
Abstract
Background:
BI 2536, a novel Polo-like kinase 1 inhibitor, was assessed in patients with unresectable advanced exocrine adenocarcinoma of the pancreas.
Methods:
The study employed a two-stage design. Randomised first-line patients received BI 2536 200 mg on day 1 (n=43) or 60 mg on days 1–3 (n=43) every 21 days. Recruitment of second-line patients was planned for a second stage dependent on an interim analysis demonstrating ⩾2 responses in the first 18 evaluable patients following 12 weeks of treatment and/or tumour control ⩾12 weeks in 5 patients per schedule. Primary end point was objective response rate (ORR).
Results:
By independent review, ORR was 2.3% (all partial) and 24.4% had stable disease as confirmed best response. The second stage was not initiated. Median overall and progression-free survivals were 149 (95% confidence interval (CI), 91–307) and 46 days (95% CI, 44–56). Most common drug-related adverse events were neutropenia (37.2%), leukopenia (29.1%), fatigue (29.1%) and nausea (22.1%); most common grade 3/4-related events were neutropenia (36.0%), leukopenia (27.9%) and thrombocytopenia (8.1%).
Conclusion:
Given the low ORR and poor survival, further development of BI 2536 monotherapy is not warranted in this population.
Similar content being viewed by others
Main
In developed countries, pancreatic cancer is the fourth leading cause of cancer-related death, accounting for approximately 266 000 deaths per year worldwide and 65 000 deaths per year in Europe (Cascinu et al, 2010; Jemal et al, 2011). In Germany, there are an estimated 14 693 new cases per year and most of these patients will die from the disease (Ferlay et al, 2011). At diagnosis, 80–85% of patients have unresectable disease (Li et al, 2004); prognosis is poor for these patients with a 5-year survival of 1.8% (Howlader et al, 2011).
Intravenous gemcitabine monotherapy is considered the standard of care for first-line treatment of advanced unresectable pancreatic cancer based on a phase III trial that demonstrated a significant improvement in survival vs 5-fluorouracil (5.65 vs 4.41 months, P=0.0025) (Burris et al, 1997). A second phase III trial examining the combination of gemcitabine with erlotinib in first-line advanced pancreatic cancer demonstrated a very small, but statistically significant, improvement in survival vs gemcitabine alone (6.24 vs 5.91 months, P=0.038) (Moore et al, 2007). Based upon these results, erlotinib in combination with gemcitabine was approved by both the United States Food and Drug Administration and the European Medicines Agency for first-line treatment of advanced pancreatic cancer (Tarceva (package insert), 2010; Tarceva (summary of product characteristics), 2011). The combination of 5-fluorouracil, leucovorin, oxaliplatin plus irinotecan (FOLFIRINOX) was recently evaluated in a European phase III trial (ACCORD/PRODIGE) of patients with metastatic pancreatic cancer. Compared with gemcitabine, FOLFIRINOX resulted in a significant improvement in survival (6.8 vs 11.1 months, P<0.0001); however, FOLFIRINOX was associated with a significantly higher incidence of grade 3/4 neutropenia, febrile neutropenia, thrombocytopenia, diarrhoea and sensory neuropathy (Conroy et al, 2011).
Given the poor prognosis and dire outcomes for patients with advanced pancreatic cancer, new therapies are an urgent medical need. A potentially promising novel approach is to target the cell cycle of rapidly dividing tumour cells via inhibition of mitotic checkpoints (Swanton, 2004). One such target is the mitotic serine/threonine kinase Polo-like kinase 1 (Plk1), which is involved in key cellular functions including passage through crucial mitosis checkpoints, organisation of the mitotic spindle, and progression through anaphase and cytokinesis (Strebhardt, 2010; Komlodi-Pasztor et al, 2011). Plk1 is overexpressed in several human cancers, including non-small cell lung cancer, prostate, ovarian, breast and colorectal cancer (Wolf et al, 1997; Takahashi et al, 2003; Schöffski, 2009). Moreover, overexpression of Plk1 and deregulation of cell-cycle control is well established in pancreatic cancer cells (Gray Jr et al, 2004).
BI 2536 is an inhibitor of the Plk family of proteins, and demonstrates potent inhibition of Plk1, leading to cell-cycle arrest and a distinctive ‘polo arrest’ phenotype (Schöffski, 2009). In mouse xenograft models, BI 2536 achieved tumour regression up to complete cure, and in a pancreatic adenocarcinoma model, BI 2536 significantly reduced growth to a greater extent than gemcitabine (Steegmaier et al, 2007). Phase I clinical studies of BI 2536 in patients with advanced cancer determined the maximum tolerated dose of BI 2536 in three different schedules: 200 mg on day 1 every 3 weeks, 100 mg on days 1 and 8 every 3 weeks and 60 mg on days 1, 2 and 3 every 3 weeks (Mross et al, 2008; Hofheinz et al, 2010; Frost et al, 2012). These studies also demonstrated that BI 2536 had preliminary evidence of efficacy and was well tolerated in this patient population. The predominant dose-limiting toxicities (DLTs) were reversible neutropenia, febrile neutropenia and thrombocytopenia (Mross et al, 2008; Hofheinz et al, 2010; Frost et al, 2012). Here we report the results of a phase II trial investigating the efficacy, safety and pharmacokinetic (PK) profile of BI 2536 as a first-line treatment for patients with advanced exocrine adenocarcinoma of the pancreas.
Materials and methods
Trial design
This was a phase II open-label, randomised, parallel-group trial of BI 2536 in patients from Germany and Austria with unresectable advanced exocrine adenocarcinoma of the pancreas, and was conducted in accordance with the Declaration of Helsinki. A two-stage design (modified Gehan), in which first-line patients were randomised to one of two BI 2536 treatment regimens in the first stage, was used (Figure 1). Progression to the second stage, which would include expansion of the first-line cohort and recruitment of second-line patients, was dependent on an interim analysis. Patients recruited to the second stage of the trial would receive treatment with the best regimen from the first stage. An interim analysis criterion determined whether the trial should continue from the first stage (first-line patients) to the second stage (second-line patients). For this criterion to be achieved in either arm, 2 out of the first 18 evaluable patients, defined as patients who had completed ⩾2 cycles, had to achieve a confirmed complete or partial response (CR or PR) following 12 weeks of treatment and/or 5 out of 18 patients had to achieve tumour control for at least 12 weeks. If this criterion was met in both arms of the trial, then the second-line regimen would be selected based on exploratory analysis of response rates, tumour-control rates, adverse event (AE) rates and severity, and practicality.

Trial design.
The primary end point of this study was objective response as determined by independent review of tumour imaging according to response evaluation criteria in solid tumours (RECIST 1.0). Secondary end points included tumour control (objective response or stable disease (SD) after the fourth treatment course), progression-free survival (PFS), overall survival (OS), CA19-9 response, safety and DLT.
Patient selection
Patients aged at least 18 years with locally advanced or metastatic, unresectable exocrine adenocarcinomas of the pancreas were enrolled. Assessments of medical history, physical examination, Karnofsky score and electrocardiogram were performed at baseline including haematology, biochemistry and coagulation parameters. In the first stage of this planned two-stage trial, first-line chemo-naïve patients were enrolled, and in the second stage, patients who had received prior gemcitabine-based chemotherapy were included. Inclusion criteria included a Karnofsky status ⩾70% for the first-line cohort and ⩾50% for the second-line cohort; the presence of at least one measureable tumour lesion and a life expectancy of at least 3 months. Exclusion criteria included prior adjuvant therapy for first-line patients, ampullary carcinoma of the pancreas, hypersensitivity to trial drug or excipients, clinically relevant toxicities persisting from prior chemotherapy, known second malignancy requiring treatment and brain metastases that were symptomatic or required therapy. Also, patients with inadequate organ function (absolute neutrophil count <1500 per mm3, platelet count ⩽100 000 per mm3 haemoglobin <9 mg dl−1, aspartate aminotransferase/alanine aminotransferase >2.5 times the upper limit of normal (or >5 times in cases of known liver metastases), bilirubin >3.0 mg dl−1 under adequate draining measures and serum creatinine >2.0 mg dl−1), concomitant illnesses (including active infection, congestive heart failure, unstable angina pectoris and cardiac arrhythmia) or who had received hormone therapy, therapy with biological response modifier, or treatment with an investigational drug in the past 4 weeks were excluded. The study was conducted in accordance with the ethical principals originating from the Declaration of Helsinki and with Good Clinical Practice as defined by the International Conference on Harmonisation. All participating patients gave written informed consent.
Treatment
Two dose regimens of BI 2536 were investigated (Figure 1): BI 2536 as a 60-min intravenous (IV) infusion given open label at a dose of 200 mg once daily on day 1 of each 21-day treatment cycle and BI 2536 as a 60-min IV infusion given open label at a dose of 60 mg once daily on days 1–3 of each 21-day treatment cycle. BI 2536 was to be discontinued in the event of DLT, defined as drug-related grade ⩾3 non-haematological toxicity (excluding non-treated nausea, vomiting or diarrhoea), drug-related grade 4 neutropenia for ⩾7 days and/or complicated by infection, or drug-related grade 4 haematological toxicity other than neutropenia. Patients could continue therapy if the DLT resolved to grades that allowed further therapy, but only at a reduced dose of BI 2536 (reduced by up to 50 mg in the 200-mg group and up to 10 mg in the 60-mg group) that was agreed upon by the clinical monitor and the investigator. Only one dose reduction per patient was allowed over the course of the trial. Per protocol, BI 2536 was to be discontinued if a second DLT occurred or if a DLT did not resolve with treatment interruption or dose reduction. Patients continuing therapy beyond the second course of treatment could undergo dose escalation (not to exceed 50 mg steps in the 200-mg cohort and 10 mg steps in the 60-mg cohort) if disease was non-progressive and treatment was well tolerated.
Investigations
Tumour response was assessed at the screening visit (or within 21 days before the start of study medication), at the end of every second treatment course, and at the conclusion of the trial. All tumour images were sent to a central imaging unit and assessed by blinded independent review according to RECIST 1.0. Confirmation of objective response required that CRs and PRs were confirmed 6 weeks after the initial criteria for response were first met. For an assessment of SD as best response, follow-up measurements must have met the SD criteria at least once after study entry at a minimum interval of 6 weeks. Best response was assessed as the best response achieved from start of treatment until disease progression/recurrence.
Safety assessments consisted of monitoring and recording of all AEs, regular monitoring of haematology and clinical chemistry measurements (laboratory evaluations), regular measurement of vital signs and performance of physical examinations. Adverse events and laboratory abnormalities were graded according to the National Cancer Institute’s Common Terminology Criteria for Adverse Events, version 3.0.
Pharmacokinetic analysis
Pharmacokinetic analysis was performed to determine plasma concentration of BI 2536. Blood samples were taken before, during and after administration (repeatedly for the first 24 h and once more 7 days post administration). The concentration of BI 2536 was determined by high-performance liquid chromatography–tandem mass spectrometry (HPLC-MS/MS).
Statistical analysis
The trial aimed to initially recruit 18 first-line patients to each treatment arm in the first stage with an interim analysis for response planned once 18 evaluable patients per schedule had received four courses of treatment (12 weeks). Patients who had not completed the second course of treatment were considered evaluable for the final analysis, but were not considered for the interim analysis. Following the interim analysis, recruitment was to be completed to 35 first-line patients to each treatment arm in the first stage for a total of 70 patients. A total of 35 second-line patients were to be recruited to the single-arm second stage.
The population for efficacy and safety analyses comprised all patients who received at least one dose of BI 2536, including patients who withdrew or discontinued for any reason. There was no per protocol population, however, protocol violations were to be described.
Results
Patient population
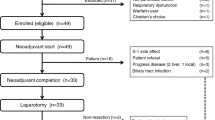
A total of 90 patients were recruited at nine study centres in Germany and one in Austria for the first stage of the trial. Of these, 89 were randomised to receive BI 2536 according to either the day 1 200-mg schedule (n=45) or the day 1–3 60-mg schedule (n=44). Details of patient recruitment and reasons for discontinuation are given in Figure 2. The first stage of the study was fully accrued prior to obtaining 18 evaluable patients for the interim analysis due to a large number of patients excluded as a result of early disease progression before completing two cycles of treatment. Thus, the pre-planned interim analysis was not conducted and the results reported herein are based on the full stage 1 study population.

CONSORT diagram. *One patient was enrolled but not randomised (screening failure).
A total of 86 patients (43 in each dosing arm) received BI 2536 and were included in the analyses. Baseline demographics were similar between the two arms (Table 1). Median age was 64.5 years and 68.6% of patients were male. Treatment arms were also well matched in terms of their pancreatic cancer history. Most (87%) patients had been diagnosed with ductal adenocarcinoma and pancreas head as the primary site (58%), one-third of patients with known differentiation grade had poorly differentiated tumours and 88% had stage 4 (metastatic) disease at screening. Per protocol, no patients had received previous chemotherapy, 1 patient had prior radiotherapy and 28 patients had undergone surgery.
Treatment exposure
The mean number of courses of BI 2536 given was similar in both treatment groups, 3.2 (range 1–14) in the day 1 200-mg group and 3.2 (1–16) in the days 1–3 60-mg group. In both groups, the majority (>65%) of patients completed <3 courses of treatment and 13 out of 86 patients received only one course, most commonly due to early disease progression. Dose reductions were required for one patient in the 200-mg group and two patients in the 60-mg group due to occurrence of DLTs. Six patients in each group had dose escalation, three of those in the days 1–3 60-mg group subsequently had dose de-escalation.
Efficacy
At the time of the interim analysis (based on investigator assessment), 5 out of 18 evaluable patients in each treatment group had PFS following 3 months of treatment. However, at the time that these patients were evaluated, a total of 79 patients had been recruited and 41 had experienced progression within two courses of treatment. Therefore, despite having met the formal criteria for the transition to stage 2, it was decided that recruitment of second-line patients into stage 2 of the trial should not proceed.
Based on independent review, no patients had a CR and two patients had confirmed PRs after treatment (response rate 2.3%). However, no corresponding PRs were observed according to the respective investigator assessments, and both patients discontinued treatment after course 4 due to clinical or non-target lesion disease progression. SD was the confirmed best response in 24.4% of patients. Tumour control at 3 months was achieved in 12.8% of patients (seven patients in the day 1 200-mg group and four patients in the days 1–3 60-mg group).
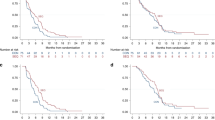
Pooled median OS was 149 days (95% confidence interval (CI), 91–307) and PFS was 46 days (95% CI, 44–56) as assessed by independent review (Figure 3). The 1-year survival and 3-month PFS rates were 17.2% and 31.1% for the day 1 200-mg arm and 13.1% and 15.4% for the days 1–3 60-mg arm, respectively. No statistically significant difference between the treatment arms was detected for any outcome.

Median OS (A) and PFS (B) for the two treatment groups.
Safety
Overall, 98.8% of patients experienced an AE of any grade, most commonly gastrointestinal (82.6%), haematological (48.8%) or general disorders (66.3%). Serious AEs irrespective of cause were experienced by 52.3% of patients. A total of 26 (30.2%) patients had an AE that qualified as a DLT (25.6% in the 200-mg group and 34.9% in the 60-mg group); 20 of these 26 patients (76.9%) had a haematological DLT, most commonly neutropenia (14 patients (16.3% of study population)). Overall incidences of other DLTs were thrombocytopenia (4.7%), anaemia (3.5%), fatigue (2.3%) and febrile neutropenia (2.3%). Diarrhoea, leukopenia, nausea, pneumonia, stomatitis and vomiting were DLTs that all occurred in single patients (frequency of 1.2%).
Overall rates of drug-related AEs of any grade were 69.8% in the day 1 200-mg group and 83.7% in the day 1–3 60-mg group (Table 2). The most common drug-related AEs were neutropenia (37.2%), leukopenia (29.1%), fatigue (29.1%) and nausea (22.1%). In general, AE rates were similar between the two dosing groups; however, nausea (27.9% vs 16.3%) and alopecia (25.6% vs 14.0%) were higher in the 60-mg group and neutropenia (41.9% vs 32.6%) and leukopenia (34.9% vs 23.3%) were higher in the 200-mg group. Fifty percent of patients had at least one grade 3/4 AE. The most common grade 3/4 drug-related AEs overall were neutropenia (36.0% of study population), leukopenia (27.9%) and thrombocytopenia (8.1%). There were 18 deaths due to AEs, none were considered to be drug-related by investigators.
Pharmacokinetic analysis
BI 2536 demonstrated a multi-compartmental PK behaviour. In general, the plasma concentrations increased up to 0.5 or 1 h after start of infusion. Following infusion, plasma concentrations decreased rapidly, indicating a fast distribution phase. By 24 h post infusion, the average plasma concentration was 18.6 ng ml−1 and further decreased to 1.70 ng ml−1 by 120 h after the 200-mg dose.
Discussion
In this randomised, open-label, exploratory study of the Plk inhibitor BI 2536, there was no indication of clinically relevant efficacy with either dosing schedule in patients with unresectable advanced pancreatic cancer. Survival was relatively short with an OS of 149 days (equivalent to 4.9 months), at the lower range of what would be expected with gemcitabine (4.9–8.2 months, based on historical data) (Eckel et al, 2006; Heinemann et al, 2008). The 1-year survival rate was 14.7%, only slightly higher than the lower limit of 11–28% reported for gemcitabine (Eckel et al, 2006; Heinemann et al, 2008). Furthermore, these efficacy data do not compare well with those recently reported from the phase III ACCORD/PRODIGE trial, in which an OS of 6.8 months with gemcitabine was improved to 11.1 months with FOLFIRINOX (Conroy et al, 2011). A modified Gehan sequential design was employed to ensure a certain level of antitumour efficacy in this trial before allowing for complete accrual to avoid exposure of patients to a potentially inactive drug. Using this design, two PRs or CRs and/or disease control for ⩾12 weeks in 5 out of 18 evaluable patients (i.e., treated for ⩾2 courses) in either arm were required before entering the second stage. In stage 2 of the trial, the number of patients per arm was planned to be 35 patients with the second-line cohort initiated after one of the two schedules had been selected. However, because of the high number of patients that were not evaluable for disease control and the late read out of disease control after 12 weeks, the two-stage design did not restrict patient recruitment before the full number of patients had already been entered into the trial. In retrospect, more stringent criteria for moving to the second stage should have been used.
As has been reported for phase I trials, BI 2536 treatment was well tolerated, with no unexpected AEs. Haematological toxicities were the most common side effects and were manageable with appropriate measures. The PK profiles of BI 2536 were comparable to those seen in phase I trials, with the drug demonstrating multi-compartmental PK behaviour with no evidence of drug accumulation following repeated infusions.
Despite a strong preclinical rationale and promising preclinical and early phase data, BI 2536 has failed to demonstrate sufficient efficacy in phase II clinical trials to date and development has been discontinued. Many of these trials were conducted in advanced, difficult-to-treat and molecularly heterogeneous tumours and these factors may have contributed to the failure of BI 2536 to demonstrate clinical efficacy. Moreover, BI 2536 has a relatively short half-life in vivo and it is possible that the failure to achieve clinical efficacy reflects an insufficient exposure of tumour cells to the active inhibitor, rather than a true resistance to the mechanism of action. As preclinical data did not indicate superiority of any tested administration schedule, four different schedules have been tested in phase 1 trials resulting in similar exposure without relevant differences in safety and efficacy. Although it is still possible that an alternate schedule of BI 2536 would result in a stronger and more prolonged inhibition of mitosis, an effective mitotic block may be difficult to achieve without significant myelosuppression in patients with solid tumours, as approximately 28% of bone marrow neutrophils are continually undergoing mitosis (Komlodi-Pasztor et al, 2011).
There still is a striking rationale for the clinical testing of Plk inhibitors in cancer patients despite the overall negative results of this trial. Preclinical studies have demonstrated that Plk inhibitors can synergise with chemotherapeutic agents to inhibit malignant cell proliferation in vitro (Gleixner et al, 2010) and tumour growth in vivo in murine xenograft models without additive toxicity (Baum et al, 2007), suggesting the potential suitability of these combinations in patients with cancer. Plk inhibitors are currently in phase I/II development in various solid tumour and haematological indications. GSK461364, an ATP-competitive inhibitor of Plk1, and HMN-214, a stilbene derivative that interferes with Plk1 subcellular localisation, demonstrated SD as best response in phase I trials of patients with advanced solid tumours (Garland et al, 2006; Olmos et al, 2011). ON 01910.Na (rigosertib), which has been claimed to interact with the Plk pathway, has also shown clinical efficacy in pancreatic cancer (Ma et al, 2011) and is currently recruiting a phase II/III trial in this indication. However, the true impact of this compound on the Plk1 pathway is currently unclear (Oussenko et al, 2011). In phase I studies in patients with advanced solid tumours, volasertib (BI 6727), an additional dihydropteridinone derivative with an improved PK profile compared with BI 2536, has provided a stronger efficacy signal than BI 2536 and a favourable safety profile (Schöffski et al, 2012). For these reasons, clinical development of BI 2536 has been terminated and volasertib has been chosen for further clinical development.
In view of the favourable safety profile observed to date, Plk inhibitors have the potential for future use in combination with other agents, including both cytotoxics and targeted agents. A phase I trial of volasertib in combination with cisplatin or carboplatin is currently ongoing and has demonstrated preliminary evidence of efficacy and tolerability in patients with advanced solid tumours (Deleporte et al, 2011) and a phase I trial of volasertib in combination with afatinib, an irreversible ErbB-family blocker, is currently ongoing. Data from these mono- and combination-therapy trials will help to determine the potential of Plk as a target in the treatment of cancer (Schöffski, 2009). In addition, there is an urgent need for biomarker studies to help identify patients who are most likely to benefit from Plk inhibition. Clinical trials including biomarker evaluations are ongoing.
Change history
03 July 2012
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Baum A, Garin-Chesa P, Gurtler U, Munzert G, Rudolf G (2007) Efficacy of BI 2536, a potent and selective inhibitor of the mitotic kinase Plk1, in models of human non-small cell lung carcinoma. J Thorac Oncol 2: s435–s436
Burris HA, Moore MJ, Andersen J, Green MR, Rothenberg M, Modiano MR, Cripps MC, Portenoy RK, Storniolo AM, Tarassoff P, Nelson R, Dorr FA, Stephens CD, Von Hoff DD (1997) Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol 15: 2403–2413
Cascinu S, Falconi M, Valentini V, Jelic S (2010) Pancreatic cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol 21 (Suppl 5): v55–v58
Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de la Fouchardiere C, Bennouna J, Bachet JB, Khemissa-Akouz F, Pere-Verge D, Delbaldo C, Assenat E, Chauffert B, Michel P, Montoto-Grillot C, Ducreux M (2011) FOLFIRINOX vs gemcitabine for metastatic pancreatic cancer. N Engl J Med 364: 1817–1825
Deleporte A, Dumez H, Awada A, Costermans J, Meeus M, Berghmans T, Ould Kaci M, Juhel N, Berge A, Taube T, Schöffski P (2011) Phase I trial of volasertib (BI 6727), a polo-like kinase 1 (Plk1) inhibitor, in combination with cisplatin or carboplatin in patients with advanced solid tumors. J Clin Oncol 29, (Abstract 3031)
Eckel F, Schneider G, Schmid RM (2006) Pancreatic cancer: a review of recent advances. Expert Opin Investig Drugs 15: 1395–1410
Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM (2011) GLOBOCAN 2008, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 10 [Internet] International Agency for Research on Cancer: Lyon, France, 2010
Frost A, Mross K, Steinbild S, Hedbom S, Unger C, Kaiser R, Trommeshauser D, Munzert G (2012) Phase I study of the Plk1 inhibitor BI 2536 IV on 3 consecutive days in advanced solid tumours. Curr Oncol 19: e28–e35
Garland LL, Taylor C, Pilkington DL, Cohen JL, Von Hoff DD (2006) A phase I pharmacokinetic study of HMN-214, a novel oral stilbene derivative with polo-like kinase-1-interacting properties, in patients with advanced solid tumors. Clin Cancer Res 12: 5182–5189
Gleixner KV, Ferenc V, Peter B, Gruze A, Meyer RA, Hadzijusufovic E, Cerny-Reiterer S, Mayerhofer M, Pickl WF, Sillaber C, Valent P (2010) Polo-like kinase 1 (Plk1) as a novel drug target in chronic myeloid leukemia: overriding imatinib resistance with the Plk1 inhibitor BI 2536. Cancer Res 70: 1513–1523
Gray PJ, Bearss DJ, Han H, Nagle R, Tsao MS, Dean N, Von Hoff DD (2004) Identification of human polo-like kinase 1 as a potential therapeutic target in pancreatic cancer. Mol Cancer Ther 3: 641–646
Heinemann V, Boeck S, Hinke A, Labianca R, Louvet C (2008) Meta-analysis of randomized trials: evaluation of benefit from gemcitabine-based combination chemotherapy applied in advanced pancreatic cancer. BMC Cancer 8: 82
Hofheinz RD, Al-Batran SE, Hochhaus A, Jager E, Reichardt VL, Fritsch H, Trommeshauser D, Munzert G (2010) An open-label, phase I study of the polo-like kinase-1 inhibitor, BI 2536, in patients with advanced solid tumors. Clin Cancer Res 16: 4666–4674
Howlader N, Noone AM, Krapcho M, Neyman N, Aminou R, Waldron W, Altekruse SF, Kosary CL, Ruhl J, Tatalovich Z, Cho H, Mariotto A, Eisner MP, Lewis DR, Chen HS, Feuer EJ, Cronin KA, Edwards BK (eds) (2011) SEER Cancer Statistics Review, 1975–2008. National Cancer Institute: Bethesda, MD http://seer.cancer.gov/csr/1975_2008/ based on November 2010 SEER data submission, posted to the SEER web site
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D (2011) Global cancer statistics. CA Cancer J Clin 61: 69–90
Komlodi-Pasztor E, Sackett D, Wilkerson J, Fojo T (2011) Mitosis is not a key target of microtubule agents in patient tumors. Nat Rev Clin Oncol 8: 244–250
Li D, Xie K, Wolff R, Abbruzzese JL (2004) Pancreatic cancer. Lancet 363: 1049–1057
Ma WW, Messersmith WA, Dy GK, Freas E, Whitworth A, Wilhelm F, Eckhardt SG, Adjei AA, Jimeno A (2011) Final results of a phase I study of the combination of a novel cell cycle inhibitor ON 01910.Na with gemcitabine in patients with advanced pancreatic and other solid tumors. J Clin Oncol 29, (Abstract 3101)
Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA, Campos D, Lim R, Ding K, Clark G, Voskoglou-Nomikos T, Ptasynski M, Parulekar W (2007) Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 25: 1960–1966
Mross K, Frost A, Steinbild S, Hedbom S, Rentschler J, Kaiser R, Rouyrre N, Trommeshauser D, Hoesl CE, Munzert G (2008) Phase I dose escalation and pharmacokinetic study of BI 2536, a novel Polo-like kinase 1 inhibitor, in patients with advanced solid tumors. J Clin Oncol 26: 5511–5517
Olmos D, Barker D, Sharma R, Brunetto AT, Yap TA, Taegtmeyer AB, Barriuso J, Medani H, Degenhardt YY, Allred AJ, Smith DA, Murray SC, Lampkin TA, Dar MM, Wilson R, de Bono JS, Blagden SP (2011) Phase I study of GSK461364, a specific and competitive Polo-like kinase 1 inhibitor, in patients with advanced solid malignancies. Clin Cancer Res 17: 3420–3430
Oussenko IA, Holland JF, Reddy EP, Ohnuma T (2011) Effect of ON 01910.Na, an anticancer mitotic inhibitor, on cell-cycle progression correlates with RanGAP1 hyperphosphorylation. Cancer Res 71: 4968–4976
Schöffski P (2009) Polo-like kinase (PLK) inhibitors in preclinical and early clinical development in oncology. Oncologist 14: 559–570
Schöffski P, Awada A, Dumez H, Gil T, Bartholomeus S, Wolter P, Taton M, Fritsch H, Glomb P, Munzert G (2012) A phase I, dose-escalation study of the novel Polo-like kinase inhibitor volasertib (BI 6727) in patients with advanced solid tumours. Eur J Cancer 48: 179–186
Steegmaier M, Hoffmann M, Baum A, Lenart P, Petronczki M, Krssak M, Gurtler U, Garin-Chesa P, Lieb S, Quant J, Grauert M, Adolf GR, Kraut N, Peters JM, Rettig WJ (2007) BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr Biol 17: 316–322
Strebhardt K (2010) Multifaceted polo-like kinases: drug targets and antitargets for cancer therapy. Nat Rev Drug Discov 9: 643–660
Swanton C (2004) Cell-cycle targeted therapies. Lancet Oncol 5: 27–36
Takahashi T, Sano B, Nagata T, Kato H, Sugiyama Y, Kunieda K, Kimura M, Okano Y, Saji S (2003) Polo-like kinase 1 (PLK1) is overexpressed in primary colorectal cancers. Cancer Sci 94: 148–152
Tarceva [package insert] (2010) OSI Pharmaceuticals, Inc.: Melville, NY; Genentech, Inc.: South San Francisco, CA
Tarceva [summary of product characteristics] (2011) Roche Registration Limited: Welwyn Garden City, UK
Wolf G, Elez R, Doermer A, Holtrich U, Ackermann H, Stutte HJ, Altmannsberger HM, Rubsamen-Waigmann H, Strebhardt K (1997) Prognostic significance of polo-like kinase (PLK) expression in non-small cell lung cancer. Oncogene 14: 543–549
Acknowledgements
This study was supported by Boehringer Ingelheim Pharma GmBH & Co, KG. Writing and editorial assistance was provided by Victoria A Robb of Geomed, which was contracted by Boehringer Ingelheim Pharma GmBH & Co, KG. The authors of this manuscript meet criteria for authorship as recommended by the International Committee of Medical Journal Editors, were fully responsible for all content and editorial decisions and were involved in all the stages of manuscript development. The authors received no compensation related to the development of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Mross, K., Dittrich, C., Aulitzky, W. et al. A randomised phase II trial of the Polo-like kinase inhibitor BI 2536 in chemo-naïve patients with unresectable exocrine adenocarcinoma of the pancreas – a study within the Central European Society Anticancer Drug Research (CESAR) collaborative network. Br J Cancer 107, 280–286 (2012). https://doi.org/10.1038/bjc.2012.257
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2012.257
Keywords
This article is cited by
-
Targeting the PLK1-FOXO1 pathway as a novel therapeutic approach for treating advanced prostate cancer
Scientific Reports (2020)
-
A new BET inhibitor, 171, inhibits tumor growth through cell proliferation inhibition more than apoptosis induction
Investigational New Drugs (2020)
-
Polo-like kinases and acute leukemia
Oncogene (2019)
-
Discovery and development of the Polo-like kinase inhibitor volasertib in cancer therapy
Leukemia (2015)
-
Polo-Like Kinases in Colorectal Cancer: Potential for Targeted Therapy
Current Colorectal Cancer Reports (2015)
