
Abstract
There remains a clear need for effective tumor cell purging in autologous stem cell transplantation (ASCT) where residual malignant cells within the autograft contribute to disease relapse. Here we propose the use of a novel Fas agonist with potent pro-apoptotic activity, termed MegaFasL, as an effective ex-vivo purging agent. MegaFasL selectively kills hematological cancer cells from lymphomas and leukemias and prevents tumor development at concentrations that do not reduce the functional capacity of human hematopoietic stem/progenitor cells both in in vitro and in in vivo transplantation models. These findings highlight the potential use of MegaFasL as an ex-vivo purging agent in ASCT.
Similar content being viewed by others


Introduction
Autologous stem cell transplantation (ASCT) has become the treatment of choice for an increasing number of selected patients with hematological malignancies. In ASCT, hematopoietic progenitor cells (HPC) are harvested from the patient before high-dose chemotherapy administered to the patient. The HPC graft is subsequently re-infused to the patient, rescuing him from hematopoietic toxicity linked to high-dose chemotherapy administration. A major drawback of ASCT is the presence of residual malignant cells within the graft re-infused to the patient. This complication has been correlated with disease relapse,1, 2, 3, 4, 5 but can be circumvented by ex-vivo purging of the graft to remove cancer cells while preserving HPC. In recent years, several technologies have been employed to purge cancerous cells from ASCT,6 including positive selection for progenitor cells,2, 7, 8 in vitro expansion of hematopoietic cells,9 depletion of tumor cells,10, 11 photodynamic purging processes,12, 13 genetic modification of tumor cells14, 15 and pulsed electric-field cell selection.16, 17 These methods are more or less weighty and variably efficient at eliminating cancer cells from the autograft in conditions that do not affect the hematopoietic activity of the autograft. This warrants the development of alternative simple methods to reduce or eliminate cancer cell contamination from grafts during ASCT.
In this context, we developed a novel soluble hexameric recombinant form of FasL, termed MegaFasL, which induces high levels of caspase-dependent apoptosis in vitro upon binding to cell surface Fas receptors.18 Indeed, Fas is expressed on almost all human tumor cells19 including hematological cancer cells. Interestingly, Fas is expressed at lower levels on some human acute leukemia progenitor cells,20 making attractive the use of a potent Fas agonist optimized for inducing cell death and able to fully eradicate hematopoietic cancers. We hypothesized that human HPC, regardless of their origin, would be resistant to MegaFasL-induced cell death as previously reported for other Fas-agonists21, 22 at concentrations that would eliminate such cancer cells.
Our study indicates that MegaFasL selectively kills hematological cancer cells and prevents tumor development at concentrations that do not reduce the functional capacity of human hematopoietic stem/progenitor cells both in an in vitro and in an in vivo transplantation model. These findings highlight the potential use of MegaFasL as an ex-vivo purging agent in ASCT.
Materials and methods
Reagent and monoclonal antibodies
MegaFasL was produced in our laboratories under current good manufacturer practice, resuspended in phosphate-buffered saline, and stored at −20 °C in aliquots. Such compound had a stable activity for at least 1 year. Information on the monoclonal antibodies and reagents used is summarized in Supplementary Table 1.
Cell origins, culture and analysis conditions
Human CD34+ cells from cord blood (CB), mobilized peripheral blood (mPB) or bone marrow (BM) were obtained unpurified (mPB) or already purified (CB, mPB, BM) from either AllCells, LLC (San Mateo, CA, USA), or purified using an anti-CD34 magnetic bead column (Miltenyi, Bergisch GladBach, Germany) from CB collected at the Obstetric Department of the CHUV after consent obtained from mothers. Aliquots of primary cells (purity >80%) from consenting patients with acute myeloid leukemia (AML 1: M2 subtype, AML 2: M3 subtype), multiple myeloma (MM, IgA-λ), marginal zone lymphoma and follicular lymphoma were collected and frozen in medium containing 10% dimethyl sulfoxide. The procedures have been accepted by the local ethics committee of our hospital. Information on cell lines used in the present manuscript is provided in Supplementary Table 2. Aliquots of cryopreserved CD34+ cells were thawed and washed twice, and their viabilities were 88±6% (by Trypan blue dye exclusion) and contained 95±2% CD34+ cells by fluorescence-activated cell sorting (FACS) analysis of immunostained cells. HPC, malignant cells from patients and cell lines incubated with or without MegaFasL were culture as previously described.18 Apoptosis was monitored using annexin-V and 7-aminoactinomycin D stainings as described by the manufacturer. The analysis was carried out using a five-color flow cytometer (Beckman Coulter Cytomics FC500, Beckman Coulter, Nyon, Switzerland).
Colony-forming cells (CFU) assay
CD34+ cells incubated with or without MegaFasL for 5 h were evaluated for their capacity to generate granulocyte/macrophage (CFU-G/M), burst-forming units-erythroid colonies and multi-lineage (CFU-Mix) colonies, by plating 0.5 to 1 × 103 CD34+ cells (from either mPB or CB) in 1-ml methylcellulose medium supplemented with human growth factors (Methocult GF+ H4435, StemCell Technologies Inc., Vancouver, BC, Canada). Colonies were scored under an inverted microscope after 2 weeks at 37 °C, in a humidified atmosphere at 5% CO2. CFU assays were similarly performed on 105 BM cells from transplanted nonobese diabetes/severe combined immunodeficiency (NOD/SCID) mice.
Long-term culture-initiating cell assay
Bulk long-term culture-initiating cell assays were performed according to the technique described elsewhere.23, 24 Bone marrow stromal cells (M2-10B4 cell line) were cultured in RPMI medium, harvested and irradiated (80 Gy). Subsequently, adherent cells (3 × 105per ml per well) were replated in 24-well plates and cultured in 1-ml Myelocult medium (StemCell Technologies Inc.) containing 1 × 10−6 M hydrocortisone. After 24 h, the stromal layers were added with 2 × 104 CD34+ cells from mPB or CB per well, previously incubated in the presence or absence of MegaFasL for 5 h. Cells were cultured for 6 weeks with weekly half-medium changes then washed, counted and plated for colony scoring as described above.
Transplantation of human cells in NOD/SCID mice and xenochimeric mouse analyses
NOD/LtSz-Prkdcscid/Prkdcscid mice were purchased from Iffa Credo (L’Arbresle, France), bred and maintained in micro-isolator cages within a specific pathogen-free room in the animal facility of the University Hospital of Lausanne. All animals were handled according to the institutional regulations after approval of the local animal ethics committee. Manipulations were performed under a laminar flow hood under sterile conditions. Mice to be transplanted were given 250-cGy total body irradiation at 6 to 8 weeks of age from a 137Cesium 4000Ci source (Robatel, Genas, France). Within 24 h of irradiation, the mice were injected with either 2–4 × 106 CD34+ cells from mPB or 5–7 × 105 CD34+ cells from CB. Alternatively, mice received a mixture of 7 × 105 CD34+ cells from CB and 1 × 104 Raji cells, or with 1 × 104 Raji cells that had been incubated with or without MegaFasL (at 50 and 200 ng/ml). In experiments in which CD34+ cells were injected into mice, the engraftment of the HPC and the multi-lineage reconstitution of human hematopoietic cells were assessed 6 weeks after transplantation. The transplanted mice were killed and BM cells were flushed from both femurs and tibias with phosphate-buffered saline containing 1% bovine serum albumin. Cells were then either analyzed by FACS using human-specific antibodies, plated for CFU assay, or processed for further DNA extraction.
Mice injected with Raji cells were followed daily for hind-leg paralysis development, a robust sign of Raji tumor growth. Mice with paresis were killed, and lymphoma presence evaluated by FACS examination of BM. Mice given a mixture of CD34+ and Raji cells without paresis were killed 6 weeks after the transplant and analyzed as above.
Polymerase chain reactions
DNAs were extracted from xenochimeric mouse BM cells, and from Raji cells diluted in NOD/SCID mouse BM cells at various percentages, using Qiamp DNA blood mini Kit (Qiagen, Hilden, Germany). DNAs were subjected to: (a) long-distance PCR for the detection of t(8;14)(q24;q32) present in Burkitt's lymphoma Raji cells exactly as described,25 and (b) PCR for amplification of human β-globin gene as described.26
Statistical analysis
The Kaplan–Meier method was used for the estimation of differences in survival between different groups of mice (mice receiving untreated cells (control) vs those given MegaFasL-treated cells). Data are expressed as mean±s.d. unless noted. Values between groups were compared using one-way analysis of variance, non-parametric test with a GraphPad Prism 4.00 software (GraphPad Software Inc., San Diego, CA, USA). A P-value less than 0.05 was considered as statistically significant.
Results
CD34+CD38low cells express low level of Fas and are resistant to MegaFasL, whereas the latter efficiently killing hematological cancer cells
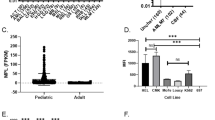
As a first step, we determined Fas expressions on CD34+CD38low cells from mPB, BM and CB, and on hematological malignant cells. CD34+CD38low cells represent a sub-population of HPC that are responsible for repopulating the hematopoietic system in human HPC transplantation models,27 and were found to express low level of cell surface Fas expression independently of their origin (Figure 1a). In accordance with our previous studies, both primary cells and cell lines from AML, MM and lymphoma express variable, yet constantly high, levels of cell surface Fas (Supplementary Figure 1).

CD34+CD38low cells express low level of Fas and are resistant to MegaFasL. (a) Cell surface Fas expression on CD34+CD38low cells determined by FACS using a fluorochrome-labelled antibody against human Fas (solid line). The numbers represent the fold increase in mean fluorescence intensity compared with that of an isotypic control antibody (filled area). (b) CD34+ cells from mPB, BM or CB cells were thawed and incubated with or without different concentrations of MegaFasL for 5 h, and apoptosis was evaluated by FACS using annexin-V and 7-aminoactinomycin D dyes. Alternatively, CD34+ cells were similarly analyzed immediately after thawing cells (pre-incub). Annexin-V+ and 7-aminoactinomycin D− cells represent early apoptotic cells (hatched bars) and annexin-V+ and 7-aminoactinomycin D+ cells represent late apoptotic cells (solid black bars). Data are representative of three independent experiments.
We next evaluated the sensitivity of CD34+ cells to MegaFasL. To investigate its cytotoxicity towards HPC, CD34+ cells from various source (mPB, BM and CB) were incubated with MegaFasL for 5 h, and apoptosis was determined by annexin and 7-aminoactinomycin D staining in early HPC (CD34+CD38low). As shown in Figure 1b, CD34+CD38low cells from various sources were resistant to killing mediated by MegaFasL, after treament with MegafasL up to 200 ng/ml, with a threshold of less than 12% HPC becoming apoptotic. This was confirmed on unpurified mPB from three preparations (Supplementary Figure 2). To extend the investigation into the effect of MegaFasL on the hematological cancer cells tested in this study, we then assessed the sensitivity of primary malignant cells and cell lines from various hematological malignancies (AML, MM and lymphoma). Cells were cultured for 5 h in presence or absence of increasing concentrations of MegaFasL (0–200 ng/ml). As shown in Supplementary Figure 3, in contrast to HPC, both primary cells and cell lines from AML, MM and lymphoma were highly sensitive to MegaFasL-induced apoptosis. Primary cells from patients with AML and MM were the most sensitive, with 90–98% and 96% becoming apoptotic after treatement with 200 ng/ml. Two lymphoma primary cells displayed moderate sensitivity to MegaFasL with 65% apoptosis (follicular lymphoma) and 59% apoptosis (marginal zone lymphoma) seen at a concentrentation of 200 ng/ml. All cancer cell lines tested in the current study, except L363 were highly sensitive to MegaFasL, with more than 80% apoptosis at 200 ng/ml. L363 showed an intermediate sensitivity to MegaFasL, with 62% becoming apoptotic after treatment with 200 ng/ml. These data also confirm our previous observations on various types of hematological cancers from 12 patients and 9 different hematotopoietic cancer cell lines.18 The variability in sensitivity to MegaFasL, particularly in primary cells can be accounted for by variability in Fas expression. Taken together, these data support that MegaFasL kills most cancer cells while preserving HPC.
MegaFasL does not affect the clonogenic and in-vivo repopulating capacities of HPC
To address this issue, we determined the clonogenic capacity of purified HPC cells after treatment with MegaFasL in vitro. CD34+ cells from mPB and CB were cultured for 5 h in presence or absence of MegaFasL and then plated in the CFU assays. Treatment with MegaFasL (up to 200 ng/ml) did not affect the numbers of erythroid (burst-forming units-erythroid colonies), granulocyte/macrophage (CFU-G/M) and mixed (CFU-mix) colonies as compared with no treatment (Figure 2a). We also investigated whether more primitive hematopoietic progenitors than CFU would be affected by treatment with MegaFasL using a bulk long-term culture-initiating cell assay. We found that the potential of HPC to persist for 6 weeks and subsequently differentiate into hematopoietic lineages was similar when treated with up 200 ng/ml MegaFasL as compared with untreated HPC, regardless of their origin (Figure 2b).

In vitro functionality and in vivo repopulating capacity of HPC are not affected by MegaFasL treatment. CD34+ cells from mPB or CB were incubated in medium with or without MegaFasL, then washed and plated on: (a) methocult medium for CFU assay, and (b) myelocult medium for long-term culture-initiating cell (LTC-IC) assay. CFU and LTC-IC data are shown as mean±s.d. of five and three independent experiments, respectively. BFU-E: burst forming units (erythroid colonies); CFU-G/M: granulocyte/macrophage colonies; and CFU-Mix are multi-lineage colonies. (c) Transplanted immediately (pre-incubation) into irradiated NOD/SCID mice or incubated for 5 h, with or without MegaFasL (post-incub) before transplantation into mice. Mice BM cells were analyzed by FACS for the presence of human (CD45+) cells. Data are from three independent experiments and each point represents the percentage of human CD45+ cells present in the BM of one mouse. The blank squares represent the percentage of human CD45+ cells present in the BM of mice engrafted with mPB, whereas filled circles represent the percentage of human CD45+ cells present in the BM of mice engrafted with CB. (d) To demonstrate the presence of HPC in the BM of mice 6 weeks after transplantation, flushed BM cells were plated on methylcellulose for a CFU assay. Results are from five mice (pre-incubation, post-incubation with 0 and 50 ng/ml MegaFasL) or four mice (post-incubation with 200 ng/ml MegaFasL) from three independent experiments. Lineage-specific colony numbers were not statistically significant between each condition. Bone marrow cells from a mouse non-injected with human CD34+ cells and similarly analyzed generated no colonies. BFU-E, CFU-G/M and CFU-Mix are as in (a).
We next evaluated the effect of MegaFasL treatment on the repopulating capacity of HPC (engraftment, persistence, differentiation) transplanted in conditioned immune-deficient mice.28, 29, 30 CD34+ cells from mPB or CB incubated with or without MegaFasL were transplanted into irradiated NOD/SCID mice and BM from xenochimeric animals were analyzed 6 weeks thereafter. Human CD45+ cells were detected in the BM of all NOD/SCID mice recipients 6 weeks after injection of CD34+ cells. First, pre-treatment with MegaFasL had no effect on their capacity to generate CD45+ cells (Figure 2c). Importantly, viability or function of HPC from mPB, which have been exposed to granulocyte colony-stimulating factor during the collection of stem cells were not readily affected by MegaFasL treatment. Second, FACS analysis using human-specific markers revealed that MegaFasL-treated HPC from CB (Supplementary Table 1 and Supplementary Figure 4) and mPB differentiated into erythroid (glycophorin A+), myeloid (CD33+/CD13+), B-lymphoid (CD19+) lineages and led to progenitors (CD34+ cells) at similar percentages to those observed in untreated HPC. Third, the capacity of xenochimeric BM cells to generate lineage-specific CFU was quantitatively similar in MegaFasL-treated and control groups (Figure 2d). FACS analysis confirmed the human origin of CFU cells. These results strongly indicate that MegaFasL does not affect the functional capacity of HPC to differentiate into the expected lineages in vivo.
Taken together, the above results strongly suggest that hematological malignant cells could be eliminated using concentrations of MegaFasL that had little effect on the function of HPC, highlighting a potential therapeutic window for the use of MegaFasL as an ex-vivo purging agent in ASCT.
MegaFasL eradicates tumor cells, whereas sparing the hematopoietic function of HPC, and prevents at long term the tumor development in NOD/SCID model of human Burkitt's lymphoma
We developed a laboratory model of a proof of concept for MegaFasL as an ex-vivo purging agent in ASCT. We used CB as source for HPC in this model, because CD34+ cells from mPB and CB are similarly resistant to MegaFasL (see above) and furthermore, CD34+ cells from CB engraft much better in NOD/SCID mice and few cells are required. We used Raji cells, because homogenous expandable population of these cells enabled to reproduce an acute lymphoblastic leukemia model with reliable and constant proliferation and mortality characteristics. As a note, in our hand primary leukemia cells do not engraft readily in animals and were not usable for defining an effect of MegaFasL (data not shown). As shown in Figure 3a, all mice (five out of five; 100%) that received the untreated HPC/Raji cell mixture developed paresis within 5 weeks post-transplantation, whereas only two out of eight mice (25%) given the cell mixture treated with MegaFasL became paretic over a 40-day examination period. There was a significant survival benefit in the MegaFasL-treated group compared with the control group (log-rank test, P<0.001). In addition, MegaFasL treatment had no significant effect on the repopulating function of HPC in mice injected with CD34+/Raji cell mixture (Supplementary Table 3). Importantly, no signs of toxicity was observed in mice (n=6) that received cell mixture treated with MegaFasL for 6 weeks, no premature death occurred in the treated group and the corresponding histopathology of liver, spleen, lung, gut, kidney and inguinal lymph nodes in this group was no different from untreated animals. This observation does not support the hypothesis that insoluble aggregates of MegaFasL remaining on the surface of treated cells cause organ damage in the recipient. FACS analysis on BM flushes from all six surviving mice injected with CD34+/Raji cell mixture treated with MegaFasL showed human hematopoiesis with cells bearing specific human markers for erythrocytes, myeloid cells, B-lymphocytes and progenitor cells in proportions very similar to those in mice injected with CD34+ cell alone (Table 1 and Supplementary Table 3). In accordance with this, CFU analyses performed on four of six BM flushes from the surviving mice showed similar numbers of burst forming units-erythroid colonies (P=0.73, analysis of variance) and CFU-G/M (P=0.55, analysis of variance) colonies when compared with analyses performed on CD34+ cell injected mice. Finally, FACS analyses of the CFUs confirmed the presence of human erythrocytes (five of six mice) and G/M (six out of six mice) in the cells flushed from the BM of mice injected with an HPC/Raji cell mixture treated with MegaFasL.

MegaFasL treatment of a cell mixture prevents in-vivo tumor development at concentrations that do not affect the hematopoietic repopulating function of CD34+ cells. (a) Mice (n=8) injected with a CD34+CB/Raji cell mixture treated with MegaFasL survived significantly longer than mice (n=5) injected with an untreated CD34+CB/Raji cell mixture. Data are expressed as the proportion of surviving mice against time. (b) PCR amplification of t(8;14)(q24;q32) present in Burkitt's lymphoma (7.6 kb) in DNA extracted from BM cells from: mice that received untreated cell mixture (lanes 1–2); mice that received a cell mixture treated with MegaFasL (lanes 3–8); pure Raji cell preparation (100) and from Raji cells diluted up to a 100-fold (10−1–10−2) with NOD/SCID mouse BM cells. Negative controls for PCR include DNA extracted from a non-injected NOD/SCID mouse (lane 9) and buffer (lane 10). DNA fragment of human-specific β-globin gene (268 bp) was used as control for PCR amplifications of human DNA. M, markers. Top: DNA ladder mix. Bottom: 50-bp DNA ladder. (c) A total of 11 animals given 200 ng/ml MegaFasL-treated Raji cells and 9 animals given 50 ng/ml MegaFasL-treated Raji cells survived significantly longer than 22 animals (controls) receiving untreated Raji cells (log rank test, P<0.0001).
Raji tumor cells express CD19, so it could not be excluded from the above that a fraction of the CD19+ cells were Raji cells remaining in the BM after MegaFasL treatment. We performed two types of experiments to address this point. First, we observed that Raji cells, but not primary B cells, grow extensively and develop large colonies upon plating on CFU medium. Application of this procedure to BM flushes from four paretic mice showed 74±21% (±s.e.m.) of CD19+ cells with the Raji phenotype, whereas similar analysis on BM flushes from four available non-paretic mice injected with a HPC/Raji cell mixture treated with MegaFasL had 2±0.35% (±s.e.m.) of CD19+ cells. Second, t(8;14) (q24;q32) Raji cell-specific DNA fragments were amplified by PCR from BM from two available specimens from recipients of an untreated cell mixture, whereas none of the six available recipients of a MegaFasL-treated cell mixture had such fragment amplified (Figure 3b). These data demonstrate that MegaFasL reduced tumor burden within animals to undetectable levels by PCR assay, and significantly delays the development of paresis at concentrations that do not impair the potential of HPC to engraft, persist and differentiate in vivo into the expected hematopoietic lineages.
The long-term in vivo tumor-preventing effect of MegaFasL on Raji tumor cells was subsequently tested in similar animals. Tumor development in 22 control animals led to a median survival of 30 days (95% CI 24–46). In contrast, NOD/SCID mice that received Raji cells treated with MegaFasL at 50 ng/ml (n=9) and 200 ng/ml (n=11), survived significantly longer (log-rank test P<0.0001 for both groups) and their median survival times were not reached at day 120 (Figure 3c). Survivals of animals injected with Raji cells treated with 50 and 200 ng/ml of MegaFasL were similar (P=0.9 between the two groups). This observation confirmed the significant tumor preventive activity at long term of MegaFasL when incubated in vitro before transplantation.
Discussion
In the present study, we exploit the concept of using MegaFasL as an ex-vivo purging agent in a model of ASCT following myeloablative high-dose chemotherapy. We showed that MegaFasL could safely eradicate leukemia and lymphoma cells without altering the function of HPC, regardless of their source. We used notably an in-vivo repopulating hematopoietic cell assay, where BM of animals have been analyzed 6 weeks after transplantation, enabling the detection of primitive HPC. The resistance to MegaFasL-induced apoptosis observed in CD34+CD38low cells from mPB, BM or CB is consistent with previous reports indicating HPC resistance to Fas-inducing apoptosis when using other Fas agonists. This resistance of HPC is most likely to be due to low level of cell surface Fas expression and to overexpression of anti-apoptotic proteins such as cFLIP21 and caspase-8L.22 In contrast, human hematological malignant cells were highly sensitive to MegaFasL treatment, with AML, MM and lymphoma cells being the most sensitive. MegaFasL sensitivity level correlates with that of Fas expression on the cell population. The high sensisitivity of most primary hematopoietic malignant cells towards MegaFasL could be explained by its potent killing effects compared with other Fas agonists. Indeed, we previously demonstrated and confirmed here that MegaFasL is able to induce apoptosis in primary malignant cells and cancer cell lines which are resistant to other Fas agonists, including sFasL, sFasL M2 and CH11.18 In an animal model of human transplantation, we showed that MegaFasL treatment of a mixed cell population of human HPC and tumor cells, before transplantation, clearly eliminated tumor cells, thus preventing tumor development at long term. This finding highlights that MegaFasL can selectively eliminate tumor cells without damping in the presence of a large amount of CD34+ cells, and the potential use of MegaFasL as an ex-vivo purging agent in ASCT, where the presence of residual tumor cells in the infused hematopoietic cells, often evidenced by gene marking, could contribute to relapse after transplantation in follicular lymphoma,1 AML,3 CML4 and MM.2 The interest of purging the autograft is also based on findings from studies reporting that patients infused with autologous hematopoietic cells, free of contaminating cancer cells as assessed by PCR, have a longer disease-free period than those patients infused with autologous cells that are positive for cancer cells by PCR assay.31, 32 Importantly, the ex-vivo use of MegaFasL allows to circumvent any potential in-vivo toxicity issues of Fas agonists in humans, because stem cell transplants would be treated with MegaFasL, then washed to remove MegaFasL before re-infusion into a patient, ensuring the safety of the procedure. Indeed, MegaFasL is not detectable in the cell supernatant after three centrifugal washes (Supplementary Figure 5). Furthermore, MegaFasL induces cell death via death receptor signaling pathways, a mechanism mostly independent of those utilized by classical chemotherapeutic agents. This could be advantageous in an ASCT setting, where residual tumor cells within the HPC graft could be resistant to conventional chemotherapy treatment due to repeated administration before cell harvesting.
In summary, the data presented here validate the principle of using MegaFasL as an ex-vivo purging agent in HPC transplantation and demonstrate its potential that could be evaluated in a clinical setting. The simplicity of purging an autograft with MegaFasL, its strong apoptotic effect on various hematological disorders, and the absence of evident HPC toxicity, underline the advantages of MegaFasL. The use of MegaFasL as an ex-vivo purging agent in ASCT may likely provide a rapid and simple, and safe method for removing contaminating cancer cells from HPC preparations. Future trials will evaluate the benefit of this strategy in ASCT in humans.
References
van Besien K, Loberiza Jr FR, Bajorunaite R, Armitage JO, Bashey A, Burns LJ et al. Comparison of autologous and allogeneic hematopoietic stem cell transplantation for follicular lymphoma. Blood 2003; 102: 3521–3529.
Gertz MA, Witzig TE, Pineda AA, Greipp PR, Kyle RA, Litzow MR . Monoclonal plasma cells in the blood stem cell harvest from patients with multiple myeloma are associated with shortened relapse-free survival after transplantation. Bone Marrow Transplant 1997; 19: 337–342.
Brenner MK, Rill DR, Moen RC, Krance RA, Mirro Jr J, Anderson WF et al. Gene-marking to trace origin of relapse after autologous bone-marrow transplantation. Lancet 1993; 341: 85–86.
Deisseroth AB, Zu Z, Claxton D, Hanania EG, Fu S, Ellerson D et al. Genetic marking shows that Ph+ cells present in autologous transplants of chronic myelogenous leukemia (CML) contribute to relapse after autologous bone marrow in CML. Blood 1994; 83: 3068–3076.
Granena A, Castellsague X, Badell I, Ferra C, Ortega J, Brunet S et al. Autologous bone marrow transplantation for high risk acute lymphoblastic leukemia: clinical relevance of ex vivo bone marrow purging with monoclonal antibodies and complement. Bone Marrow Transplant 1999; 24: 621–627.
Freedman AS, Neuberg D, Mauch P, Soiffer RJ, Anderson KC, Fisher DC et al. Long-term follow-up of autologous bone marrow transplantation in patients with relapsed follicular lymphoma. Blood 1999; 94: 3325–3333.
Hildebrandt M, Serke S, Meyer O, Ebell W, Salama A . Immunomagnetic selection of CD34+ cells: factors influencing component purity and yield. Transfusion 2000; 40: 507–512.
Tricot G, Gazitt Y, Leemhuis T, Jagannath S, Desikan KR, Siegel D et al. Collection, tumor contamination, and engraftment kinetics of highly purified hematopoietic progenitor cells to support high dose therapy in multiple myeloma. Blood 1998; 91: 4489–4495.
Lundell BI, Vredenburgh JJ, Tyer C, DeSombre K, Smith AK . Ex vivo expansion of bone marrow from breast cancer patients: reduction in tumor cell content through passive purging. Bone Marrow Transplant 1998; 22: 153–159.
Anderson KC, Andersen J, Soiffer R, Freedman AS, Rabinowe SN, Robertson MJ et al. Monoclonal antibody-purged bone marrow transplantation therapy for multiple myeloma. Blood 1993; 82: 2568–2576.
Lee NS, Cheong HJ, Kim SJ, Kim SE, Kim CK, Lee KT et al. Ex vivo purging of leukemia cells using tumor-necrosis-factor-related apoptosis-inducing ligand in hematopoietic stem cell transplantation. Leukemia 2003; 17: 1375–1383.
Gulliya KS, Fay JW, Dowben RM, Berkholder S, Matthews JL . Elimination of leukemic cells by laser photodynamic therapy. Cancer Chemother Pharmacol 1988; 22: 211–214.
Mulroney CM, Gluck S, Ho AD . The use of photodynamic therapy in bone marrow purging. Semin Oncol 1994; 21: 24–27.
Teoh G, Chen L, Urashima M, Tai YT, Celi LA, Chen D et al. Adenovirus vector-based purging of multiple myeloma cells. Blood 1998; 92: 4591–4601.
Thirukkumaran CM, Luider JM, Stewart DA, Cheng T, Lupichuk SM, Nodwell MJ et al. Reovirus oncolysis as a novel purging strategy for autologous stem cell transplantation. Blood 2003; 102: 377–387.
Eppich HM, Foxall R, Gaynor K, Dombkowski D, Miura N, Cheng T et al. Pulsed electric fields for selection of hematopoietic cells and depletion of tumor cell contaminants. Nature Biotechnol 2000; 18: 882–887.
Craiu A, Saito Y, Limon A, Eppich HM, Olson DP, Rodrigues N et al. Flowing cells through pulsed electric fields efficiently purges stem cell preparations of contaminating myeloma cells while preserving stem cell function. Blood 2005; 105: 2235–2238.
Greaney P, Nahimana A, Lagopoulos L, Etter AL, Aubry D, Attinger A et al. A Fas agonist induces high levels of apoptosis in haematological malignancies. Leuk Res 2006; 30: 415–426.
Peter ME, Budd RC, Desbarats J, Hedrick SM, Hueber AO, Newell MK et al. The CD95 receptor: apoptosis revisited. Cell 2007; 129: 447–450.
Costello RT, Mallet F, Gaugler B, Sainty D, Arnoulet C, Gastaut JA et al. Human acute myeloid leukemia CD34+/CD38- progenitor cells have decreased sensitivity to chemotherapy and Fas-induced apoptosis, reduced immunogenicity, and impaired dendritic cell transformation capacities. Cancer Res 2000; 60: 4403–4411.
Kim H, Whartenby KA, Georgantas III RW, Wingard J, Civin CI . Human CD34+ hematopoietic stem/progenitor cells express high levels of FLIP and are resistant to Fas-mediated apoptosis. Stem Cells 2002; 20: 174–182.
Mohr A, Zwacka RM, Jarmy G, Buneker C, Schrezenmeier H, Dohner K et al. Caspase-8 L expression protects CD34+ hematopoietic progenitor cells and leukemic cells from CD95-mediated apoptosis. Oncogene 2005; 24: 2421–2429.
Hogge DE, Lansdorp PM, Reid D, Gerhard B, Eaves CJ . Enhanced detection, maintenance, and differentiation of primitive human hematopoietic cells in cultures containing murine fibroblasts engineered to produce human steel factor, interleukin-3, and granulocyte colony-stimulating factor. Blood 1996; 88: 3765–3773.
Berardi AC, Meffre E, Pflumio F, Katz A, Vainchenker W, Schiff C et al. Individual CD34+CD38lowCD19-CD10- progenitor cells from human cord blood generate B lymphocytes and granulocytes. Blood 1997; 89: 3554–3564.
Basso K, Frascella E, Zanesco L, Rosolen A . Improved long-distance polymerase chain reaction for the detection of t(8;14)(q24;q32) in Burkitt′s lymphomas. Am J Pathol 1999; 155: 1479–1485.
Duchosal MA, Eming SA, McConahey PJ, Dixon FJ . Characterization of hu-PBL-SCID mice with high human immunoglobulin serum levels and graft-versus-host disease. Am J Pathol 1992; 141: 1097–1113.
Dick JE, Bhatia M, Gan O, Kapp U, Wang JCY . Assay of human stem cells by repopulation of NOD/SCID mice. Stem Cells 1997; 15: 199–203.
Vormoor J, Lapidot T, Pflumio F, Risdon G, Patterson B, Broxmeyer HE et al. Immature human cord blood progenitors engraft and proliferate to high levels in severe combined immunodeficient mice. Blood 1994; 83: 2489–2497.
Cashman J, Bockhold K, Hogge DE, Eaves AC, Eaves CJ . Sustained proliferation, multi-lineage differentiation and maintenance of primitive human haemopoietic cells in NOD/SCID mice transplanted with human cord blood. Br J Haematol 1997; 98: 1026–1036.
Hogan CJ, Shpall EJ, McNulty O, McNiece I, Dick JE, Shultz LD et al. Engraftment and development of human CD34+-enriched cells from umbilical cord blood in NOD/LtSz-scid/scid mice. Blood 1997; 90: 85–96.
Fields KK, Elfenbein GJ, Trudeau WL, Perkins JB, Janssen WE, Moscinski LC . Clinical significance of bone marrow metastases as detected using the polymerase chain reaction in patients with breast cancer undergoing high-dose chemotherapy and autologous bone marrow transplantation. J Clin Oncol 1996; 14: 1868–1876.
Freedman AS, Neuberg D, Mauch P, Soiffer RJ, Anderson KC, Fisher DC et al. Long-term follow-up of autologous bone marrow transplantation in patients with relapsed follicular lymphoma. Blood 1999; 94: 3325–3333.
Acknowledgements
We thank Professor P Hohlfeld at the Department of Obstetrics, Division of Fetal Medicine, University Hospital of Lausanne, for coordinating cord blood collection; Dr Bady within the clinical epidemiological center at CHUV for help with statistical analyses; and Kuen-Mooi Béguin for animal care. We are also indebted to all members of the Bone Marrow Laboratory, CHUV, for kindly helping us in collecting clinical samples. This work was supported by a contract (number 7244.1;4 LSPP-LS) from the innovation promotion agency at the Swiss Federal Office for Professional Education and Technology.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
PG, AA, SD, KMD and MD were employed by Apoxis, (now TopoTarget Switzerland SA). All other authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Blood Cancer Journal website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Nahimana, A., Aubry, D., Lagopoulos, L. et al. A novel potent Fas agonist for selective depletion of tumor cells in hematopoietic transplants. Blood Cancer Journal 1, e47 (2011). https://doi.org/10.1038/bcj.2011.47
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bcj.2011.47
Keywords
This article is cited by
-
Therapeutic approaches targeting CD95L/CD95 signaling in cancer and autoimmune diseases
Cell Death & Disease (2022)
-
Highly efficient, In-vivo Fas-mediated Apoptosis of B-cell Lymphoma by Hexameric CTLA4-FasL
Journal of Hematology & Oncology (2014)
