findings in patients with dementia. Acta Neuropathol 1995; 90: 239–43.
Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A 1993; 90: 1977–81.
Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, et al. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J Neurosci 2007; 27: 796–807.
Reitz C . Dyslipidemia and the risk of Alzheimer's disease. Curr Atheroscler Rep 2013; 15: 307.
Intlekofer KA, Cotman CW . Exercise counteracts declining hippocampal function in aging and Alzheimer's disease. Neurobiol Dis 2013; 57: 47–55.
Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT . Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med 2011; 1: a006189.
Xu TH, Yan Y, Kang Y, Jiang Y, Melcher K, Xu HE . Alzheimer's disease-associated mutations increase amyloid precursor protein resistance to gamma-secretase cleavage and the Abeta42/Abeta40 ratio. Cell Dis 2016; 2: 16026.
Pimplikar SW, Nixon RA, Robakis NK, Shen J, Tsai LH . Amyloid-independent mechanisms in Alzheimer's disease pathogenesis. J Neurosci 2010; 30: 14946–54.
Robakis NK . Mechanisms of AD neurodegeneration may be independent of Abeta and its derivatives. Neurobiol Aging 2011; 32: 372–9.
Zhu J, Li W, Mao Z . Cdk5: mediator of neuronal development, death and the response to DNA damage. Mech Ageing Dev 2011; 132: 389–94.
Liraz O, Boehm-Cagan A, Michaelson DM . ApoE4 induces Abeta42, tau, and neuronal pathology in the hippocampus of young targeted replacement apoE4 mice. Mol Neurodegener 2013; 8: 16. Doi: 10.1186/1750-1326-8-16.
Mann DM, Brown A, Wilks DP, Davies CA . Immunocytochemical and lectin histochemical studies of plaques and tangles in Down's syndrome patients at different ages. Prog Clin Biol Res 1989; 317: 849–56.
Puzzo D, Gulisano W, Arancio O, Palmeri A . The keystone of Alzheimer pathogenesis might be sought in a beta physiology. Neuroscience 2015; 307: 26–36.
Ghosal K, Stathopoulos A, Pimplikar SW . APP intracellular domain impairs adult neurogenesis in transgenic mice by inducing neuroinflammation. PLoS One 2010; 5: e11866.
Ghosal K, Vogt DL, Liang M, Shen Y, Lamb BT, Pimplikar SW . Alzheimer's disease-like pathological features in transgenic mice expressing the APP intracellular domain. Proc Natl Acad Sci U S A 2009; 106: 18367–72.
Kepp KP . Ten challenges of the amyloid hypothesis of Alzheimer's disease. J Alzheimer's Disease 2017; 55: 447–57.
Shen ZX . Brain cholinesterases: II. The molecular and cellular basis of Alzheimer's disease. Med Hypotheses 2004; 63: 308–21.
Wenk GL . Neuropathologic changes in Alzheimer's disease. J Clin Psychiatry 2003; 64 Suppl 9: 7–10.
Francis PT, Palmer AM, Snape M, Wilcock GK . The cholinergic hypothesis of Alzheimer's disease: a review of progress. J Neurol Neurosurg Psychiatry 1999; 66: 137–47.
Waring SC, Rosenberg RN . Genome-wide association studies in Alzheimer disease. Arch Neurol 2008; 65: 329–34.
Mahley RW, Weisgraber KH, Huang Y . Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer's disease. Proc Natl Acad Sci U S A 2006; 103: 5644–51.
Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, et al. Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med 2013; 368: 107–16.
Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet 2013; 45: 1452–8.
Goedert M, Spillantini MG, Crowther RA . Tau proteins and neurofibrillary degeneration. Brain Pathol 1991; 1: 279–86.
Deane R, Zlokovic BV . Role of the blood-brain barrier in the pathogenesis of Alzheimer's disease. Curr Alzheimer Res 2007; 4: 191–7.
Sondag CM, Dhawan G, Combs CK . Beta amyloid oligomers and fibrils stimulate differential activation of primary microglia. J Neuroinflamm 2009; 6: 1.
Hayden EY, Teplow DB . Amyloid beta-protein oligomers and Alzheimer's disease. Alzheimers Res Ther 2013; 5: 60.
White JA, Manelli AM, Holmberg KH, Van Eldik LJ, Ladu MJ . Differential effects of oligomeric and fibrillar amyloid-beta 1-42 on astrocyte-mediated inflammation. Neurobiol Dis 2005; 18: 459–65.
Paris D, Town T, Parker TA, Tan J, Humphrey J, Crawford F, et al. Inhibition of Alzheimer's beta-amyloid induced vasoactivity and proinflammatory response in microglia by a cGMP-dependent mechanism. Exp Neurol 1999; 157: 211–21.
Lee M, You HJ, Cho SH, Woo CH, Yoo MH, Joe EH, et al. Implication of the small GTPase Rac1 in the generation of reactive oxygen species in response to beta-amyloid in C6 astroglioma cells. Biochem J 2002; 366: 937–43.
Soscia SJ, Kirby JE, Washicosky KJ, Tucker SM, Ingelsson M, Hyman B, et al. The Alzheimer's disease-associated Amyloid beta-protein is an antimicrobial peptide. PLoS One 2010; 5.
Kumar DK, Choi SH, Washicosky KJ, Eimer WA, Tucker S, Ghofrani J, et al. Amyloid-beta peptide protects against microbial infection in mouse and worm models of Alzheimer's disease. Sci Transl Med 2016; 8: 340ra72.
Namzaric - A Combination of 2 Old Drugs for Alzheimer's Disease. Med Lett Drugs Ther 2015; 57: 105–6.
Rogers SL, Friedhoff LT, Apter JT, Richter RW, Hartford JT, Walshe TM, et al. The efficacy and safety of donepezil in patients with Alzheimer's disease: Results of a US multicentre, randomized, double-blind, placebo-controlled trial. Dementia 1996; 7: 293–303.
Bryson HM, Benfield P . Donepezil. Drug Aging 1997; 10: 234–9.
Woodruff-Pak DS, Tobia MJ, Jiao XL, Beck KD, Servatius RJ . Preclinical investigation of the functional effects of memantine and memantine combined with galantamine or donepezil. Neuropsychopharmacology 2007; 32: 1284–94.
Riepe MW, Adler G, Ibach B, Tracik F . Adding memantine to therapy with rivastigmine in patients with mild to moderate Alzheimer's disease: Results of a 12-week pilot study. Neurology 2005; 64: A363–A64.
De Felice FG, Vieira MN, Saraiva LM, Figueroa-Villar JD, Garcia-Abreu J, Liu R, et al. Targeting the neurotoxic species in Alzheimer's disease: inhibitors of Abeta oligomerization. FASEB J 2004; 18: 1366–72.
Caruana M, Hogen T, Levin J, Hillmer A, Giese A, Vassallo N . Inhibition and disaggregation of alpha-synuclein oligomers by natural polyphenolic compounds. FEBS Lett 2011; 585: 1113–20.
Lashuel HA, Petre BM, Wall J, Simon M, Nowak RJ, Walz T, et al. alpha-synuclein, especially the Parkinson's disease-associated mutants, forms pore-like annular and tubular protofibrils. J Mol Biol 2002; 322: 1089–102.
Visioli F, Poli A, Galli C . Antioxidant and other biological activities of phenols from olives and olive oil. Med Res Rev 2002; 22: 65–75.
Visioli F, Bellosta S, Galli C . Oleuropein, the bitter principle of olives, enhances nitric oxide production by mouse macrophages. Life Sci 1998; 62: 541–6.
Andreadou I, Iliodromitis EK, Mikros E, Constantinou M, Agalias A, Magiatis P, et al. The olive constituent oleuropein exhibits anti-ischemic, antioxidative, and hypolipidemic effects in anesthetized rabbits. J Nutr 2006; 136: 2213–9.
Fu Z, Aucoin D, Ahmed M, Ziliox M, Van Nostrand WE, Smith SO . Capping of abeta42 oligomers by small molecule inhibitors. Biochemistry 2014; 53: 7893–903.
Granic I, Nyakas C, Luiten PGM, Eisel ULM, Halmy LG, Gross G, et al. Calpain inhibition prevents amyloid-beta-induced neurodegeneration and associated behavioral dysfunction in rats. Neuropharmacology 2010; 59: 334–42.
Ladiwala ARA, Mora-Pale JM, Dordick JS, Tessier PM . Aromatic small molecules remodel toxic soluble oligomers of amyloid beta through three independent pathways. Abstr Pap Am Chem S 2011; 241.
Garai K, Sengupta P, Sahoo B, Maiti S . Selective destabilization of soluble amyloid beta oligomers by divalent metal ions. Biochem Biophys Res Commun 2006; 345: 210–5.
Scherzer-Attali R, Shaltiel-Karyo R, Adalist YH, Segal D, Gazit E . Generic inhibition of amyloidogenic proteins by two naphthoquinone-tryptophan hybrid molecules. Proteins 2012; 80: 1962–73.
Scherzer-Attali R, Farfara D, Cooper I, Levin A, Ben-Romano T, Trudler D, et al. Naphthoquinone-tyrptophan reduces neurotoxic Abeta*56 levels and improves cognition in Alzheimer's disease animal model. Neurobiol Dis 2012; 46: 663–72.
Soto-Ortega DD, Murphy BP, Gonzalez-Velasquez FJ, Wilson KA, Xie F, Wang QA, et al. Inhibition of amyloid-beta aggregation by coumarin analogs can be manipulated by functionalization of the aromatic center. Bioorgan Med Chem 2011; 19: 2596–602.
Granic I, Nyakas C, Luiten PG, Eisel UL, Halmy LG, Gross G, et al. Calpain inhibition prevents amyloid-beta-induced neurodegeneration and associated behavioral dysfunction in rats. Neuropharmacology 2010; 59: 334–42.
Zhao W, Wang J, Ho L, Ono K, Teplow DB, Pasinetti GM . Identification of antihypertensive drugs which inhibit Amyloid-beta protein oligomerization. J Alzheimers Dis 2009; 16: 49–57.
McKoy AF, Chen J, Schupbach T, Hecht MH . A novel inhibitor of amyloid beta (Abeta) peptide aggregation: from high throughput screening to efficacy in an animal model of Alzheimer disease. J Biol Chem 2012; 287: 38992–9000.
Godyn J, Jonczyk J, Panek D, Malawska B . Therapeutic strategies for Alzheimer's disease in clinical trials. Pharmacol Rep 2016; 68: 127–38.
Frisardi V, Solfrizzi V, Imbimbo PB, Capurso C, D'Introno A, Colacicco AM, et al. Towards disease-modifying treatment of Alzheimer's disease: drugs targeting beta-amyloid. Curr Alzheimer Res 2010; 7: 40–55.
Eketjall S, Janson J, Kaspersson K, Bogstedt A, Jeppsson F, Falting J, et al. AZD3293: a novel, orally active BACE1 inhibitor with high potency and permeability and markedly slow off-rate kinetics. J Alzheimers Dis 2016; 50: 1109–23.
Molecule of the month. Semagacestat. Drug News Perspect 2008; 21: 390.
Cooke CE, Lee HY, Xing S . Adherence to antiretroviral therapy in managed care members in the United States: a retrospective claims analysis. J Manage Care Pharm 2014; 20: 86–92.
Poli G, Corda E, Lucchini B, Puricelli M, Martino PA, Dall'ara P, et al. Therapeutic effect of CHF5074, a new gamma-secretase modulator, in a mouse model of scrapie. Prion 2012; 6: 62–72.
Eriksen JL, Sagi SA, Smith TE, Weggen S, Das P, McLendon DC, et al. NSAIDs and enantiomers of flurbiprofen target gamma-secretase and lower Abeta 42 in vivo. J Clin Invest 2003; 112: 440–9.
Mueller-Steiner S, Zhou Y, Arai H, Roberson ED, Sun B, Chen J, et al. Antiamyloidogenic and neuroprotective functions of cathepsin B: implications for Alzheimer's disease. Neuron 2006; 51: 703–14.
Fluhrer R, Multhaup G, Schlicksupp A, Okochi M, Takeda M, Lammich S, et al. Identification of a beta-secretase activity, which truncates amyloid beta-peptide after its presenilin-dependent generation. J Biol Chem 2003; 278: 5531–8.
Jia QT, Deng YL, Qing H . Potential therapeutic strategies for Alzheimer's disease targeting or beyond. Beta-Amyloid: insights from clinical trials. BioMed Res Int 2014; 2014: 837157. Doi: 10.1155/2014/837157.
Lannfelt L, Relkin NR, Siemers ER . Amyloid-ss-directed immunotherapy for Alzheimer's disease. J Int Med 2014; 275: 284–95.
Crespi GA, Hermans SJ, Parker MW, Miles LA . Molecular basis for mid-region amyloid-beta capture by leading Alzheimer's disease immunotherapies. Sci Rep 2015; 5: 9649.
Novakovic D, Feligioni M, Scaccianoce S, Caruso A, Piccinin S, Schepisi C, et al. Profile of gantenerumab and its potential in the treatment of Alzheimer's disease. Drug Des Devel Ther 2013; 7: 1359–64.
O'Nuallain B, Acero L, Williams AD, Koeppen HP, Weber A, Schwarz HP, et al. Human plasma contains cross-reactive Abeta conformer-specific IgG antibodies. Biochemistry 2008; 47: 12254–6.
Dodel R, Hampel H, Depboylu C, Lin S, Gao F, Schock S, et al. Human antibodies against amyloid beta peptide: a potential treatment for Alzheimer's disease. Ann Neurol 2002; 52: 253–6.
Nishiyama Y, Taguchi H, Hara M, Planque SA, Mitsuda Y, Paul S . Metal-dependent amyloid beta-degrading catalytic antibody construct. J Biotechnol 2014; 180: 17–22.
Lee EB, Zhang B, Liu K, Greenbaum EA, Doms RW, Trojanowski JQ, et al. BACE overexpression alters the subcellular processing of APP and inhibits Abeta deposition in vivo. J Cell Biol 2005; 168: 291–302.
Abdul-Hay SO, Sahara T, McBride M, Kang D, Leissring MA . Identification of BACE2 as an avid beta-amyloid-degrading protease. Mol Neurodegen 2012; 7.
Taguchi H, Planque S, Nishiyama Y, Szabo P, Weksler ME, Friedland RP, et al. Catalytic antibodies to amyloid beta peptide in defense against Alzheimer disease. Autoimmun Rev 2008; 7: 391–7.
Taguchi H, Planque S, Sapparapu G, Boivin S, Hara M, Nishiyama Y, et al. Exceptional amyloid beta peptide hydrolyzing activity of nonphysiological immunoglobulin variable domain scaffolds. J Biol Chem 2008; 283: 36724–33.
Soto C, Sigurdsson EM, Morelli L, Kumar RA, Castano EM, Frangione B . beta-sheet breaker peptides inhibit fibrillogenesis in a rat brain model of amyloidosis: Implications for Alzheimer's therapy. Nat Med 1998; 4: 822–6.
Ramirez BG, Blazquez C, Gomez del Pulgar T, Guzman M, de Ceballos ML . Prevention of Alzheimer's disease pathology by cannabinoids: neuroprotection mediated by blockade of microglial activation. J Neurosci 2005; 25: 1904–13.
Soto C, Kascsak RJ, Saborio GP, Aucouturier P, Wisniewski T, Prelli F, et al. Reversion of prion protein conformational changes by synthetic beta-sheet breaker peptides. Lancet 2000; 355: 192–7.
Arispe N, Pollard HB, Rojas E . Giant multilevel cation channels Formed by Alzheimer-disease Amyloid Beta-Protein [A-Beta-P-(1-40)] in bilayer-membranes. Proc Natl Acad Sci U S A 1993; 90: 10573–7.
Ohyagi Y . A drug targeting intracellular amyloid-beta and oxidative stress: apomorphine. Rinsho Shinkeigaku 2011; 51: 884–7.
Pappolla M, Bozner P, Soto C, Shao H, Robakis NK, Zagorski M, et al. Inhibition of Alzheimer beta-fibrillogenesis by melatonin. J Biol Chem 1998; 273: 7185–8.
Sylla T, Pouysegu L, Da Costa G, Deffieux D, Monti JP, Quideau S . Gallotannins and tannic acid: first chemical syntheses and in vitro inhibitory activity on Alzheimer's amyloid beta-peptide aggregation. Angew Chem Int Ed Engl 2015; 54: 8217–21.
Wang P, Liao W, Fang J, Liu Q, Yao J, Hu M, et al. A glucan isolated from flowers of Lonicera japonica Thunb. inhibits aggregation and neurotoxicity of Abeta42. Carbohydrate polymers 2014; 110: 142–7.
Luo Y, Smith JV, Paramasivam V, Burdick A, Curry KJ, Buford JP, et al. Inhibition of amyloid-beta aggregation and caspase-3 activation by the Ginkgo biloba extract EGb761. Proc Natl Acad Sci U S A 2002; 99: 12197–202.
Chakrabortee S, Liu Y, Zhang L, Matthews HR, Zhang H, Pan N, et al. Macromolecular and small-molecule modulation of intracellular Abeta42 aggregation and associated toxicity. Biochem J 2012; 442: 507–15.
Zhou Y, Jiang C, Zhang Y, Liang Z, Liu W, Wang L, et al. Structural optimization and biological evaluation of substituted bisphenol A derivatives as beta-amyloid peptide aggregation inhibitors. J Med Chem 2010; 53: 5449–66.
Huang SM, Mouri A, Kokubo H, Nakajima R, Suemoto T, Higuchi M, et al. Neprilysin-sensitive synapse-associated amyloid-beta peptide oligomers impair neuronal plasticity and cognitive function. J Biol Chem 2006; 281: 17941–51.
Hafez D, Huang JY, Huynh AM, Valtierra S, Rockenstein E, Bruno AM, et al. Neprilysin-2 is an important beta-amyloid degrading enzyme. Am J Pathol 2011; 178: 306–12.
Huang JY, Bruno AM, Patel CA, Huynh AM, Philibert KD, Glucksman MJ, et al. Human membrane metallo-endopeptidase-like protein degrades both beta-amyloid 42 and beta-amyloid 40. Neuroscience 2008; 155: 258–62.
Eckman EA, Watson M, Marlow L, Sambamurti K, Eckman CB . Alzheimer's disease beta-amyloid peptide is increased in mice deficient in endothelin-converting enzyme. J Biol Chem 2003; 278: 2081–4.
Tucker HM, Kihiko-Ehmann M, Wright S, Rydel RE, Estus S . Tissue plasminogen activator requires plasminogen to modulate amyloid-beta neurotoxicity and deposition. J Neurochem 2000; 75: 2172–7.
Yamin R, Bagchi S, Hildebrant R, Scaloni A, Widom RL, Abraham CR . Acyl peptide hydrolase, a serine proteinase isolated from conditioned medium of neuroblastoma cells, degrades the amyloid-beta peptide. J Neurochem 2007; 100: 458–67.
Yamin R, Zhao C, O'Connor PB, Mckee AC, Abraham CR . Acyl peptide hydrolase degrades monomeric and oligomeric amyloid-beta peptide. Mol Neurodegener 2009; 4: 33. Doi: 10.1186/1750-1326-4-33.
Mueller-Steiner S, Zhou Y, Arai H, Roberson ED, Sun BG, Chen J, et al. Antiamyloidogenic and neuroprotective functions of cathepsin B: implications for Alzheimer's disease. Neuron 2006; 51: 703–14.
Abdul-Hay SO, Sahara T, McBride M, Kang D, Leissring MA . Identification of BACE2 as an avid beta-amyloid-degrading protease. Mol Neurodegeneration 2012; 7: Article No. 46.
Zhang B, Maiti A, Shively S, Lakhani F, McDonald-Jones G, Bruce J, et al. Microtubule-binding drugs offset tau sequestration by stabilizing microtubules and reversing fast axonal transport deficits in a tauopathy model. Proc Natl Acad Sci U S A 2005; 102: 227–31.
Gruninger F . Invited review: drug development for tauopathies. Neuropathol Appl Neurobiol 2015; 41: 81–96.
Godyn J, Jonczyk J, Panek D, Malawska B . Therapeutic strategies for Alzheimer's disease in clinical trials. Pharmacol Rep 2016; 68: 127–38.
Reger MA, Watson GS, Green PS, Baker LD, Cholerton B, Fishel MA, et al. Intranasal insulin administration dose-dependently modulates verbal memory and plasma amyloid-beta in memory-impaired older adults. J Alzheimers Dis 2008; 13: 323–31.
Qi L, Ke L, Liu X, Liao L, Ke S, Liu X, et al. Subcutaneous administration of liraglutide ameliorates learning and memory impairment by modulating tau hyperphosphorylation via the glycogen synthase kinase-3beta pathway in an amyloid beta protein induced alzheimer disease mouse model. Eur J Pharmacol 2016; 783: 23–32.
Soto C, Sigurdsson EM, Morelli L, Kumar RA, Castano EM, Frangione B . Beta-sheet breaker peptides inhibit fibrillogenesis in a rat brain model of amyloidosis: implications for Alzheimer's therapy. Nat Med 1998; 4: 822–6.
Soto C . Alzheimer's and prion disease as disorders of protein conformation: implications for the design of novel therapeutic approaches. J Mol Med 1999; 77: 412–8.
Diaz JC, Simakova O, Jacobson KA, Arispe N, Pollard HB . Small molecule blockers of the Alzheimer Abeta calcium channel potently protect neurons from Abeta cytotoxicity. Proc Natl Acad Sci U S A 2009; 106: 3348–53.
Fantini J, Di Scala C, Yahi N, Troadec JD, Sadelli K, Chahinian H, et al. Bexarotene blocks calcium-permeable ion channels formed by neurotoxic Alzheimer's beta-amyloid peptides. ACS Chem Neurosci 2014; 5: 216–24.
Anekonda TS, Quinn JF, Harris C, Frahler K, Wadsworth TL, Woltjer RL . L-type voltage-gated calcium channel blockade with isradipine as a therapeutic strategy for Alzheimer's disease. Neurobiol Dis 2011; 41: 62–70.
Marcos B, Chuang TT, Gil-Bea FJ, Ramirez MJ . Effects of 5-HT6 receptor antagonism and cholinesterase inhibition in models of cognitive impairment in the rat. Br J Pharmacol 2008; 155: 434–40.
Jiang HL, Luo XM, Bai DL . Progress in clinical, pharmacological, chemical and structural biological studies of huperzine A: A drug of traditional Chinese medicine origin for the treatment of Alzheimer's disease. Curr Med Chem 2003; 10: 2231–52.
Tao Y, Fang L, Yang Y, Jiang H, Yang H, Zhang H, et al. Quantitative proteomic analysis reveals the neuroprotective effects of huperzine A for amyloid beta treated neuroblastoma N2a cells. Proteomics 2013; 13: 1314–24.
Zhu ZY, Li CJ, Wang X, Yang ZY, Chen J, Hu LH, et al. 2,2′,4′-Trihydroxychalcone from glycyrrhiza glabra as a new specific BACE1 inhibitor efficiently ameliorates memory impairment in mice. J Neurochem 2010; 114: 374–85.
Li WM, Mak M, Jiang HL, Wang QW, Pang YP, Chen KX, et al. Novel anti-Alzheimer's dimer Bis(7)-cognitin: cellular and molecular mechanisms of neuroprotection through multiple targets. Neurotherapeutics 2009; 6: 187–201.
Zhu Z, Yan J, Jiang W . Yao X-g, Chen J, Chen L, et al. Arctigenin effectively ameliorates memory impairment in Alzheimer's disease model mice targeting both beta-amyloid production and clearance. J Neurosci 2013; 33: 13138–49.
Bush AI . The metallobiology of Alzheimer's disease. Trends Neurosci 2003; 26: 207–14.
Burstein AH, Grimes I, Galasko DR, Aisen PS, Sabbagh M, Mjalli AM . Effect of TTP488 in patients with mild to moderate Alzheimer's disease. BMC Neurol 2014; 14: 12.
Prickaerts J, van Goethem NP, Chesworth R, Shapiro G, Boess FG, Methfessel C, et al. EVP-6124, a novel and selective alpha7 nicotinic acetylcholine receptor partial agonist, improves memory performance by potentiating the acetylcholine response of alpha7 nicotinic acetylcholine receptors. Neuropharmacology 2012; 62: 1099–110.
Lawlor B, Kennelly S, O'Dwyer S, Cregg F, Walsh C, Coen R, et al. NILVAD protocol: a European multicentre double-blind placebo-controlled trial of nilvadipine in mild-to-moderate Alzheimer's disease. BMJ Open 2014; 4: e006364.
Howie AJ, Brewer DB, Howell D, Jones AP . Physical basis of colors seen in Congo red-stained amyloid in polarized light. Lab Invest 2008; 88: 232–42.
Aterman K . A historical note on the iodine-sulphuric acid reaction of amyloid. Histochemistry 1976; 49: 131–43.
Naiki H, Higuchi K, Kitagawa K, Shimada A, Chen WH, Hosokawa M, et al. Fluorometric examination of tissue amyloid fibrils in murine senile amyloidosis–use of the fluorescent indicator, thioflavine-T. Lab Invest 1990; 62: 768–73.
Elghetany MT, Saleem A . Methods for staining amyloid in tissues–a review. Stain Technol 1988; 63: 201–12.
Vassar PS, Culling CFA . Fluorescent stains, with special reference to amyloid and connective tissues. Arch Pathol 1959; 68: 487–98.
Toyama H, Ye D, Ichise M, Liow JS, Cai L, Jacobowitz D, et al. PET imaging of brain with the beta-amyloid probe, [11C]6-OH-BTA-1, in a transgenic mouse model of Alzheimer's disease. Eur J Nucl Med Mol Imaging 2005; 32: 593–600.
Dezutter NA, Dom RJ, de Groot TJ, Bormans GM, Verbruggen AM . 99mTc-MAMA-chrysamine G, a probe for beta-amyloid protein of Alzheimer's disease. Eur J Nucl Med 1999; 26: 1392–9.
Younan ND, Viles JH . A comparison of three fluorophores for the detection of amyloid fibers and prefibrillar oligomeric assemblies. ThT (Thioflavin T); ANS (1-anilinonaphthalene-8-sulfonic acid); and bisANS (4,4′-dianilino-1,1′-binaphthyl-5,5′-disulfonic acid). Biochemistry 2015; 54: 4297–306.
Mishra R, Sjolander D, Hammarstrom P . Spectroscopic characterization of diverse amyloid fibrils in vitro by the fluorescent dye Nile red. Mol Biosyst 2011; 7: 1232–40.
Zhang YH, Mann D, Raymick J, Sarkar S, Paule MG, Lahiri DK, et al. K114 inhibits A-beta aggregation and inflammation in vitro and in vivo in AD/Tg mice. Curr Alzheimer Res 2014; 11: 299–308.
Sato K, Higuchi M, Iwata N, Saido TC, Sasamoto K . Fluoro-substituted and 13C-labeled styrylbenzene derivatives for detecting brain amyloid plaques. Eur J Med Chem 2004; 39: 573–8.
Maiti P, Hall TC, Paladugu L, Kolli N, Learman C, Rossignol J, et al. A comparative study of dietary curcumin, nanocurcumin, and other classical amyloid-binding dyes for labeling and imaging of amyloid plaques in brain tissue of 5x-familial Alzheimer's disease mice. Histochem Cell Biol 2016; 146: 609–25.
Kosik KS . The neuronal microRNA system. Nat Rev Neurosci 2006; 7: 911–20.
Vilardo E, Barbato C, Ciotti M, Cogoni C, Ruberti F . MicroRNA-101 regulates amyloid precursor protein expression in hippocampal neurons. J Biol Chem 2010; 285: 18344–51.
Ling S, Zhou J, Rudd JA, Hu Z, Fang M . The recent updates of therapeutic approaches against abeta for the treatment of Alzheimer's disease. Anat Record 2011; 294: 1307–18.
Linkins KW, Lloyd LR, Hjelmstad GO, Strausbaugh HJ . Potential savings in the cost of caring for Alzheimer's disease treatment with rivastigmine. Pharmacoeconomics 2000; 18: 609–10.
Lopez-Miranda V, Soto-Montenegro ML, Vera G, Herradon E, Desco M, Abalo R . Resveratrol: a neuroprotective polyphenol in the Mediterranean diet. Rev Neurol 2012; 54: 349–56.
Winblad B, Andreasen N, Minthon L, Floesser A, Imbert G, Dumortier T, et al. Safety, tolerability, and antibody response of active Abeta immunotherapy with CAD106 in patients with Alzheimer's disease: randomised, double-blind, placebo-controlled, first-in-human study. Lancet Neurol 2012; 11: 597–604.
Wang CM, Devries S, Camboni M, Glass M, Martin PT . Immunization with the SDPM1 peptide lowers amyloid plaque burden and improves cognitive function in the APPswePSEN1(A246E) transgenic mouse model of Alzheimer's disease. Neurobiol Dis 2010; 39: 409–22.
Baruch K, Deczkowska A, Rosenzweig N, Tsitsou-Kampeli A, Sharif AM, Matcovitch-Natan O, et al. PD-1 immune checkpoint blockade reduces pathology and improves memory in mouse models of Alzheimer's disease. Nat Med 2016; 22: 135–7.
Tucker HM, Kihiko-Ehmann M, Wright S, Rydel RE, Estus S . Tissue plasminogen activator requires plasminogen to modulate amyloid-beta neurotoxicity and deposition. J Neurochem 2000; 75: 2172–7.
Yamin R, Bagchi S, Hildebrant R, Scaloni A, Widom RL, Abraham CR . Acyl peptide hydrolase, a serine proteinase isolated from conditioned medium of neuroblastoma cells, degrades the amyloid-beta peptide. J Neurochem 2007; 100: 458–67.
Yamin R, Zhao C, O'Connor PB, McKee AC, Abraham CR . Acyl peptide hydrolase degrades monomeric and oligomeric amyloid-beta peptide. Mol Neurodegener 2009; 4: 33. doi: 10.1186/1750-1326-4-33.
Avramopoulos D . Genetics of Alzheimer's disease: recent advances. Genome Med 2009; 1: 34.
Southan C, Hancock JM . A tale of two drug targets: the evolutionary history of BACE1 and BACE2. Front Genet 2013; 4: 293.
Acknowledgements
This work was supported by the Postdoctoral Science Foundation of China (No Y512031078), by grants from the National Natural Science Foundation of China (No 31300607) and the Shanghai Science and Technology Committee (No 13ZR1447600), as well as in part by the Jay and Betty Van Andel Foundation, Amway (China), and the Ministry of Science and Technology of China (No 2012CB910403, 2013CB910601 and XDB08020303).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Chen, Gf., Xu, Th., Yan, Y. et al. Amyloid beta: structure, biology and structure-based therapeutic development. Acta Pharmacol Sin 38, 1205–1235 (2017). https://doi.org/10.1038/aps.2017.28
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2017.28
Keywords
This article is cited by
-
Histone acetylation in an Alzheimer’s disease cell model promotes homeostatic amyloid-reducing pathways
Acta Neuropathologica Communications (2024)
-
Death-associated protein kinase 1 as a therapeutic target for Alzheimer's disease
Translational Neurodegeneration (2024)
-
A one-two punch targeting reactive oxygen species and fibril for rescuing Alzheimer’s disease
Nature Communications (2024)
-
Neuropathogenesis-on-chips for neurodegenerative diseases
Nature Communications (2024)
-
Design of amyloidogenic peptide traps
Nature Chemical Biology (2024)
