
Abstract
Background:
Previous analyses from a randomised trial in women aged 24–45 years have shown the quadrivalent human papillomavirus (qHPV) vaccine to be efficacious in the prevention of infection, cervical intraepithelial neoplasia (CIN), and external genital lesions (EGLs) related to HPV 6/11/16/18. In this report, we present end-of-study efficacy, safety, and immunogenicity data with a median follow-up time of 4.0 years.
Methods:
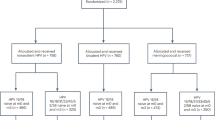
We enrolled 3819 24–45-year-old women with no history of cervical disease or genital warts in the past 5 years. Women received quadrivalent vaccine or placebo at day 1, and at months 2 and 6. Ascertainment of CIN/EGL was accomplished through Pap testing, genital inspection, and cervicovaginal sampling (every 6 months). The main analysis was conducted in a per-protocol efficacy population (that received three doses, was naive to the relevant HPV types at day 1, and remained free of infection through month 7). Efficacy was also estimated in other naive and non-naive populations.
Results:
Vaccine efficacy against the combined incidence of persistent infection, CIN/EGL related to HPV6/11/16/18 in the per-protocol population was 88.7% (95% CI: 78.1, 94.8). Efficacy for women who were seropositive and DNA negative for the relevant vaccine HPV type at the time of enrolment who received at least 1 dose was 66.9% (95% CI: 4.3, 90.6). At month 48, 91.5, 92.0, 97.4, and 47.9% of vaccinated women were seropositive to HPV 6/11/16/18, respectively. No serious vaccine-related adverse experiences were reported.
Conclusions:
The qHPV vaccine demonstrated high efficacy, immunogenicity, and acceptable safety in women aged 24–45 years, regardless of previous exposure to HPV vaccine type.
Similar content being viewed by others


Main
Persistent infection of the uterine cervix by 15–20 carcinogenic human papillomavirus (HPV) genotypes leads to the vast majority of cervical cancers (Walboomers et al, 1999; Muñoz et al, 2003) and related precursor lesions (International Agency for Research on Cancer Working Group, 2007). Although most sexually active women are at risk of HPV infection, the incidence of HPV infection peaks soon after the onset of sexual activity in most populations (Jacobs et al, 2000; Schiffman and Kjaer, 2003; Dunne et al, 2007). Although incidence rates tend to decline thereafter, women older than age 25 years also remain at risk for acquisition of new HPV infections (Castellsague et al, 2009; Munoz et al, 2009).
Data from Colombia show that the 5-year cumulative risk of incident cervical HPV infection decreased from 42.5% in females aged 15–19 years to 30% in those aged 25–29 years, and to 22% in those aged 30–44 years (Munoz et al, 2004). However, a second peak in HPV DNA prevalence has been observed in women in the fourth and fifth decades of life (de Sanjose et al, 2007). Whether this second peak is due to new infections, viral reactivation, waning immunity, or another mechanism is unclear. The cohort study from Colombia supports the possibility of new infections, as the curve of incident high-risk HPV infections is also bimodal with a first peak in women under 25 years of age and a second peak after menopause (Munoz et al, 2004). Conflicting evidence with respect to a bimodal infection peak is provided byRodriguez et al (2010), although these two studies are not directly comparable.
Previous studies have demonstrated that the prophylactic quadrivalent HPV (qHPV) vaccine is highly effective in preventing HPV 6-, 11-, 16-, or 18-related high-grade cervical, vulvar, or vaginal intraepithelial neoplasia (CIN, VIN, or VaIN, respectively), as well as adenocarcinoma in situ (AIS) in women aged 16–26 years, who are negative to the respective vaccine HPV types at the time of enrolment (Garland et al, 2007; The FUTURE II Study Group, 2007b). High efficacy against HPV 6- and 11-related genital warts was also seen (Garland et al, 2007). Data from these trials indicated that women who are negative to all four vaccine HPV types (negative by both serological and DNA testing) before vaccination derive full benefit (i.e., protection from disease caused by all four vaccine HPV types). Women who have evidence of infection with ⩾1 vaccine HPV types before vaccination derive only partial benefit (i.e., protection from the types which subjects were not infected with at the time of vaccination) (The FUTURE II Study Group, 2007a).
In addition, recently published interim data support the efficacy of the qHPV vaccine in susceptible women aged 24–45 years. Among per-protocol populations of women aged 24–45 years (85.2% of all vaccinated subjects were included in the per-protocol population), vaccine efficacy against the combined incidence of persistent infection, cervical, and external genital disease related to HPV 6, 11, 16, and 18 was 90.5% (95% CI: 73.7–97.5) (Munoz et al, 2009). Vaccine efficacy against the co-primary end point (combined incidence of persistent infection of ⩾6 months duration, cervical, and external genital disease related to HPV 16 and 18) was 83.1% (95% CI: 50.6, 95.8) (Munoz et al, 2009). These interim data were based on a mean follow-up time of 2.2 years. As this trial is now complete, a reanalysis of all data gathered during the trial is presented, inclusive of the data originally published by Munoz et al.
In this report, we further evaluate the efficacy, safety, and immunogenicity of the prophylactic qHPV vaccine in 24–45-year-old women, using end-of-study data obtained from an international randomised, placebo-controlled phase III clinical trial with a median follow-up time of 4.0 years (mean of 3.8 years). These final data help define the benefit women 24–45 years of age will get from prophylactic HPV vaccination.
Materials and methods
Study design
Between 18 June 2004 and 30 April 2005, 3819 women between the ages of 24 and 45 years were enrolled at 38 international study sites into an ongoing randomised, placebo-controlled, double-blind safety, immunogenicity, and efficacy study (Protocol 019; NCT00090220). Subjects were enrolled from community health centres, academic health centres, and primary health-care providers in Colombia, France, Germany, Philippines, Spain, Thailand, and the United States. The current report represents the end-of-study analysis, after a median follow-up time of 4.0 years.
Women were eligible to participate in the study if they were not pregnant and if they had not undergone hysterectomy. Subjects were asked to use effective contraception through month 7 of the study. Women with a history of genital warts or current/past cervical disease (CIN or cancer) were not eligible for enrolment. Those with previous cervical definitive therapy and those having undergone a cervical biopsy within the past 5 years were also excluded. In addition, those subjects infected with HIV (human immunodeficiency virus) and those who were otherwise immunocompromised were not eligible for enrolment. Lifetime number of sexual partners was not an inclusion or exclusion criterion.
The Institutional Review Board at each participating centre approved the protocol, and informed consent was obtained from all subjects. Studies were conducted in conformance with applicable country or local requirements regarding ethics committee review, informed consent, and other statutes or regulations regarding the protection of the rights and welfare of human subjects participating in the biomedical research.
Vaccine
Subjects were stratified into two age strata (⩽34 and ⩾35 years) and randomised in an ∼1 : 1 ratio to receive either qHPV (types 6, 11, 16, 18) L1 VLP vaccine (GARDASIL/SILGARD, Merck & Co. Inc., Whitehouse Station, NJ, USA) or visually indistinguishable adjuvant-containing placebo at day 1, and at months 2 and 6. Details of the qHPV vaccine have been published previously (Olsson et al, 2007).
A computer-generated allocation schedule was generated by the sponsor's Clinical Biostatistics department. Following informed consent and determination that all entry criteria were met, eligible subjects were randomised to a vaccination group using an Interactive Voice Response System. Recruitment was designed to approximately achieve equal proportions between the two age strata study-wide. All study-site investigators and personnel, study participants, monitors, and central laboratory personnel, remained blinded to treatment allocation throughout the study.
Efficacy end points and case definition
The co-primary efficacy end points were (1) the combined incidence of HPV 6-, 11-, 16-, or 18-related persistent infection (⩾6 months duration), cervical, and external genital disease (includes CIN, VIN, VaIN, AIS, cervical, vulvar, or vaginal cancer, and genital warts) and (2) the combined incidence of HPV 16- or 18-related persistent infection, cervical, and external genital disease. The secondary efficacy end point was the combined incidence of HPV 6- or 11-related persistent infection, cervical, and external genital disease. The first co-primary efficacy hypothesis was to be tested with ⩾25 cases of the first co-primary efficacy end point and ⩾14 cases of the second co-primary efficacy end point were observed (i.e., fixed event design). The second primary hypothesis was to be tested only if the first co-primary hypothesis test was successful. The secondary hypothesis was to be tested when ⩾19 cases of the secondary efficacy end point were observed, and only if both co-primary hypothesis tests were successful. Under an assumed incidence of 0.6, 0.2, and 0.6 per 100 person-years (Muñoz et al, 2006; Insinga et al, 2007), corresponding to HPV 16-, HPV 18-, and HPV 6/11-related persistent infection and disease, respectively, the study with 3819 subjects has 87 and 80% power to achieve success on the co-primary and secondary hypotheses, respectively, if the vaccine is at least 80% efficacious.
Persistent infection was defined as detection of the same HPV type in cervicovaginal/anogenital swabs at ⩾2 consecutive visits spaced ⩾6 months apart (±1 month visit window), or the presence of cervical/genital disease associated with the relevant type with type-specific HPV DNA detected in cervicovaginal or anogenital swabs at the visit directly before or after biopsy. Disease was defined as a tissue sample diagnosed by a four-member pathology panel as CIN, AIS, VIN, VaIN, genital warts, or cervical, vulvar, or vaginal cancer with type-specific HPV DNA detected in tissue from the same lesion, as described previously (Garland et al, 2007).
Safety and immunogenicity objectives
The primary safety objective was to demonstrate that a three-dose regimen of qHPV vaccine is generally well tolerated in women aged 24–45 years. The primary immunogenicity objectives included (1) evaluation of the kinetics and age dependence of anti-HPV 6, 11, 16, and 18 responses after administration of a three-dose regimen of qHPV vaccine and (2) comparison of anti-HPV 6, 11, 16, and 18 responses after administration of a three-dose regimen of qHPV vaccine among HPV-negative women aged 24–45 years.
Clinical follow-up
For ascertainment of disease, a complete gynaecological examination was performed at day 1 and at months 7, 12, 24, 36, and 48, which included a pelvic exam (both speculum and bimanual exams). External genital inspections were performed using a magnifying glass at day 1 and at months 7, 12, 18, 24, 30, 36, 42, and 48. Labial/vulvar/perineal, and perianal swabs, as well as endo/ecto cervical swabs for HPV Multiplex PCR testing were obtained at day 1 and at months 7, 12, 18, 24, 30, 36, 42, and 48. ThinPrep (Cytyc, Boxborough, MA, USA) Pap tests were also collected during these visits. Once received, cytology specimens were evaluated using the Bethesda System-2001 (Solomon et al, 2002). Referral for colposcopy was algorithm based. Histological specimens were first interpreted for clinical management by pathologists at a central laboratory (Diagnostic Cytology Laboratories, Indianapolis, IN, USA), and then evaluated for end point determination by a blinded panel of four pathologists. If indicated, definitive therapy was performed.
Serum for immunogenicity testing was collected before vaccination on day 1, and at months 7, 12, 24, 36, and 48. Antibody responses and the proportion of subjects seroconverting for vaccine-type epitope-specific neutralising anti-HPV antibodies were analysed using a competitive Luminex-based immunoassay (cLIA) developed by Merck Research Laboratories as described previously (Opalka et al, 2003).
Adverse experiences were solicited from subjects by general questioning at study visits and using a vaccine report card (VRC). The VRC was provided to the subject at each vaccination visit to record temperatures, as well as local and systemic adverse experiences.
Statistical analysis
No tests of hypothesis were conducted in the current analyses. The final tests relating to the study co-primary and secondary efficacy hypotheses were conducted previously, the results of which were published in the study by Munoz et al (2009). The results presented in this report are the updated estimates of efficacy of the qHPV vaccine against the co-primary and secondary efficacy end points and against selected exploratory efficacy end points.
Populations studied
Efficacy analysis was conducted in the per-protocol efficacy (PPE) population, that is, subjects who were seronegative at day 1 and PCR-negative (swab and biopsy specimens) from day 1 through month 7 to the relevant vaccine HPV type(s) and did not violate the protocol. The PPE-eligible participants received all 3 vaccinations within 1 year, and had 1 or more follow-up visits after month 7. Case counting commenced at month 7.
Analyses were also conducted in an intention-to-treat (ITT) population consisting of subjects who received ⩾1 dose of vaccine or placebo and returned for follow-up. These subjects could have been seropositive and/or PCR positive to vaccine HPV types at enrolment, and represent a general non-vaccinated population. In addition, this population also includes subjects who were baseline positive to non-vaccine HPV types. Case counting in the ITT population commenced after day 1.
Additional analyses were conducted in a population of subjects deemed ‘naive to the relevant HPV type’ (NRT). This population consisted of subjects who received ⩾1 dose of vaccine or placebo, returned for follow-up, and were DNA negative at enrolment for the HPV type of interest. In addition, these subjects were seronegative at enrolment for the HPV type of interest, if it was one of the four vaccine HPV types. The NRT case counting began after day 1.
Role of the funding source
The studies were designed by the sponsor (Merck & Co. Inc.) in collaboration with external investigators and an external data and safety monitoring board. The sponsor collated data, monitored the conduct of the study, performed statistical analysis, and coordinated the writing of the manuscript with all authors. The authors were actively involved in the collection, analysis, or interpretation of data, the revising of the manuscript for intellectual content, and approved the final manuscript. All authors had access to data and participated in the decision on where to submit the paper for publication.
Results
Overall, 3819 women (mean age 34.3±6.3 years) were enrolled into the study and randomised to receive either qHPV vaccine or placebo. A total of 3692 women (96.7%) received all three vaccinations and entered the follow-up period. Key baseline characteristics were generally similar between subjects in the vaccine and placebo groups (Table 1). Almost all women were non-virgins at enrolment (99.9%); and the mean age of sexual debut was 19 (±3.7) years. Detailed information on enrolment, baseline characteristics, and sexual history of the study population can be seen in the study by Munoz et al (2009).
One-third of women (33.2%) were positive to HPV 6, 11, 16, or 18 at baseline by serology or DNA testing, but most of these women (91%) were positive to none or only 1 of the 4 vaccine types. Geometric mean antibody titres in response to qHPV vaccine peaked at month 7 and declined to relative stability between months 24 and 48 (Figure 1). A vast majority of previously HPV-negative vaccinated women seroconverted for HPV 6 (98.4%), 11 (98.1%), 16 (98.8%), and 18 (97.3%) by month 7. At month 48, 91.5, 92.0, 97.4, and 47.9% of vaccinated women were still considered seropositive to HPV 6, 11, 16, and 18, respectively (as defined by the cLIA assay). Antibody titres for the placebo group were within the margin of error for the assay and were not plotted. In general, women seropositive to a particular vaccine HPV type at enrolment (who were subsequently vaccinated) had higher antibody titres for that type throughout follow-up than did women who were negative to that type at enrolment (Supplementary Figure 1). As compared with seronegative women, month 48 geometric mean titre levels were greater in women seropositive and DNA negative for HPV 6, 11, 16, and 18 at enrolment by 7.8-, 13.2-, 4.2-, and 15.1-fold, respectively.

Summary of the persistence of anti-HPV geometric mean titres (GMTs) in women receiving the qHPV vaccine (also see Supplementary Figure 1 stratified by baseline vaccine-type serostatus). The dots shown the in plot correspond to estimates of GMTs. The vertical bars superimposed to each dot represent the 95% confidence interval associated with the estimated GMT. The mini-vertical axes immediately to the left of the vertical bars show the range of the 95% confidence interval specific to each GMT estimate. The length of each vertical bar is relative only to the mini-scale immediately to the left of the vertical bar. The length of the vertical bars across time points is not comparable. Comparisons of statistically significant difference or absence of statistically significant difference between GMTs over time should not be made on the basis of comparisons of the length of the vertical bars. The antibody titres for the placebo group were within the margin of error for the assay and were not plotted.
In the PPE population, an additional 53 cases related to vaccine HPV types were observed (7 vaccine and 46 placebo) subsequent to the original analyses of data previously published (Table 2) (Munoz et al, 2009). All seven additional cases observed in the vaccine group were cases of persistent infection. The one case of CIN 2/3 in the vaccine group at the end of study was observed in the original study follow-up. The HPV DNA types detected among subjects with cases of HPV 6/11/16/18-related persistent infection, CIN, or external genital lesions EGL in the PPE population vaccine group can be seen in Supplementary Table 1. In the NRT and ITT populations (ITT), 60 and 68 additional cases of infection and/or disease related to vaccine HPV types were observed, respectively.
In the PPE population, there were 10 cases of infection and/or disease related to vaccine HPV types in the vaccine group as shown in Table 2. Three cases were observed during the 2007 end point-driven analysis and seven in the additional follow-up period. Only 1 of the 10 cases had HPV 16-persistent infection alone. The HPV 6, 11, 16, and 18 antibody titres at month 7 for this potential breakthrough case were 2513, 3775, 7850, and 1532 (uMU ml−1), respectively. Six other cases had persistent infections with both HPV 16 and other high-risk non-vaccine types, one case had HPV 16- and 18-persistent infections with other high-risk non-vaccine types, and two cases had HPV 6-persistent infection with other high-risk non-vaccine types.
Vaccine efficacy against the combined incidence of persistent infection, CIN, or EGL related to vaccine HPV types in the PPE, NRT, and ITT populations was 88.7% (95% CI: 78.1, 94.8), 79.9% (95% CI: 69.4, 87.3), and 47.2% (95% CI: 33.5, 58.2), respectively (Table 2). Efficacy against the combined incidence of persistent infection, CIN, or EGL in the PPE population related to vaccine HPV types was 91.3% (95% CI: 78.4, 97.3) in women aged 24–34 years and 83.8% (95% CI: 57.9, 95.1) in women aged 35–45 years (data not shown). Efficacy against any grade CIN related to vaccine HPV types was statistically significant for all three analysis populations; however, efficacy estimates for CIN 2/3 or worse in the PPE, NRT, and ITT analysis populations did not reach statistical significance. Vaccine efficacy against the combined incidence of persistent infection, CIN, or EGL in the PPE and ITT populations related to HPV 16/18 was 84.7% (95% CI: 67.5, 93.7) and 41.6% (95% CI: 24.3–55.2), respectively (Table 2). Vaccine efficacy against the combined incidence of persistent infection, CIN, or EGL in the PPE and ITT populations related to HPV 6/11 was 94.8% (95% CI: 79.9, 99.4) and 61.3% (95% CI: 38.8, 76.2), respectively (data not shown). Prevalent HPV 6/11/16/18 infection, which is present at baseline, was largely responsible for the lower estimate of efficacy in the ITT population against the composite end point. Supplementary Figure 2 illustrates the marked differences in incidence by baseline infection status. Overall efficacy estimates were somewhat lower in the older age group (35–45 years), but differences were not statistically significant. Efficacy estimates for HPV infection of at least 6- and 12-month duration were comparable.
In the ITT analysis of high-grade CIN regardless of HPV type, there was an imbalance of baseline infection and disease at randomisation that resulted in a higher number of cases of CIN 2/3 or worse in the vaccine group as compared with the placebo group. As shown in Table 3, this excess number of CIN 2/3 cases was mainly due to non-vaccine HPV types. Table 1 shows that HPV types 33, 39, 51, and 58 accounted for a substantial imbalance of proportion of the cases of infection at baseline. The high level of background HPV infection due to these 4 HPV types accounted for 20 cases of CIN 2/3 or worse in the vaccine group vs 5 cases in the placebo group. Of the 20 cases of CIN 2/3 or worse, 11 were diagnosed within the first 4 months of the study (Table 4).
Vaccine efficacy in reducing the incidence of any abnormal Pap smear related to any vaccine HPV types during follow-up was 97.4% (95% CI: 84.5, 99.9) in the PPE population and 50.1% (95% CI: 24.2, 67.8) in the ITT population (Table 5). Efficacy point estimates in the PPE population did not differ substantially by age group, HPV type, or severity of Pap abnormality. Efficacy against the incidence of abnormal Pap smears related to HPV 16 or 18 was 96.3% (95% CI: 77.7, 99.9) in the PPE population and 47.5% (95% CI: 16.9, 67.4) in the ITT population (data not shown).
Among vaccinated women in the PPE population, there was a 12.6, 16.3, and 30.9% reduction seen in the occurrence of colposcopy, biopsy, and any definitive therapy, respectively (Supplementary Table 2). None of these reductions were statistically significant.
Vaccine efficacy estimates against HPV6/11/16/18-related persistent infection (there were no cases of CIN) in women with serological evidence of previous HPV infection, but without evidence of current infection (seropositive/DNA negative) were calculated (Table 6). Overall efficacy in these women was 66.9% (95% CI: 4.3, 90.6). Women aged 35–45 years showed higher efficacy than did women aged 24–34 years (81.3 vs 27.4%, respectively); however this difference was not statistically significant.
Vaccine safety and tolerability data are summarised in Table 7. There was a slightly higher occurrence of overall adverse experiences and vaccine-related adverse experiences in the vaccinated group. This is largely due to the higher incidence of overall and vaccine-related injection-site adverse experiences, which were reported. No serious vaccine-related adverse experiences were reported in either the vaccine or the placebo cohorts. Reasons for discontinuation due to a vaccine-related adverse experience in the vaccine group included hypersensitivity, urticaria, mouth ulceration, injection-site swelling, and facial oedema. Reasons for discontinuation due to a vaccine-related adverse experience in the placebo group included fatigue and overdose. Reasons for death in the vaccine group (n=7) included two cardiac arrests (one secondary to breast cancer metastasis and the other secondary to cerebrovascular accident), acute liver disease secondary to nasopharyngeal cancer, breast cancer, tuberculosis, pulmonary embolism, and pericarditis. Death in the placebo group resulted from pulmonary embolism.
Discussion
Data from this clinical trial firmly confirm that the qHPV vaccine is highly efficacious at preventing lower genital tract disease in women up to age 45 years as based on the final 4-year data. Efficacy estimates against the combined incidence of persistent infection, CIN, or EGL related to vaccine HPV types in the PPE and NRT populations were 88.7% (95% CI: 78.1, 94.8) and 79.9% (95% CI: 69.4, 87.3), respectively. Although the observed efficacy of most end points was largely comparable with previous evaluations in this population (Munoz et al, 2009), there were exceptions, particularly for the HPV 16- and 18-related end points in the ITT population (Table 2). These newly significant efficacy results are likely the result of additional follow-up and end point accrual. The extra end points accrued during the 1.6 additional years of follow-up increased precision of the efficacy estimates when compared with those previously reported (Munoz et al, 2009). These data reaffirm previous conclusions that prophylactic administration of qHPV vaccine to 24- to 45-year-old women is highly efficacious in preventing HPV 6-, 11-, 16-, and 18-related infection and disease (genital warts, AIS, any grade CIN, VIN2/3, VaIN 2/3).
Consistent with trial data from younger women, this end-of-study analysis among women aged 24–45 years confirms that the qHPV vaccine is also highly efficacious in women with evidence of previous exposure to HPV 6/11/16/18 infection but with no evidence of current infection (seropositive and DNA negative). Efficacy against the combined end points was 66.9% overall and reached 81.3% in the older age group (35–45 years). These data suggest that women who had had previous infections or previous exposure to HPV may benefit from vaccination with the qHPV vaccine.
Vaccination with qHPV vaccine produced robust antibody responses in women 24–45 years of age. Seropositivity to each of the 4 vaccine HPV types exceeded 97% at 4 weeks after dose 3 (Munoz et al, 2009). At month 48, 91.5, 92.0, 97.4, and 47.9% of women were still seropositive for HPV 6, HPV 11, HPV 16, and HPV 18, respectively. Although these data indicate that HPV 18 seropositivity declined more quickly than did other vaccine HPV types, no cases of infection or disease related to HPV 18 were seen among vaccinees in the PPE population, similar to previous results seen in younger women (Joura et al, 2008).
Administration of the qHPV vaccine was generally well tolerated. The proportion of subjects who reported serious adverse experiences was comparable between the qHPV vaccine group and the placebo group. Few subjects discontinued study participation because of an adverse experience. Although there was a slightly higher incidence of adverse experiences in the vaccine group, these results are largely due to increases in injection-site reactions, as seen in other studies of the qHPV vaccine in women (Garland et al, 2007; The FUTURE II Study Group, 2007b; Munoz et al, 2009). Although there were no serious vaccine-related adverse experiences, there was an imbalance in the number of women discontinuing because of adverse experiences, likely due to increased injection-site adverse experiences. Moreover, considering the entire study follow-up, there were more women who died in the vaccine group (n=7) than in the placebo group (n=1). However, the reasons for death in these subjects were varied and without a noticeable trend (Table 7). No study deaths were deemed by investigators as related to vaccination.
The current study has several limitations. First, certain restrictions to study entry may indicate that women in our study were at somewhat lower risk of acquiring HPV than were women in the general population (disease history, etc.). However, the baseline prevalence and incidence of HPV 16/18 infection in our placebo group were similar of those reported in the literature among women of similar age (data not shown) (Clifford et al, 2005). Second, because only 50–70% of HPV infections result in detectable anti-HPV responses, the baseline serology test may have underestimated previous exposure to HPV 6/11/16/18 in the study population. Third, the lack of baseline serology measurements with respect to non-vaccine HPV types affected our ability to define the HPV-negative population with respect to non-vaccine HPV types. This may have affected our CIN 2/3 efficacy results in the ‘HPV-naive population’ with respect to non-vaccine HPV types. Finally, this study was not powered to demonstrate statistically significant efficacy for high-grade disease end points, and randomisation was not designed to achieve treatment group balance with respect to baseline prevalent infections on a by-HPV type basis. Thus, the study was not able to demonstrate statistically significant efficacy against CIN 2/3 in women aged 24–45 years. The treatment group imbalance with respect to baseline prevalent infections further negatively impacted the study's ability to demonstrate positive vaccine efficacy on high-grade CIN regardless of HPV type, as well as on cervical and external genital procedures.
The implications of these findings are relevant to the construction of accurate recommendations on cervical cancer-preventative strategies in adult women. On the basis of the reported efficacy estimates of the qHPV vaccine and other trial data, mathematical modellers are now provided with new data to perform more precise cost-benefit and cost-effectiveness analyses assessing various single and combined strategies for cervical cancer prevention in adult women. It is clear that HPV vaccination is likely to be beneficial to sexually active adult women as they are at a continuous risk of acquiring new HPV infections and of developing abnormal Pap smears, CIN, and cervical cancer (Castellsague et al, 2009). However, public health recommendations for mass vaccination must take into consideration the cost-effectiveness of vaccination programmes. Current vaccine and implementation cost modelling studies have shown that vaccination becomes less cost-effective with the increasing age of the target vaccination group. This is the main reason why the key priority of all public health recommendations concerning HPV vaccination is to target girls and young women for routine vaccination, with or without catch-up programmes up to ages 25 or 26 years. As the overall cost benefit becomes progressively less favourable with increasing age, most health authorities have not widely recommended routine vaccination of older women. Nevertheless, as documented in this trial, sexually active women over the age of 26 years also have the potential to benefit from vaccination and should be allowed the opportunity to choose to be vaccinated on an individual basis.
In summary, the data presented in this report strengthen previous results on the immunogenicity, safety, and high efficacy of the qHPV vaccine in adult women up to 45 years of age, regardless of previous exposure to HPV vaccine types.
References
Castellsague X, Schneider A, Kaufmann AM, Bosch FX (2009) HPV vaccination against cervical cancer in women above 25 years of age: key considerations and current perspectives. Gynecol Oncol 115 (Suppl 3): S15–S23
Clifford GM, Gallus S, Herrero R, Munoz N, Snijders PJ, Vaccarella S, Anh PT, Ferreccio C, Hieu NT, Matos E, Molano M, Rajkumar R, Ronco G, de Sanjose S, Shin HR, Sukvirach S, Thomas JO, Tunsakul S, Meijer CJ, Franceschi S (2005) Worldwide distribution of human papillomavirus types in cytologically normal women in the International Agency for Research on Cancer HPV prevalence surveys: a pooled analysis. Lancet 366 (9490): 991–998
de Sanjose S, Diaz M, Castellsague X, Clifford G, Bruni L, Munoz N, Bosch FX (2007) Worldwide prevalence and genotype distribution of cervical human papillomavirus DNA in women with normal cytology: a meta-analysis. Lancet Infect Dis 7 (7): 453–459
Dunne EF, Unger ER, Sternberg M, McQuillan G, Swan DC, Patel SS, Markowitz LE (2007) Prevalence of HPV infection among females in the United States. JAMA 297: 813–819
Garland SM, Hernandez-Avila M, Wheeler CM, Perez G, Harper DM, Leodolter S, Tang GWK, Ferris DG, Steben M, Bryan JT, Taddeo FJ, Railkar R, Esser MT, Sings HL, Nelson M, Boslego J, Sattler C, Barr E, Koutsky LA (2007) Quadrivalent vaccine against human papillomavirus to prevent anogenital diseases. N Engl J Med 356 (19): 1928–1943
Insinga RP, Dasbach EJ, Elbasha EH, Liaw KL, Barr E (2007) Progression and regression of incident cervical HPV 6, 11, 16 and 18 infections in young women. Infect Agent Cancer 2: 15
International Agency for Research on Cancer Working Group. Human Papillomaviruses. [90]. (2007). IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. IARC: Lyon, France, IARC.
Jacobs MV, Walboomers JMM, Snijders PJF, Voorhorst FJ, Verheijen RHM, Fransen-Daalmeijer N, Meijer CJLM (2000) Distribution of 37 mucosotropic HPV types in women with cytologically normal cervial smears: the age-related patterns for high-risk and low-risk types. Int J Cancer 87: 221–227
Joura EA, Kjaer SK, Wheeler CM, Sigurdsson K, Iversen OE, Hernandez-Avila M, Perez G, Brown DR, Koutsky LA, Tay EH, Garcia P, Ault KA, Garland SM, Leodolter S, Olsson SE, Tang GW, Ferris DG, Paavonen J, Lehtinen M, Steben M, Bosch X, Dillner J, Kurman RJ, Majewski S, Munoz N, Myers ER, Villa LL, Taddeo FJ, Roberts C, Tadesse A, Bryan J, Lupinacci LC, Giacoletti KE, Lu S, Vuocolo S, Hesley TM, Haupt RM, Barr E (2008) HPV antibody levels and clinical efficacy following administration of a prophylactic quadrivalent HPV vaccine. Vaccine 26 (52): 6844–6851
Muñoz N, Bosch FX, de Sanjosé S, Herrero R, Castellsaguë X, Shah KV, Snijders PJF, Meijer CJLM, for the International Agency for Research on Cancer Multicenter Cervical Cancer Study Group (2003) Epidemiologic classification of human papillomavirus types associated with cervical cancer. N Engl J Med 348 (6): 518–527
Muñoz N, Castellsague X, de Gonzalez AB, Gissmann L (2006) HPV in the etiology of human cancer. Vaccine 24 (Suppl 3): S1–S10
Munoz N, Manalastas R, Pitisuttihum P, Tresukosol D, Monsonego J, Ault K, Clavel C, Luna J, Myers E, Hood S, Bautista O, Bryan J, Taddeo F, Esser M, Vuocolo S, Haupt R, Barr E, Saah A (2009) Safety, immunogenicity, and efficacy of quadrivalent HPV (types 6, 11, 16, 18) recombinant vaccine in adult women between 24 and 45 years of age: a randomized, double-blind trial. Lancet 373: 1921–1922
Munoz N, Mendez F, Posso H, Molano M, van den Brule AJ, Ronderos M, Meijer C, Munoz A, Instituto Nacional de Cancerologia HPV study group (2004) Incidence, duration, and determinants of cervical human papillomavirus infection in a cohort of Colombian women with normal cytological results. J Infect Dis 15 (190): 2077–2087
Olsson S-E, Villa LL, Costa R, Petta C, Andrade R, Malm C, Iversen O-E, Hoye J, Steinwall M, Riis-Johannessen G, Andersson-Ellstrom A, Elfgren K, von Krogh G, Lehtinen M, Paavonen J, Tamms G, Giacoletti K, Lupinacci L, Esser M, Vuocolo S, Saah AJ, Barr E (2007) Induction of immune memory following administration of a prophylactic quadrivalent human papillomavirus (HPV) types 6/11/16/18 L1 virus-like-particle vaccine. Vaccine 25: 4931–4939
Opalka D, Lachman CE, MacMullen SA, Jansen KU, Smith JF, Chirmule N, Esser MT (2003) Simultaneous quantitation of antibodies to neutralizing epitopes on virus-like particles for human papillomavirus types 6, 11, 16 and 18 by a multiplexed luminex assay. Clin Diagn Lab Immunol 10 (1): 108–115
Rodriguez AC, Schiffman M, Herrero R, Hildesheim A, Bratti C, Sherman ME, Solomon D, Guillen D, Alfaro M, Morales J, Hutchinson M, Katki H, Cheung L, Wacholder S, Burk RD (2010) Longitudinal study of human papillomavirus persistence and cervical intraepithelial neoplasia grade 2/3: critical role of duration of infection. J Natl Cancer Inst 102 (5): 315–324
Schiffman M, Kjaer SK (2003) Chapter 2: Natural history of anogenital human papillomavirus infection and neoplasia. J Natl Cancer Inst Monogr 31: 14–19
Solomon D, Davey D, Kurman R, Moriarty A, O’Connor D, Prey M, Raab S, Sherman M, Wilbur D, Wright Jr T, Young N (2002) The 2001 Bethesda system: terminology for reporting results of cervical cytology. JAMA 287 (16): 2114–2119
The FUTURE II Study Group (2007a) Prophylactic efficacy of a quadrivalent human papillomavirus (HPV) vaccine in women with virologic evidence of HPV infection. J Infect Dis 196 (10): 1438–1446
The FUTURE II Study Group (2007b) Quadrivalent vaccine against human papillomavirus to prevent high-grade cervical lesions. N Engl J Med 356 (19): 1915–1927
Walboomers JMM, Jacobs MV, Manos MM, Bosch FX, Kummer A, Shah KV, Snijders PJF (1999) Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol 189 (1): 12–19
Acknowledgements
We thank all study participants. Merck Research Laboratories, a Division of Merck & Company, Inc., funded this study in its entirety. Study Investigators: Colombia – Nathalia Amaya, Alfonso Correa, Ivette Maldonato, Natascha Ortiz, Manuel Antonio PlataFrance – Joseph Monsonego, Jean-Paul Bory, Elisabeth Rohmer-Meyer, Gilles Mirguet, Didier Riethmuller; Germany – Elisabeth Barthell, Anna-Margarete Funke, Fritz Jaenicke, Werner Lichtenegger, Silke Schmidt-Petruschkat; Philippines – Ricardo M Manalastas, Jr Efren Domingo; Spain – Xavier Castellsague; Thailand – Punnee Pitisuttithum, Damrong Tresukosol, Saowanee Karnplumjid, Jittima Manonai, Suwanit Therasakvichya; United States – David Baker, Karl Beutner, Stan Block, Ramon Cestero, Daron Ferris, Larry Glazerman, Sharmila Makhija, William Nebel, Larry Seidman, Malcolm Sperling, Suzanne Trupin, Molly Yardley, Peter Zedler, Wallace Wilkerson, Darron Brown.
Authors Contributions
RMH, SM, and AS managed the sponsor's operations. JM, XC, DF, KA, JL, and PP set up study sites and enrolled participants into the study. KA, EM, JL, and NM helped draft the protocol. JB developed the PCR-based HPV 6/11/16/18 detection assays and tested the genital swab and biopsy samples using the assays. OMB developed and instituted the data analysis plan. XC and SV drafted the manuscript, to which all others contributed and approved before submission.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
XC has received travel and speaker honoraria and investigator grants from Merck & Co. Inc., GlaxoSmithKline, and Sanofi-Pasteur MSD. NM has received honoraria from Merck & Co. Inc. and Sanofi-Pasteur MSD and is a member of the Merck global advisory board for HPV vaccine, as well as a member of Sanofi-Pasteur MSD HPV steering committee. JL has received travel and speaker and investigator grants from Sanofi-Pasteur MSD. JM has conducted HPV vaccine studies for Merck & Co. Inc. and GlaxoSmithKline, and is on the medical advisory board for GlaxoSmithKline, Geneprobe, Sanofi-Pasteur MSD, Roche, and Abbott diagnostics. KA has conducted HPV vaccine studies for Merck & Co. Inc. and GlaxoSmithKline, and as acted as a consultant to Merck & Co. Inc. EM has served as a consultant to Merck & Co. Inc. NM, JL, KA, JM, and EM are members of the Merck & Co. Inc. HPV steering committee. OMB, SM, JB, SV, AS, and RMH are employees of Merck & Co. Inc. and potentially own stock and/or stock options in the company. DF has conducted HPV vaccine studies for Merck & Co. Inc. and GlaxoSmithKline, and is a speaker and on the medical advisory board for Merck & Co. Inc. Funding: Merck & Co. Inc.
Additional information
Supplementary Information accompanies the paper on British Journal of Cancer website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Castellsagué, X., Muñoz, N., Pitisuttithum, P. et al. End-of-study safety, immunogenicity, and efficacy of quadrivalent HPV (types 6, 11, 16, 18) recombinant vaccine in adult women 24–45 years of age. Br J Cancer 105, 28–37 (2011). https://doi.org/10.1038/bjc.2011.185
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2011.185
Keywords
This article is cited by
-
Effect of Health Education on Awareness of HPV Vaccination and its Acceptance Among Postpartum Women
Indian Journal of Gynecologic Oncology (2022)
-
Sexual Behavior Among US Adults: New Sex Partners and Number of Lifetime Sex Partners, NHANES 2013–2016 and NSFG 2011–2015
Sexuality Research and Social Policy (2022)
-
Regression rate of high-grade cervical intraepithelial lesions in women younger than 25 years
Archives of Gynecology and Obstetrics (2022)
-
Barriers to Human Papillomavirus Vaccine Uptake Among Racial/Ethnic Minorities: a Systematic Review
Journal of Racial and Ethnic Health Disparities (2021)
-
High Prevalence of Cervical High-Risk Human Papillomavirus Harboring Atypical Genotypes in Human Immunodeficiency Virus -Infected and -Uninfected First-Generation Adult Immigrant Women Originating from Sub-Saharan Africa and Living in France
Journal of Immigrant and Minority Health (2021)
