
Abstract
Accumulated evidence shows that EZH2 is deregulated in a wide range of cancer types, and it has a crucial role in stem cell maintenance and tumour development. Therefore, blocking EZH2 expression or activity may represent a promising strategy for anticancer treatment. In this review, we address the current understanding of the mechanisms underlying EZH2 regulation alongside the function of EZH2 gene targets that are involved in cancer progression. Finally, we will describe cancer therapies that target EZH2 or its downstream cascades, which could potentially reverse the oncogenic and stemness properties of the tumour cells to suppress cancer progression and recurrence.
Similar content being viewed by others


Main
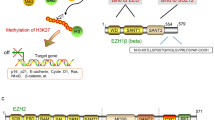
EZH2 is the catalytic core protein in the Polycomb Repressor Complex 2 (PRC2), which catalyses the trimethylation of histone3 lysine27 (H3K27) and mediates gene silencing of the target genes that are involved in fundamental cellular processes, such as cell fate decision, cell cycle regulation, senescence, cell differentiation and cancer (Sauvageau and Sauvageau, 2010). Recent findings implicate that EZH2 deregulation could be an important driver of tumour development and progression, and that inactivation of EZH2 may be therapeutically effective in many cancers.
Tumourigenic role of EZH2
EZH2 is highly expressed in a wide range of cancer types, including breast, prostate, bladder, colon, lung, pancreatic cancer, sarcoma and lymphomas Overexpression of EZH2 is often correlated with advanced stages of human cancer progression and poor prognosis (Sauvageau and Sauvageau, 2010). Enforced expression of EZH2 in cell lines shows increased proliferation and oncogenic capacity. Overexpressing EZH2 in mammary epithelial cells of the tumourigenic mouse model using mammary tumour virus long-terminal repeat (MMTV-EZH2) leads to epithelial hyperplasia phenotype (Li et al, 2009). Furthermore, somatic EZH2 mutations and deletions are found in 22% of germinal-centre diffuse large B-cell lymphomas, 7% of follicular lymphomas, and 12–23% of patients with myelodysplasic and myeloproliferative disorders. Most of the mutations are known to inactivate the enzymatic activity of the PRC2 complex (measured by the methylation status of an unmodified histone peptide substrate), which firstly seems to contradict the oncogenic properties of EZH2 (Piunti and Pasini, 2011). For example, in lymphoma, a heterozygous missense mutation frequently occurs at amino acid Y641, within the SET domain of EZH2. It was observed that wild-type EZH2 displays highest catalytic activity for the first mono-methylation of H3K27, but relatively weak capability for the subsequent di- and tri-methylation. On the contrary, the Y641 mutant displays limited ability in catalysing mono-methylation, but acquires higher catalytic efficiency for the subsequent reactions. Unsurprisingly, it was shown that the mutant Y641 allele is always found associated with a wild-type allele (heterozygous) in B-cell lymphoma cells. Heterozygous Y641 mutant, thus, can work in conjunction with wild-type EZH2 to augment H3K27 methylation, which may be functionally equivalent to EZH2 overexpression (Chase and Cross, 2011). Similarly, mutations reported in some epigenetic regulators that are functionally related to EZH2 are also shown to be involved in oncogenesis. For example, inactivating mutations of an H3K27 demethylase, ubiquitously transcribed tetratricopeptide repeat gene on X chromosome (UTX), recurrently occur in a wide range of malignancies, which may be functionally equivalent to EZH2 overexpression (Chase and Cross, 2011).
In contrast to the mutations seen in lymphoma, EZH2 mutations in myeloid neoplasms in SET domain are mostly nonsense and stop codon mutations, and thus are deficient in their histone methyltransferase activity (Chase and Cross, 2011). As either activating or inactivating mutations of EZH2 can be associated with certain types of cancer, the complex role of EZH2 mutants in cancer needs to be further investigated in terms of whether these EZH2 mutants exert differential regulation of a specific cohort of target genes that contribute to oncogenesis.
EZH2 function and recruitment on chromatin
When the PRC2 complex is recruited to chromatin, the histone methyltransferase EZH2 catalyses the trimethylation of the lysine 27 of histone H3 (H3K27me3), which leads to subsequent recruitment of the PRC1 complex that monoubiquitylates the lysine 119 of histone H2A (H2AK119ub1) to prevent RNA polymerase II-dependent transcriptional elongation and consolidate transcriptional repression (Sauvageau and Sauvageau, 2010). Several reports suggest that EZH2 directly interacts with DNA methyltransferases (DNMT1, DNMT3A, DNMT3B) and that EZH2 is necessary for the maintenance of DNA methylation and stable repression of specific genes, including many tumour suppressors (Sauvageau and Sauvageau, 2010).
In Drosophila, Polycomb group proteins (PcGs) are recruited to specific DNA sequences, Polycomb response elements (PREs). The vertebrate murine PRE was identified as a palindromic double PHO (mammalian ortholog as YY1)-binding site. In human embryonic stem cells, a potential 1.8-kb PRE that contains YY1 binding sites was recently found between the HOXD11 and HOXD12 loci, where YY1, PRC1 and PRC2 components are recruited to this PRE (Morey and Helin, 2010).
Some studies show that DNA binding factors are also involved in recruitment of PcGs to specific target genes. For example, JARID2 is able to bind to PcG target genes and the interaction is required to recruit PRC2 to these target genes in ES cells. Furthermore, in acute promyelocytic leukaemia, a fusion oncoprotein, promyelocytic leukaemia-retinoic acid receptor alpha (PML-RARα), recruits PRC2, nucleosome-remodelling complex and DNMTs to the target promoters, whereas knocking down PRC2 component results in promoter reactivation and granulocytic differentiation (Villa et al, 2007). Similarly, two other leukaemic fusion proteins, PLZF-RARα and TMPRSS2-ERG (Boukarabila et al, 2009; Yu et al, 2010), can also recruit PRC2 and PRC1 to specific target genes, suggesting that oncogenes may target PcGs as a key step for carcinogenesis.
It was indicated that 20% of the large intervening noncoding RNAs (lincRNA) are associated with PRC2. Among these lincRNAs, HOTAIR is found to recruit the PRC2 complex to the Hox loci. HOTAIR is often overexpressed in the metastatic breast tumours (Gupta et al, 2010). Loss of HOTAIR impairs cancer cell invasiveness, whereas ectopic expression of HOTAIR relocalises PRC2 complex to bind to the target genes that are signified in embryonic fibroblasts (Gupta et al, 2010). These data suggest a critical role of lincRNAs in the regulation of PRC2/EZH2 recruitment to specific target genes that contribute to cancer progression.
EZH2 linking stem cells to cancer
Numerous studies indicate the role of EZH2 in the maintenance of self-renewal of adult and ES cells. A genome-wide integrative analysis shows that a significant subset of the PRC2 target genes in aggressive prostate cancer are also targets of PRC2 in embryonic stem (ES) cells, and their repression in tumours is associated with poor prognosis (Yu et al, 2007). A shared gene expression signature enriched in PRC2 target genes and Oct4/Sox2/Nanog target genes further reveals a direct link between poorly differentiated human tumour cells and ES cells (Piunti and Pasini, 2011). Overexpression of EZH2 has also been linked to cancer initiation and progression. It is speculated that during oncogenesis, a plastic chromatin state could progress towards permanent silencing through acquisition of DNA methylation or Polycomb-mediated histone methylation. As EZH2 expression is expected to be low in differentiated tissue cells, overexpression of EZH2 could reinforce or promote dormant progenitors or more differentiated cells to an aggressive stem cell-like state. Furthermore, high-grade tumours are enriched with a high content of cancer stem cells, and it was proposed that a more aggressive secondary cancer stem/progenitor cell population may arise from a primary cancer stem cell population through acquisition of additional genetic mutations that deregulate cancer stem/progenitor homeostasis and drive cancer progression (Visvader and Lindeman, 2008). The involvement of EZH2 overexpression in cancer stem cells and cancer progression was consolidated by recent finding, which elucidates that EZH2 contributing not only to cancer stem cell formation but also to expansion of an aggressive cancer stem cell population that promotes cancer progression (Chang et al, 2011). In this study, Chang et al. identify a mechanism by which EZH2 expression-mediated downregulation of DNA damage repair leads to accumulation of recurrent RAF1 gene amplification in breast tumour-initiating cells (BTICs), which activates downstream signalling to promote BTIC expansion in aggravated breast cancer.
Regulation of EZH2 in cancer
Accumulated evidence indicates that EZH2 can be regulated in different types of human cancers at transcriptional, post-transcriptional and post-translational levels. It was shown that transcription factors E2Fs bind to the PRC2- Ezh2 and Eed promoters and transactivate their expression, which is required for E2F mediated-cell proliferation (Bracken et al, 2003) (Table 1). EZH2 expression can also be transcriptionally activated by a fusion oncoprotein EWS-FLI1 in Ewing's sarcoma, and induced EZH2 expression has a key role in endothelial/neuroectodermal differentiation and tumour growth (Richter et al, 2009; Table 1). On the contrary, SNF5, a chromatin-remodelling subunit, was shown to directly repress Ezh2 transcription, and deregulated EZH2 expression is required for SNF5-deficiency-induced lymphoma (Wilson et al, 2010; Table 1).
The microenvironment of solid tumours contains regions of poor oxygenation as a result of hypoxia. It is noteworthy that hypoxia/HIF1α activation is associated with high-grade basal breast cancer and poor prognosis. A recent study further reveals that EZH2 expression in the BTIC population is particularly enhanced by hypoxia through HIF1α-mediated transactivation, which in turn promotes the expansion of BTICs and cancer progression (Chang et al, 2011; Table 1).
In addition to transcriptional regulation, Ezh2 transcript is known to be regulated by several micro-RNAs. For example, miR-26a binds to and inhibits Ezh2 transcript expression in lymphoma (Sander et al, 2008;Table 1). Of note, miR-101, which is frequently lost in metastatic prostate tumours, targets the 3′UTR of Ezh2 mRNA and promotes its degradation (Varambally et al, 2008; Table 1).
EZH2 can also be modulated by a variety of post-translational modifications. EZH2 is phosphorylated by AKT on Ser21, which results in decreased PRC2 histone methyltransferase (HMT) activity and contributes to tumour development (Cha et al, 2005; Table 1). Recent discoveries from several groups indicate that EZH2 is regulated by cell-cycle-dependent signalling through phosphorylation at Thr350/487 by CDK1 or CDK2 (Chen et al, 2010; Wei et al, 2011; Table 1). It was shown that CDK1 phosphorylation of EZH2 at Thr487 leads to reduced HMT activity due to disrupted interaction of EZH2 with other PRC2 components (Wei et al, 2011; Table 1), whereas another report indicates that phosphorylation of EZH2 at Thr345/487 promotes EZH2 ubiquitination and subsequent degradation (Wu and Zhang, 2011; Table 1). However, there are other studies demonstrating that CDK1 phosphorylation of EZH2 promotes recruitment of EZH2 to specific target loci (Kaneko et al, 2010; Table 1), and that inhibiting CDK1 could enhance EZH2 target gene expression, potentially by increasing the binding of EZH2 to lincRNAs (please also refer to the previous section). These functional discrepancies of CDK1-mediated EZH2 phosphorylation may be the result of target gene- or cell type-specific manners and are expected to be resolved by future studies.
In addition, exposure to tobacco smoke condensate (TSC) induces recruitment of EZH2 to Wnt-antagonist Dkk1 promoter, leading to activated oncogenic Wnt signalling in lung cancer cells (Hussain et al, 2009; Table 1). However, the mechanism by which TSC promotes EZH2 association to Dkk1 promoter is unclear and also needs to be further investigated.
Direct EZH2 targets in cancer
Accumulated evidence shows that EZH2 contributes to various aspects of cancer by regulating a myriad of target genes. It was shown that EZH2-containing PRC2 transcriptionally represses cell cycle suppressor INK-ARF to drive cell cycle progression, prevent cell senescence and also exhaustion of stem/cancer stem cells (Bracken et al., 2007; Table 2). EZH2 can also inhibit astroglial differentiation and promote glioma tumourigenecity by repressing BMP Receptor1-Beta (BMPR1B) expression and BMPR1B-mediated differentiation signalling (Lee et al, 2008; Table 2).
In addition, EZH2 promotes epithelial–mesenchymal transition (EMT), a process that is associated with cancer progression and metastasis, by interacting with transcription factor SNAIL1 and suppressing expression of epithelial marker E-cadherin (CDH1) (Cao et al, 2008; Table 2). EZH2 was also shown to trigger metastasis directly through epigenetic silencing of a RasGAP gene, Disabled Homolog2-Interacting Protein (DAB2IP). This gene functions as a signalling scaffold that coordinately regulates Ras and NFκB. By suppressing DAB2IP, EZH2 activates Ras and NF-κB to promote prostate tumour initiation and metastasis (Min et al, 2010; Table 2). Integrative genomics analysis also reveals another critical EZH2 target gene, adrenergic receptor beta-2 (ADRB2), in metastatic prostate cancer. Inhibition of ADRB2 by EZH2 induces cell invasiveness and transformation of prostate epithelial cells (Yu et al, 2007; Table 2).
Moreover, EZH2 is implicated in promoting tumour angiogenesis. It shows that VEGF, which stimulates angiogenesis, can upregulate E2F1/3 transcription factors to transactivate EZH2 expression. Increased EZH2 expression by VEGF silences expression of a negative regulator of angiogenesis, Vasohibin1 (VASH1), and subsequently enhances angiogenesis (Lu et al, 2010; Table 2).
A recent study further identified that under hypoxia insult, induced EZH2 expression downregulates DNA damage repair protein RAD51 expression, which leads to genomic aberrations, such as RAF1 gene amplification, to promote RAF1-ERK-β-catenin signalling and expansion of BTICs (Chang et al, 2011; Table 2). This finding further reveals that AZD6244, a clinical trial drug that inhibits RAF1-ERK signalling, is effective in preventing breast cancer progression by eliminating BTICs (Chang et al, 2011; Table 2).
Together, these studies indicate that EZH2 has an essential and multi-faceted role in cancer. Blocking EZH2 expression or activity may represent a promising strategy for anticancer treatment targeting tumour cells, tumour endothelial cells and tumour stem cells.
Potential cancer therapeutics targeting Polycomb function or downstream signalling
It is known that EZH2 is crucial in stem cell maintenance and tumour development. Reducing EZH2 expression using siRNA or treatment of a small-molecule-S-adenosylhomocysteine hydrolase inhibitor 3-deazaneplanocin (DZNep) that inhibits methyltransferases and induces degradation of EZH2 was shown to result in cell growth inhibition and reduced tumour formation in various cancers (Piunti and Pasini, 2011). For example, disruption of EZH2 by DZNep or by short-hairpin RNAs (shRNAs) was shown to significantly impair self-renewal and the tumour-initiating capacity of glioblastoma cancer stem cells (Suva et al, 2009) or ovarian cancer stem cell-like populations (Rizzo et al, 2011). Downregulation of EZH2 by shRNAs in vivo significantly decreased breast xenograft tumour growth and improved survival (Gonzalez et al, 2009). Similarly, DZNep was shown to be effective in inhibiting prostate cancer cell growth and its anti-tumour activity is in part mediated by suppressing the tumourigenic potential of the prostate cancer stem cell (Crea et al, 2011). However, the therapeutic value of DZNep needs to be further assessed, as it is not a specific EZH2 inhibitor; methylation of other histone lysines and arginines can be globally inhibited by DZNep.
In addition to the direct regulation of EZH2 activity, cancer therapy that blocks EZH2-mediated oncogenic signalling is warranted. A recent study illustrates that EZH2 can induce RAF1-ERK-β-catenin signalling, which in turn enhances the survival and proliferation of BTICs (Chang et al, 2011). This finding further reveals a previously unidentified therapeutic effect of RAF1-ERK signalling inhibitors in preventing breast cancer progression by eliminating BTICs and elucidates important clinical implications.
Future Perspectives
As mentioned above, EZH2 can be regulated through various mechanisms. Also, it is plausible that modulation of these EZH2-regulatory mechanisms could significantly impact EZH2 activity and be therapeutically effective in many cancers. For example, it was shown that EZH2 expression is highly enhanced by hypoxia through HIF1α-mediated transactivation in the BTICs and contributes to cancer progression (Chang et al, 2011). Hypoxia inducible factor (HIF) inhibitors have been used in clinical trials and have shown marked effects in inhibiting tumour growth (Semenza, 2003). As suggested in Chang et al., HIF inhibitors could also potentially be effective in suppressing EZH2 oncogenic function in tumour stem cells to prevent cancer recurrence. In addition, it was found that aberrant activation of CDK1/2 decreases tumour suppressor gene DAB2IP expression and contributes to the aggressiveness of cancer cells by phosphorylating EZH2 at Thr350 (Chen et al, 2010). Thus, using CDK1/2 inhibitors to dephosphorylate EZH2 is also likely to disable EZH2-mediated oncogenesis.
Furthermore, it was found that, similar to the effects of knocking down EZH2, overexpression of miR-101 inhibits proliferation and invasiveness of cancer cells in vitro. It will be appealing to test whether expressing miR-101 using microRNA therapy can cause therapeutic effects in vivo as microRNA therapy has been exploited in preclinical and clinical trials as a potential cancer treatment regimen (ClinicalTrials.gov).
However, previous work demonstrated that conditional inactivation of Ezh2 in mouse adult stem cells (from hematopoietic, brain or pancreatic tissues) could yield minor defects in normal organ development or functions (Abdel-Wahab and Levine, 2010). Therefore, administering EZH2 inhibitors by a tumour-specific delivery system may be necessary to avoid the side effects in normal (stem) cells. Furthermore, better characterising and specifically blocking EZH2-mediated oncogenic targets/signalling pathways can be more efficient and effective.
Together, understanding the regulatory mechanisms of EZH2 and the function of EZH2 gene targets will shed light on the development of novel cancer treatments targeting EZH2/Polycomb mediated-tumourigenesis to suppress oncogenic and stemness-associated pathways, which will ultimately achieve the goal of cancer prevention and remission.
Change history
29 March 2012
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Abdel-Wahab O, Levine RL (2010) EZH2 mutations: mutating the epigenetic machinery in myeloid malignancies. Cancer Cell 18: 105–107
Boukarabila H, Saurin AJ, Batsche E, Mossadegh N, van Lohuizen M, Otte AP, Pradel J, Muchardt C, Sieweke M, Duprez E (2009) The PRC1 Polycomb group complex interacts with PLZF/RARA to mediate leukemic transformation. Genes Dev 23: 1195–1206
Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K (2003) EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J 22: 5323–5335
Cao Q, Yu J, Dhanasekaran SM, Kim JH, Mani RS, Tomlins SA, Mehra R, Laxman B, Cao X, Kleer CG, Varambally S, Chinnaiyan AM (2008) Repression of E-cadherin by the polycomb group protein EZH2 in cancer. Oncogene 27: 7274–7284
Cha TL, Zhou BP, Xia W, Wu Y, Yang CC, Chen CT, Ping B, Otte AP, Hung MC (2005) Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science 310: 306–310
Chang CJ, Yang JY, Xia W, Chen CT, Xie X, Chao CH, Woodward WA, Hsu JM, Hortobagyi GN, Hung MC (2011) EZH2 promotes expansion of breast tumor initiating cells through activation of RAF1-beta-catenin signaling. Cancer Cell 19: 86–100
Chase A, Cross NC (2011) Aberrations of EZH2 in cancer. Clin Cancer Res 17: 2613–2618
Chen S, Bohrer LR, Rai AN, Pan Y, Gan L, Zhou X, Bagchi A, Simon JA, Huang H (2010) Cyclin-dependent kinases regulate epigenetic gene silencing through phosphorylation of EZH2. Nat Cell Biol 12: 1108–1114
Crea F, Hurt EM, Mathews LA, Cabarcas SM, Sun L, Marquez VE, Danesi R, Farrar WL (2011) Pharmacologic disruption of Polycomb Repressive Complex 2 inhibits tumorigenicity and tumor progression in prostate cancer. Mol Cancer 10: 40
Gonzalez ME, Li X, Toy K, DuPrie M, Ventura AC, Banerjee M, Ljungman M, Merajver SD, Kleer CG (2009) Downregulation of EZH2 decreases growth of estrogen receptor-negative invasive breast carcinoma and requires BRCA1. Oncogene 28: 843–853
Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, Tsai MC, Hung T, Argani P, Rinn JL, Wang Y, Brzoska P, Kong B, Li R, West RB, van de Vijver MJ, Sukumar S, Chang HY (2010) Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464: 1071–1076
Hussain M, Rao M, Humphries AE, Hong JA, Liu F, Yang M, Caragacianu D, Schrump DS (2009) Tobacco smoke induces polycomb-mediated repression of Dickkopf-1 in lung cancer cells. Cancer Res 69: 3570–3578
Kaneko S, Li G, Son J, Xu CF, Margueron R, Neubert TA, Reinberg D (2010) Phosphorylation of the PRC2 component Ezh2 is cell cycle-regulated and up-regulates its binding to ncRNA. Genes Dev 24: 2615–2620
Lee J, Son MJ, Woolard K, Donin NM, Li A, Cheng CH, Kotliarova S, Kotliarov Y, Walling J, Ahn S, Kim M, Totonchy M, Cusack T, Ene C, Ma H, Su Q, Zenklusen JC, Zhang W, Maric D, Fine HA (2008) Epigenetic-mediated dysfunction of the bone morphogenetic protein pathway inhibits differentiation of glioblastoma-initiating cells. Cancer Cell 13: 69–80
Li X, Gonzalez ME, Toy K, Filzen T, Merajver SD, Kleer CG (2009) Targeted overexpression of EZH2 in the mammary gland disrupts ductal morphogenesis and causes epithelial hyperplasia. Am J Pathol 175: 1246–1254
Lu C, Han HD, Mangala LS, Ali-Fehmi R, Newton CS, Ozbun L, Armaiz-Pena GN, Hu W, Stone RL, Munkarah A, Ravoori MK, Shahzad MM, Lee JW, Mora E, Langley RR, Carroll AR, Matsuo K, Spannuth WA, Schmandt R, Jennings NB, Goodman BW, Jaffe RB, Nick AM, Kim HS, Guven EO, Chen YH, Li LY, Hsu MC, Coleman RL, Calin GA, Denkbas EB, Lim JY, Lee JS, Kundra V, Birrer MJ, Hung MC, Lopez-Berestein G, Sood AK (2010) Regulation of tumor angiogenesis by EZH2. Cancer Cell 18: 185–197
Min J, Zaslavsky A, Fedele G, McLaughlin SK, Reczek EE, De Raedt T, Guney I, Strochlic DE, Macconaill LE, Beroukhim R, Bronson RT, Ryeom S, Hahn WC, Loda M, Cichowski K (2010) An oncogene-tumor suppressor cascade drives metastatic prostate cancer by coordinately activating Ras and nuclear factor-kappaB. Nat Med 16: 286–294
Morey L, Helin K (2010) Polycomb group protein-mediated repression of transcription. Trends Biochem Sci 35: 323–332
Piunti A, Pasini D (2011) Epigenetic factors in cancer development: polycomb group proteins. Future Oncol 7: 57–75
Richter GH, Plehm S, Fasan A, Rossler S, Unland R, Bennani-Baiti IM, Hotfilder M, Lowel D, von Luettichau I, Mossbrugger I, Quintanilla-Martinez L, Kovar H, Staege MS, Muller-Tidow C, Burdach S (2009) EZH2 is a mediator of EWS/FLI1 driven tumor growth and metastasis blocking endothelial and neuro-ectodermal differentiation. Proc Natl Acad Sci USA 106: 5324–5329
Rizzo S, Hersey JM, Mellor P, Dai W, Santos-Silva A, Liber D, Luk L, Titley I, Carden CP, Box G, Hudson DL, Kaye SB, Brown R (2011) Ovarian cancer stem cell-like side populations are enriched following chemotherapy and overexpress EZH2. Mol Cancer Ther 10: 325–335
Sander S, Bullinger L, Klapproth K, Fiedler K, Kestler HA, Barth TF, Moller P, Stilgenbauer S, Pollack JR, Wirth T (2008) MYC stimulates EZH2 expression by repression of its negative regulator miR-26a. Blood 112: 4202–4212
Sauvageau M, Sauvageau G (2010) Polycomb group proteins: multi-faceted regulators of somatic stem cells and cancer. Cell Stem Cell 7: 299–313
Semenza GL (2003) Targeting HIF-1 for cancer therapy. Nat Rev Cancer 3: 721–732
Suva ML, Riggi N, Janiszewska M, Radovanovic I, Provero P, Stehle JC, Baumer K, Le Bitoux MA, Marino D, Cironi L, Marquez VE, Clement V, Stamenkovic I (2009) EZH2 is essential for glioblastoma cancer stem cell maintenance. Cancer Res 69: 9211–9218
Varambally S, Cao Q, Mani RS, Shankar S, Wang X, Ateeq B, Laxman B, Cao X, Jing X, Ramnarayanan K, Brenner JC, Yu J, Kim JH, Han B, Tan P, Kumar-Sinha C, Lonigro RJ, Palanisamy N, Maher CA, Chinnaiyan AM (2008) Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science 322: 1695–1699
Villa R, Pasini D, Gutierrez A, Morey L, Occhionorelli M, Vire E, Nomdedeu JF, Jenuwein T, Pelicci PG, Minucci S, Fuks F, Helin K, Di Croce L (2007) Role of the polycomb repressive complex 2 in acute promyelocytic leukemia. Cancer Cell 11: 513–525
Visvader JE, Lindeman GJ (2008) Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer 8: 755–768
Wei Y, Chen YH, Li LY, Lang J, Yeh SP, Shi B, Yang CC, Yang JY, Lin CY, Lai CC, Hung MC (2011) CDK1-dependent phosphorylation of EZH2 suppresses methylation of H3K27 and promotes osteogenic differentiation of human mesenchymal stem cells. Nat Cell Biol 13: 87–94
Wilson BG, Wang X, Shen X, McKenna ES, Lemieux ME, Cho YJ, Koellhoffer EC, Pomeroy SL, Orkin SH, Roberts CW (2010) Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell 18: 316–328
Wu SC, Zhang Y (2011) CDK1-mediated phosphorylation of Ezh2 regulates its stability. J Biol Chem
Yu J, Cao Q, Mehra R, Laxman B, Tomlins SA, Creighton CJ, Dhanasekaran SM, Shen R, Chen G, Morris DS, Marquez VE, Shah RB, Ghosh D, Varambally S, Chinnaiyan AM (2007) Integrative genomics analysis reveals silencing of beta-adrenergic signaling by polycomb in prostate cancer. Cancer Cell 12: 419–431
Yu J, Mani RS, Cao Q, Brenner CJ, Cao X, Wang X, Wu L, Li J, Hu M, Gong Y, Cheng H, Laxman B, Vellaichamy A, Shankar S, Li Y, Dhanasekaran SM, Morey R, Barrette T, Lonigro RJ, Tomlins SA, Varambally S, Qin ZS, Chinnaiyan AM (2010) An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell 17: 443–454
Zeidler M, Varambally S, Cao Q, Chinnaiyan AM, Ferguson DO, Merajver SD, Kleer CG (2005) The Polycomb group protein EZH2 impairs DNA repair in breast epithelial cells. Neoplasia 7: 1011–1019
Acknowledgements
The authors are grateful for the support of the National Breast Cancer Foundation, Inc.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Chang, CJ., Hung, MC. The role of EZH2 in tumour progression. Br J Cancer 106, 243–247 (2012). https://doi.org/10.1038/bjc.2011.551
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2011.551
Keywords
This article is cited by
-
Current and future therapeutic strategies for high-grade gliomas leveraging the interplay between epigenetic regulators and kinase signaling networks
Journal of Experimental & Clinical Cancer Research (2024)
-
EZH2-mediated epigenetic silencing of tumor-suppressive let-7c/miR-99a cluster by hepatitis B virus X antigen enhances hepatocellular carcinoma progression and metastasis
Cancer Cell International (2023)
-
Diverse activity of miR-150 in Tumor development: shedding light on the potential mechanisms
Cancer Cell International (2023)
-
Photodynamic therapy in cancer stem cells — state of the art
Lasers in Medical Science (2023)
-
Depletion of enhancer zeste homolog 2 (EZH2) directs transcription factors associated with T cell differentiation through epigenetic regulation of Yin Yang 1(YY1) in combating non-small cell lung cancer (NSCLC)
Medical Oncology (2023)
