
Abstract
Background:
The DNA-repair gene DNA-dependent kinase catalytic subunit (DNA-PKcs) favours or inhibits carcinogenesis, depending on the cancer type. Its role in human hepatocellular carcinoma (HCC) is unknown.
Methods:
DNA-dependent protein kinase catalytic subuni, H2A histone family member X (H2AFX) and heat shock transcription factor-1 (HSF1) levels were assessed by immunohistochemistry and/or immunoblotting and qRT–PCR in a collection of human HCC. Rates of proliferation, apoptosis, microvessel density and genomic instability were also determined. Heat shock factor-1 cDNA or DNA-PKcs-specific siRNA were used to explore the role of both genes in HCC. Activator protein 1 (AP-1) binding to DNA-PKcs promoter was evaluated by chromatin immunoprecipitation. Kaplan–Meier curves and multivariate Cox model were used to study the impact on clinical outcome.
Results:
Total and phosphorylated DNA-PKcs and H2AFX were upregulated in HCC. Activated DNA-PKcs positively correlated with HCC proliferation, genomic instability and microvessel density, and negatively with apoptosis and patient’s survival. Proliferation decline and massive apoptosis followed DNA-PKcs silencing in HCC cell lines. Total and phosphorylated HSF1 protein, mRNA and activity were upregulated in HCC. Mechanistically, we demonstrated that HSF1 induces DNA-PKcs upregulation through the activation of the MAPK/JNK/AP-1 axis.
Conclusion:
DNA-dependent protein kinase catalytic subunit transduces HSF1 effects in HCC cells, and might represent a novel target and prognostic factor in human HCC.
Similar content being viewed by others


Main
Human hepatocellular carcinoma (HCC) is one of the most common and lethal tumours worldwide (Villanueva et al, 2007). Better understanding of pathogenetic mechanisms may hasten identification of new prognostic markers and development of new diagnostic and therapeutic strategies against this deadly disease (Villanueva et al, 2007; Calvisi et al, 2012).
In recent years, we investigated the prognostic role of deregulated signalling pathways in HCC differently prone to progression, induced in rodent strains with different susceptibility to hepatocarcinogenesis, and human HCC subtypes, selected according to their clinicopathological features (Calvisi et al, 2012). We found highest upregulation of iNOS/NF-kB (inducible nitric oxide synthase/nuclear factor-κB) and RAS/ERK (extracellular signal-regulated kinase) cascades, ubiquitination of the ERK inhibitor DUSP1 (dual-specificity phosphatase 1), and deregulation of FOXM1 (Forkhead box M1B), MYBL2 (v-Myb avian myeloblastosis viral oncogene homolog-like2; Frau et al, 2011; Calvisi et al, 2012), and cell cycle key genes (Pascale et al, 2002, Calvisi et al, 2012) in rapidly progressing HCC of F344 rats, genetically susceptible to hepatocarcinogenesis, and in a human HCC subtype with poorer prognosis (<3 years survival, after partial liver resection, HCCP). These changes were lower/absent in slow progressing HCC of resistant BN rats, and human HCC with better outcome (>3 years survival; HCCB). Furthermore, highest ubiquitination and proteasome degradation of cell cycle inhibitors of WAF/Kip families, P130, RASSF1A (Ras-associated factor-1), and FOXO1 (forkhead box O1) contribute to cell cycle progression in F344 HCC and HCCP (Calvisi et al, 2009).
DNA-dependent protein kinase catalytic subunit (DNA-PKcs) has a pivotal role in the non-homologous end-joining pathway for DNA double-strand break (DSB) repair and is required for cellular resistance to ionising radiation (Weterings and Chen, 2008; Hsu et al, 2012). In response to DSB formation, DNA-PKcs is recruited to DSBs by the Ku70/80 heterodimer and rapidly phosphorylated at multiple serine and threonine residues (Weterings and Chen, 2008; Hsu et al, 2012). In vivo, DNA-PKcs phosphorylation at threonine 2609 (Thr2609) is critical for DSB repair and radioresistance (Ding et al, 2003; Yajima et al, 2006; Chen et al, 2007). In addition to its key function in DNA damage repair, DNA-PKcs has an important role in cell cycle control by orchestrating mitosis, microtubule dynamics and proper chromosomal segregation (Lee et al, 2011; Hsu et al, 2012). Furthermore, DNA-PKcs activity impedes the mitotic catastrophe in response to DNA damage (Shang et al, 2010). Based on these data, a tumour suppressor function of DNA-PKcs has been envisaged, due to its capability to safeguard the genome integrity (Hsu et al, 2012). In accordance with this hypothesis, loss of DNA-PKcs expression was found to be a frequent event along gastric carcinogenesis, and was associated with lymphatic invasion, lymph node metastasis and poor patient survival (Lee et al, 2005). Attenuated expression of DNA-PKcs has also been found in a subset of ovarian cancers, whereas DNA-PKcs is ubiquitously expressed in normal ovaries and in benign ovary tumours (Shao et al, 2007). Also, reduced activity of DNA-PKcs was associated with increased risk to develop lung cancer in a population-based study (Auckley et al, 2001). In addition, high expression of DNA-PKcs was shown to be an important predictor for better response to radiation therapy in oesophageal cancer and early breast cancer (Noguchi et al, 2002; Söderlund Leifler et al, 2010). On the other hand, as rapidly proliferating tumour cells are exposed to various stressors, including telomere shortening, oncogene-induced DNA replication stress and generation of reactive oxidative and nitroxidative species by the tumour microenvironment, it is plausible to hypothesise that DNA-PKcs might also support carcinogenesis by protecting tumour cells from DNA damage-induced death (Hsu et al, 2012). In accordance with the latter hypothesis, increased protein levels of DNA-PKcs have been detected in various tumour types, including colorectal, oesophageal and lung tumours, and were strongly associated with advanced clinical stages and poor survival (Hosoi et al, 2004; Tonotsuka et al, 2006; Parsons et al, 2008; Xing et al, 2008). Furthermore, increased activity of DNA-PKcs might protect cancer cells from DNA-damaging agents such as chemotherapeutic drugs and radiotherapy. Higher DNA-PKcs activity was in fact detected in glioma patients who were resistant to cisplatin-based chemotherapy (Mukherjee et al, 2009). Also, in oral squamous cell carcinoma, a strong upregulation of DNA-PKcs and Ku proteins was detected after radiotherapy in the residual cancer cells (Shintani et al, 2003). The latter finding suggests that induction of DNA-PKcs and Ku following radiotherapy could provide a selective survival advantage to tumour clones against radiation-induced damage and might be implicated in tumour recurrence (Shintani et al, 2003). Thus, it has been hypothesised that inhibitors of DNA-PKcs should be highly useful in sensitising tumours to chemo- and radio-therapy as well as to prevent tumour recurrence (Shintani et al, 2003; Hsu et al, 2012).
Altogether, these observations indicate that DNA-PKcs might have paradoxically opposing roles in cancer, either favoring or inhibiting carcinogenesis, depending on the cell context or the tissue type (Hsu et al, 2012). In HCC, a tumour type characterised by intrinsic resistance to chemotherapeutic drugs (Bruix et al, 2004), the role of DNA-PKcs has never been investigated and remains elusive. Only one previous study suggests an oncogenic potential of DNA-PKcs in liver cancer due its ability to stabilise the c-Myc oncoprotein in the HepG2 cell line (An et al, 2008). Furthermore, the upstream regulators of DNA-PKcs in HCC and other solid tumours have been poorly defined. Here, we provide evidence that DNA-PKcs has a pro-oncogenic, pleiotropic role on HCC cells, significantly influencing their proliferation, survival and DNA-repair properties. Also, we identified the heat shock factor-1 (HSF1)/mitogen-activated protein kinase (MAPK)/c-Jun N-terminal kinase (JNK) axis as a major inducer of DNA-PKcs in HCC cells.
Materials and methods
Human tissue samples
Six normal livers, 62 HCCs and corresponding surrounding non-tumour liver tissues were used. Tumours were divided in HCC with shorter/poor (HCCP; n=32) and longer/better (HCCB; n=30) survival, characterised by <3 and >3 years survival following partial liver resection, respectively (Lee et al, 2004). Patients’ features are reported in Supplementary Table 2. Liver tissues were kindly provided by Dr Snorri S. Thorgeirsson (National Cancer Institute, Bethesda, MD, USA) and collected at the Pietro Valdoni Surgery Department (University of Rome La Sapienza, Rome, Italy). Institutional Review Board approval was obtained at participating hospitals and the National Institutes of Health.
Proliferation and apoptotic indices, microvessel density
Proliferation and apoptotic indices were determined in human HCC by counting Ki-67-positive cells immunostained with mouse monoclonal anti-Ki67 antibody (clone MIB-1; Dako Deutschland, Hamburg, Germany) and apoptotic figures stained with the ApoTag peroxidase in situ apoptosis kit (Millipore, Billerica, MA, USA), respectively, on at least 3000 hepatocytes. Microvessel density (MVD) was evaluated in HCC slides immunostained with mouse monoclonal anti-CD34 antibody (Vector Laboratories, Burlingame, CA, USA) as previously described (Calvisi et al, 2009). In brief, the four highest MVD areas for each tumour were photographed at high power ( × 200) and the size of each area was standardised using the ImageJ software (http://rsb.info.nih.gov/ij/). Microvessel density was expressed as the percentage of the total CD34-stained spots per section area (0.94 mm2).
Random amplified polymorphic DNA (RAPD) analysis
Twenty-two previously designed primers (Calvisi et al, 2004) were used to score genomic alterations in human HCCs, and the RAPD reaction was performed as described (Luceri et al, 2000). Differences from corresponding non-tumourous livers were scored in the case of a change in the intensity, absence of a band or appearance of a new band in HCC. The frequency of altered RAPD profiles was calculated for each liver lesion as reported (Luceri et al, 2000).
Cell lines and treatments
The Chang and WRL-68 non-transformed liver cell lines, and the Huh6, HepG2 (harbouring wild-type p53 gene), Huh7 and Hep3B, and SNU-423 (harbouring mutated p53 gene) HCC cell lines were used for the studies in vitro. Cell lines were maintained as monolayer cultures in Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum. Cells were plated at 2.0 × 103/well in 96-well plate and grown for 12 h, then serum-deprived for 24 h and treated with siRNA against DNA-PKcs (Santa Cruz Biotechnology, Santa Cruz, CA, USA) or, alternatively, with the DNA-PKcs inhibitor, NU7441 (Santa Cruz Biotechnology; 5 μ M), the MEK/MAPK inhibitor U0126 (Santa Cruz Biotechnology; 20 μ M) or the JNK inhibitor SP600125 (Santa Cruz Biotechnology; 10 μ M) following the manufacturer’s recommendations. Transient transfection experiments of HCC cell lines with HSF1 cDNA in pCMV6-AC vector (OriGene Technologies, Rockville, MD, USA) was performed following the manufacturer's protocol using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) for transfection procedures. Transiently transfected cells were incubated for 24 and 48 h. For the induction of DNA damage, cells were treated with 2 μmol/l doxorubicin (Sigma-Aldrich, St Louis, MO, USA) for 2 h, washed twice with phosphate-buffered saline (PBS) and returned to normal growth medium for the indicated time periods. To assess cell proliferation, HCC cell lines were plated at the concentration of 2.0 × 103/well in 96-well plates, allowed to attach and adjust for the next 12 h (which corresponds to the 0 h time point in our graphs), and grown for the additional 24 and 48 h. Proliferation was assessed with the Brdu Cell Proliferation Assay Kit (Cell Signaling Technology, Danvers, MA, USA) by measuring the absorbance at 450 nm following the manufacturer’s protocol. To measure apoptosis, HCC cell lines were plated at the concentration of 2.0 × 103/well in 96-well plates, incubated for 12 h and then subjected to 24-h serum deprivation, which corresponded to the 0-h time point in the apoptosis graphs. HCC cells continue to growth in serum-free medium for additional 24 and 48 h. Apoptosis was assessed with the Cell Death Detection Elisa Plus Kit (Roche Molecular Biochemicals, Indianapolis, IN, USA) by measuring the absorbance at 405 nm following the manufacturer’s protocol. DNA damage of cultured HCC cells was determined by the DNA Damage Quantification Kit (Biovision, Mountain View, CA, USA) following the manufacturer's instructions. All experiments were repeated at least three times per each cell line.
Western blot analysis, chromatin immunoprecipitation (ChIP)
Normal livers, HCC and respective non-neoplastic surrounding livers were homogenised in lysis buffer (30 mM Tris (pH 7.5), 150 mM NaCl, 1% NP-40, 0.5% Na deoxycholate, 0.1% SDS, 10% glycerol and 2 mM EDTA) containing the Complete Protease Inhibitor Cocktail (Roche Molecular Biochemicals), and sonicated. Protein concentrations were determined with the Bio-Rad Protein Assay Kit (Bio-Rad, Hercules, CA, USA) using bovine serum albumin as standard. For western blotting, aliquots of 80 μg were denatured by boiling in Tris-Glycine SDS Sample Buffer (Invitrogen), separated by SDS–PAGE, and transferred onto nitrocellulose membranes (Invitrogen) by electroblotting. Membranes were blocked in 5% non-fat dry milk in Tris-buffered saline containing 0.1% Tween 20 for 1 h and probed with specific antibodies listed in Supplementary Table 1. Each primary antibody was followed by incubation with horseradish peroxidase-secondary antibody diluted 1:5000 for 1 h and then revealed with the Super Signal West Pico (Pierce Chemical Co.,New York, NY, USA). For each protein, densities were calculated by ImageQuaNT 5.1 software (GE Healthcare, Piscataway, NJ, USA), normalised to β-actin (Chemicon International, Temecula, CA, USA; dilution 1 : 20,000) levels and mean values evaluated for statistical significance. For the identification of potential HSF1- and activator protein 1 (AP-1)-binding sites in the human DNA-PKcs promoter, the TESS transcriptional element search tool was used (http://www.cbil.upenn.edu/cgi-bin/tess/tess?RQ=WELCOME). ChIP was performed using the ChIP Assay Kit (Millipore) following the manufacturer’s protocol. The rabbit polyclonal anti-c-Jun antibody or the rabbit polyclonal anti-c-Fos (Santa Cruz Biotechnology) antibody was used to immunoprecipitate chromosomal DNA in cross-linked chromatin prepared from exponentially growing HuH6, HuH7, WRL-68, SNU-423, and HLE cell lines. Immunoprecipitated chromatin was subjected to PCR analysis using the primers DNA-PKcs Fw (TAGGGAGCGTGTGCTTCTTT) and DNA-PKcs Rv (GGGGCCTAGGTCAAGAGTTC), which produce an amplicon of 274 base pairs encompassing some of the putative AP-1-binding sites in the human DNA-PKcs promoter.
Quantitative reverse transcription-polymerase chain reaction (qRT–PCR). Primers for human DNA-PKcs, HSF1 and RNR18 genes were purchased from Applied Biosystems (Foster City, CA, USA). PCR reactions were performed with 100 ng of cDNA on the whole sample collection and cell lines, using an ABI Prism 7000 Sequence Detection System and TaqMan Universal PCR Master Mix (Applied Biosystems). Cycling conditions were: 10 min of denaturation at 95 °C and 40 cycles at 95 °C for 15 s and at 60 °C for 1 min. Quantitative values were calculated by using the PE Biosystems Analysis software and expressed as N target (NT). NT=2−ΔCt, wherein ΔCt value of each sample was calculated by subtracting the average Ct value of the target gene from the average Ct value of the RNR18 gene.
Immunohistochemistry
Immunohistochemical staining was performed on 10% formalin-fixed, paraffin-embedded sections. Deparaffinised sections were incubated in 3% H2O2 dissolved in PBS 1 × for 30 min to quench the endogenous peroxidase. For antigen retrieval, slides were microwaved in 10 mM citrate buffer (pH 6.0) for 15 min. The mouse monoclonal anti-phospho-DNA-PKcs (Thr2609; Santa Cruz Biotechnology) and the rabbit polyclonal anti-phospho-HSF1 antibody (Ser326; Bioss, Woburn, MA, USA) were used at 1 : 500 and 1 : 2000 dilution, respectively. The immunoreactivity was visualised with the Vectastain Elite ABC kit (Vector Laboratories), using Vector NovaRED (Vector Laboratories) as the chromogen. Slides were counterstained with Mayer’s hematoxylin. Phospho-DNA-PKcs index was determined by counting phospho-DNA-PKcs positive nuclei over 3000 hepatocyte nuclei.
DNA-PKcs and HSF1 activity assays
The activity of DNA-PKcs and HSF1 was assessed by using the SignaTECT DNA-Dependent Protein Kinase Assay System (Promega Corporation, Madison, WI, USA) and the [pSer326]HSF1 ELISA (Enzo Life Sciences Inc., Farmingdale, NY, USA) kits, respectively, on nuclear extracts from normal, non-tumourous and tumourous livers, following the manufacturer’s instructions.
Statistical Analysis
GraphPad Prism 5.01 (www.graphpad.com) was used to evaluate statistical significance by Tukey–Kramer, Student's t and Mann–Whitney tests and linear regression analyses. Overall survival was estimated according to Kaplan–Meier and Log-rank (Mantel–Cox) test. Univariate and multivariate Cox analysis was used to estimate hazard ratios for risk factors using STATA 9 statistical software (Stata Corporation, College Station, TX, USA). Values of P<0.05 were considered significant. Data are expressed as means±s.d.
Results
DNA-PKcs is upregulated in HCC
DNA-PKcs levels were assessed in human normal livers, HCCs and respective non-neoplastic surrounding livers by western blot analysis and real-time RT–PCR. A progressive upregulation of total and activated/phosphorylated (Thr2609) levels of DNA-PKcs as well as DNA-PKcs mRNA expression and activity occurred in non-tumourous tissues and HCC when compared with normal livers, with the highest levels being detected in HCCP (Figure 1A–D). An equivalent pattern was detected when assessing the total and activated/phosphorylated levels (Ser139) of H2A histone family member X (H2AFX) protein, a DNA-PKcs downstream effector involved in DNA damage response (Fernandez-Capetillo et al, 2003; Park et al, 2003; An et al, 2010). Subsequent immunohistochemical assessment of phosphorylated DNA–PKcs (Thr2609) showed that nuclear DNA-PKcs increased in surrounding livers and HCC with respect to normal liver (Figure 2A and I). No differences occurred between surrounding liver subtypes, whereas the percentage of phosphorylated DNA–PKcs-positive nuclei was significantly higher in HCC than surrounding liver counterparts, and progressively increased from HCCB to HCCP (Figure 2A and I).

Expression and activity of DNA-PKcs and expression of H2AFX and their phosphorylated forms in human neoplastic liver lesions. Liver tumours were divided in two distinct prognostic categories based on the length of patient survival after partial liver resection: HCC with survival >3 years following partial hepatectomy (HCCB), and HCC with survival <3 years (HCCP). (A) Representative western blot analysis of DNA-PKs and H2AFX. Hepatic tissue lysates were subjected to western blotting with specific antibodies. (B) Chemiluminescence analysis: optical densities of the peaks were normalised to β-Actin values and expressed in arbitrary units. (C) DNA-PKcs mRNA levels were determined by quantitative RT–PCR. N Target (NT)=2−ΔCt; ΔCt=Ct RNR18 - Ct target gene. (D) Specific activity of DNA-PKcs: pmol [32]P/min/μl. Data are means (s.d.) of six normal livers (NL), 30 HCCB and 32 HCCP, and corresponding surrounding livers (SL) with better or poorer prognosis. Dot: different from NL for at least P=0.0005; asterisk: different from SL for P<0.0001; dagger: different from SLB and HCCB for at least P=0.002.

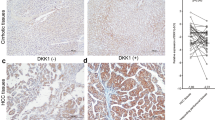
(A – H ): Liver histology (stained with hematoxylin and eosin; HE) and representative immunohistochemistry of activated/phosphorylated (Thr2609) DNA-PKcs in HCC subtypes with better (HCCB; E, F ) or poorer (HCCP; G, H ) outcome, and respective non-tumourous surrounding livers (SL; A, B). Note the progressive increase of DNA-PKcs from SL to HCCP (C, D). The HE shows a typical macrotrabecular HCC in the left part of the picture and a rim of surrounding non-neoplastic liver tissue at the right (G, H). Poorly differentiated HCCP showing a strong p-DNA-PKcs immunoreactivity in most of the malignant hepatocytes. Original magnification × 200 in C and D; × 400 in A, B, E–H. (I): Box plots showing the dispersion of the results of six normal livers (NL), 22 HCCB, 22 HCCP and corresponding SLs evaluated for p-DNA-PKcs nuclear immunoreactivity. Mann–Whitney test: *Indicates difference from NL: at least P=0.0004 for all lesions. •Indicates difference from HCC: P<0.0001 for HCCB and HCCP. §Indicates difference from HCCB: P<0.0001.
Correlation of DNA-PKcs activity with HCC clinicopathological parameters
Next, we evaluated the relationship of DNA-PKcs activity with clinicopathological features of HCC patients. Proliferation index, genomic instability and microvessel density values positively correlated with DNA-PKcs activity, whereas the latter was inversely correlated with apoptosis (Figure 3A and D). Moreover, Kaplan–Meier survival curves of human HCC with high and low DNA-PKcs activity clearly showed a significant association of shorter survival with high DNA-PKcs activity (Figure 3E). Remarkably, in the multivariate analysis model (Supplementary Table 3), DNA-PKcs activity was significantly associated with overall survival, together with patients’ sex, serum AFP level and HCC apoptosis and microvessel density.

Relationship between DNA-PKcs-specific activity and biological features of human HCC. The relationships of DNA-PKcs specific activity (SA; as assessed by ELISA) and proliferation index (expressed as the percentage of Ki67-positive nuclei with respect to total cells counted) (A); apoptotic index (percentage of apoptotic cells) (B); microvessel density (MVD; percentage of CD34-positive cells) (C) and genomic instability (RAPD; expressed as the percentage of primers showing altered RAPD profiles) (D); is shown. (E) Kaplan–Meier survival curves of human HCC with high and low DNA-PKcs SA. Two-phase decay curves were used to calculate the cutoff value of DNA-PKcs (204–230) in correspondence to curve intersection in the correlation analysis of DNA-PKcs with proliferation, apoptosis, MVD and RAPD. Mantel–Cox statistical analysis: differences between survival curves, P<0.0001.
DNA-PKcs regulates proliferation, survival and DNA-repair of HCC cells independent of p53
The role of DNA-PKcs in human liver cancer was further investigated by assessing the consequences of DNA-PKcs inactivation via siRNA in vitro. For silencing experiments, we selected two non-transformed liver cell lines, Chang and WRL-68, and four HCC cell lines: Huh6, HepG2 (harbouring wild-type p53), Huh7 and HLE (harbouring mutant p53). Specific siRNA showed an equivalent efficiency in the inhibition of DNA-PKcs in the six cell lines (Supplementary Figure 1). At the cellular level, DNA-PKcs silencing reduced proliferation and induced apoptosis in the six cell lines (Figure 4). Importantly, the effects on growth restraint were significantly higher in HCC cell lines when compared with non-transformed liver cell lines. Furthermore, when silencing of DNA-PKcs was associated with treatment with the DNA-damaging agent doxorubicin, very strong growth restraint, elevated apoptosis and massive DNA damage were detected in all HCC cell lines, irrespective of p53 status (Figure 4). In contrast, only a slight decline in proliferation and increase in apoptosis and DNA damage was detected in Chang and WRL-68 non-transformed cell lines under the same treatment (Figure 4). Equivalent results were obtained following administration to the cell lines of the DNA-PKcs inhibitor, NU7441 (Supplementary Figure 2). Altogether, the present data indicate that DNA-PKcs provides multiple growth advantages to HCC cell lines independent of p53.

Effect of inhibition of DNA-PKcs expression by specific siRNA against DNA-PKcs (sD) either alone or associated with doxorubicin administration (DR) on viability (OD 450 nm), apoptosis (OD 405 nm) and DNA damage (absolute number of apurinic sites per 105 base pairs) of normal liver (Chang, WRL-68) and HCC cell lines with wild-type (Huh6, HepG2) and mutated (Huh7, HLE) p53 gene. Data are means±s.d. of nine experiments. Effect of inhibition of DNA-PKcs expression by specific siRNA against DNA-PKcs (sD) either alone or associated with doxorubicin administration (DR) on viability (OD 450 nm), apoptosis (OD 405 nm) and DNA damage (absolute number of apurinic sites per 105 base pairs) of normal liver (Chang, WRL-68) and HCC cell lines with wild-type (Huh6, HepG2) and mutated (Huh7, HLE) p53 gene. Data are means±s.d. of nine experiments. Abbreviation: S, scrambled siRNA. Asterisk: different from S for at least P<0.001.
HSF1 is a major inducer of DNA-PKcs in HCC cell lines
Previous findings indicate that HSF1 has a role in the activation of DNA-PKcs (Nueda et al, 1999), by direct interaction (Peterson et al, 1995). Thus, we investigated this issue in HLE and HepG2 cell lines treated with doxorubicin. We found that doxorubicin treatment resulted in increased HSF1 and DNA-PKcs mRNA levels as well as HSF1-DNA-PKcs binding in HLE (Figure 5A and C) and HuH6 (not shown) cells. Heat shock transcription factor-1-DNA-PKcs binding was paralleled by increased levels of total and activated/phosphorylated DNA-PKcs (Thr2609), HSF1 (Ser326) and H2AFX (Ser139) (Figure 5A). Interestingly, the increase in DNA-PKcs mRNA levels following doxorubicin treatment was significantly reduced when HSF1 silencing was associated with doxorubicin administration (Figure 5C). The latter observation suggests a transcriptional upregulation of DNA-PKcs by HSF1. Thus, we analysed the human DNA-PKcs promoter for HSF1-binding sites (Supplementary Figure 3). Although one putative binding site for HSF1 was identified, the subsequent ChIP experiment conducted in WRL-68, HuH6 and HLE cells failed to confirm the interaction between HSF1 and DNA-PKcs (data not shown). On the other hand, the DNA-PKcs promoter harboured several putative binding sites for AP-1, a pivotal downstream effector of MAPK and JNK pathways (Supplementary Figure 3). The MAPK cascade has been previously shown to be a major target of HSF1 (Dai et al, 2007). Noticeably, inhibition of either MAPK or JNK activity by U0126- or SP600125-specific inhibitors, respectively, strongly reduced the rise of DNA-PKcs mRNA induced by doxorubicin treatment in HuH6 cells (Figure 6A). DNA-PKcs upregulation driven by doxorubicin was further decreased when U0126 and SP600125 inhibitors were administered simultaneously. Furthermore, when HSF1 forced overexpression in SNU-423 cells was coupled to combined U0126 and SP600125 treatment, HSF1-induced upregulation of DNA-PKcs mRNA was almost completely abolished by inhibitor treatment (Figure 6B). Subsequent ChIP analysis showed a functional interaction between c-Jun and c-Fos (members of the AP-1 complex) and the DNA-PKcs promoter (Figure 6C and E). Altogether, these data strongly suggest that the AP-1 mediates DNA-PKcs mRNA upregulation driven by HSF1. The relationship between HSF1 and DNA-PKcs was further investigated in human HCC specimens. A similar pattern to that described for p-DNA-PKcs was detected when assessing the total and activated/phosphorylated levels (Ser326) of HSF1. Indeed, total and activated/phosphorylated HSF1 protein levels as well as HSF1 mRNA and activity were progressively upregulated from non-tumourous tissues to HCC when compared with normal livers, with the highest levels being detected in HCCP (Figure 7A and D). Furthermore, a significant, direct correlation was found between the levels of HSF1 activity and that of DNA-PKcs in the sample collection by Spearman’s correlation test (Figure 7E). Also, an equivalent staining pattern for p-HSF1 and p-DNA-PKcs in normal livers, HCC and non-tumourous surrounding liver tissues was detected by immunohistochemistry (Figure 8).

Relationship between DNA-PKcs and its putative target, HSF1, in human HCC cell lines. (A) Induction of HSF1, DNA-PKcs and H2A histone family, member X (H2AFX) and HSF1-DNA-PKcs complex in the HuH6 cell line following doxorubicin (DR) treatment, as assessed by western blotting and immunoprecipitation (BP, blocking peptide). mRNA levels of HSF1 (B) and DNA-PKcs (C) following DR treatment and HSF1 silencing by siRNA (sH) as assessed by real-time PCR in the HuH6 cell line. N Target (NT)=2−ΔCt; ΔCt=Ct RNR18 - Ct target gene. Data are means±s.d. of nine experiments. Abbreviations: C, control (untreated); D, DMSO (vehicle). Statistical analysis: asterisk, different from C, D<0.001; dagger, different from DR, P<0.001.

The MAPK and JNK pathways promote transcriptional activation of DNA-PKcs in human HCC cell lines. (A) Effect of Doxorubicin (DR) treatment and concomitant inhibition of the ERK cascade by the MEK/MAPK inhibitor U0126 (U0; 20 μ M) and/or the JNK inhibitor SP600125 (SP; 10 μ M) in the SNU-423 cell line on DNA-PKcs mRNA levels, as assessed by real-time RT–PCR. (B) Effect of HSF1 (H) gene overexpression by cDNA transfection and concomitant inhibition of the MAPK cascade by the MEK inhibitor U0126 (U0) and/or the JNK inhibitor SP600125 (SP) in the SNU-423 cell line on the DNA-PKcs mRNA levels, as assessed by real-time RT–PCR. N Target (NT)=2−ΔCt; ΔCt=Ct RNR18 - Ct target gene. Data from non-transfected cells and from cells transfected with empty vector did not show any significant difference and were, thus, merged. Abbreviations: (C), not transfected/empty vector; H, HSF1 transfected; U0, U0126; SP, SP600125. Statistical analysis: a, different from C, P<0.001; b, different from DR, P<0.001; c, different from DR+U0, P<0.001; d, different from DR+SP, P<0.001. (C–E) Interaction of c-Fos and c-Jun proteins, members of the AP-1 complex, with the DNA-PKcs promoter in liver and HCC cell lines as detected by ChIP. Chromatin was immunoprecipitated with either an anti-c-Fos or an anti-c-Jun antibody and subjected to PCR analysis of the DNA-PKcs promoter. (C) Doxorubicin (DR) treatment induces the binding of c-Fos with the DNA-PKcs promoter in the SNU-423 cell line. Twenty-four and 48 h time points were analysed. Arrow indicates the 500 bp band. (M) 100 bp DNA marker; (A) negative control (absence of DNA in the PCR reaction); (I) input (aliquot of chromatin before immunoprecipitation). (D, E) Binding of c-Fos (D) and c-Jun (E) antibodies to the DNA-PKcs promoter of HUH6, HuH7 and WRL-68 cell lines. Arrow indicates the 500 bp band. (M) 100 bp DNA marker; (A) negative control (absence of DNA in the PCR reaction); (I) input (aliquot of chromatin before immunoprecipitation); 1, cross-linked chromatin from the HuH6 cell line immunoprecipitated with the anti-β-actin antibody (which does not interact with DNA-PKcs); 2–4, cross-linked chromatin immunoprecipitated with the anti-c-Fos or anti-c-Jun antibody in the HUH6, HuH7 and WRL-68 cell lines, respectively.

Expression and activity of HSF1 in human non-tumourous liver and hepatocellular carcinomas (HCC). HCC were divided in two categories based on patient survival length after partial liver resection: HCC with survival >3 years (HCCB), and HCC with survival <3 years (HCCP). (A) Representative western blot analysis of HSF1 and its activated/phosphorylated form at serine 326 (Ser326). Hepatic tissue lysates were subjected to western blotting with specific antibodies. (B) Chemiluminescence analysis: optical densities of the peaks were normalised to β-actin values and expressed in arbitrary units. (C) HSF1 mRNA levels were determined by quantitative RT–PCR. N Target (NT)=2−ΔCt; ΔCt=Ct RNR18 - Ct target gene. (D) Specific activity of HSF1: ng/ml of HSF1 (pSer326). Data are means (s.d.) of six normal livers (NL), 30 HCCB and 32 HCCP, and corresponding surrounding livers (SL) with better or poorer prognosis. Dot: different from NL for at least P=0.003; asterisk: different from SL for P<0.0001; dagger: different from SLB and HCCB for P<0.0001. (E) Relationships between HSF1 and DNA-PKcs-specific activity (SA) in HCCs.

Representative immunohistochemical patterns of activated/phosphorylated HSF1 at serine 326 (p-HSF1 Ser326) and activated/phosphorylated DNA-PKcs at threonine 2609 (p-DNA-PKcs Thr2609) in HCC subtypes with better (HCCB) or poorer (HCCP) outcome, and non-tumourous surrounding liver tissues (SL). Note the progressive and parallel increase of both p-HSF1 (Ser326) and p-DNA-PKcs (Thr2609) nuclear immunoreactivity from SL to HCC. Original magnification × 200.
Finally, to delineate the importance of DNA-PKcs in HSF1-driven growth, we transiently overexpressed HSF1 gene in the SNU-423 cell line (exhibiting low basal levels of HSF1 mRNA) and evaluate the effects of DNA-PKcs silencing (Supplementary Figure 4). Of note, suppression of DNA-PKcs by siRNA led to a remarkable decline in proliferation as well as increase in apoptosis and DNA damage in SNU-423 cells transfected with HSF1 (Supplementary Figure 4).
Discussion
Previous studies have shown that DNA-PKcs has either an oncogenic or tumour suppressive role in different tumour types (Hsu et al, 2012). In the present investigation, we show that DNA-PKcs is aberrantly expressed and possesses oncogenic properties in human HCC. Indeed, we found a significant upregulation of DNA-PKcs and of its target H2AX, an earliest and specific marker of DSB (Garcia-Canton et al, 2012), in HCC when compared with normal livers and non-tumourous surrounding livers. These findings suggest that induction of DNA-PKcs is involved in liver malignant transformation. Moreover, a marked increase of DNA-PKcs levels was detected in HCCP when compared with HCCB, implying a role of DNA-PKcs also in HCC biological aggressiveness and patient’s outcome. Accordingly, it has been previously shown that high levels of DNA-PKcs directly correlate with an unfavourable prognosis, tumour recurrence and resistance to radiotherapy in various cancer entities (Shintani et al, 2003; Hosoi et al, 2004; Tonotsuka et al, 2006; Parsons et al, 2008; Xing et al, 2008; Mukherjee et al, 2009). Therefore, the present data strongly support an important prognostic role of DNA-PKcs in human liver cancer.
Based on the present investigation, DNA-PKcs levels directly correlate with genomic instability and proliferation index, and inversely with the apoptotic index and patient’s survival length, suggesting that DNA-PKcs contributes to hepatocarcinogenesis via multiple mechanisms. In accordance with the latter hypothesis, we found that DNA-PKcs silencing significantly reduced the growth and increased apoptosis in HCC cells. This disadvantageous effect on cell growth was strikingly augmented by the treatment with the DNA-damaging agent doxorubicin. Of note, the effects on in vitro growth following suppression of DNA-PKcs were much more pronounced in HCC than in non-transformed liver cell lines, implying an addiction of liver tumour cells to DNA-PKcs in order to proliferate and survive. Furthermore, a strong reduction of proliferation and induction of apoptosis accompanied the downregulation of DNA-PKcs in HCC cell lines irrespective of the status of the tumour suppressor gene p53 in these cells. This observation suggests that DNA-PKcs suppression might be universally detrimental for the growth of HCC cells. In addition, the different sensitivity exhibited by liver tumour and non-transformed cell lines toward depletion of DNA-PKcs, might have important therapeutic implications. Indeed, based on our findings in vitro, inhibiting DNA-PKcs activation could provide a multifaceted and effective chemotherapeutic strategy against liver cancer while sparing normal hepatocytes.
Although it seems counterintuitive that a pathway that normally serves to restrain proliferation would be beneficial for HCC cells, the DNA-repair and maintenance of genomic stability afforded by DNA-PKcs could save HCC cells from DNA damage-induced death. In support of this notion, it has been shown that breast cancer cells from BRCA2-deficient tumours show a strong dependency on the DNA stress protein poly-ADP-ribose polymerase (PARP), as PARP inhibition impaired the DNA-damage response to the point of lethality (Bryant et al, 2005). Similarly, the present data imply an addiction of HCC cell lines to DNA-PKcs for their proliferation and survival, especially when subjected to DNA-damaging insults. In addition, a DNA-repair signature identified by microarray analysis characterises HCC with dismal prognosis (Lee et al, 2004). Thus, these data suggest that suppression of tumour DNA-damage response might be highly beneficial for the treatment of human liver cancer, especially in the biologically aggressive subset.
At the molecular level, the putative upstream effector of DNA-PKcs, HSF1 (Peterson et al, 1995; Nueda et al, 1999), was investigated. Our results indicate that HSF1 induces upregulation of DNA-PKs both via direct interaction and through the MAPK/JNK/AP-1 axis in HCC cells. To the best of our knowledge, this is the first study showing the MAPK/JNK/AP-1 axis as an inducer of DNA-PKcs in cancer. In addition, we found that suppression of both MAPK and JNK pathway is more effective than inhibition of each single pathway alone in downregulating DNA-PKcs levels. Furthermore, we detected a synergistic effect of MAPK and JNK concomitant inhibition on HCC cell growth in vitro following the administration of doxorubicin (Supplementary Figure 5). The latter observations suggest that the MAPK and JNK cascades promote the resistance to growth restraint induced by DNA damage in HCC cells by upregulating DNA-PKcs. Furthermore, we show that DNA-PKcs is a major factor responsible for transducing the effects of HSF1 in HCC cells. Indeed, silencing of DNA-PKcs resulted in a remarkable inhibition of cell proliferation in HCC cell lines overexpressing HSF1.
Although DNA-PKcs has been originally identified as a crucial member of the DNA-damage response via its ability to promote non-homologous end joining and homologous recombinant repair (Ding et al, 2003; Chen et al, 2007; Weterings and Chen, 2008; Hsu et al, 2012), emerging evidence indicates that DNA-PKcs function on cell biology might be broader than previously assumed (Hsu et al, 2012). Indeed, it has been shown that DNA-PKcs is involved in the transcriptional regulation of lipogenic genes and activation of the AKT protooncogene via phosphorylation mechanisms (Feng et al, 2004; Dragoi et al, 2005; Wong et al, 2009). Of note, de novo lipogenesis and aberrant AKT activation are major oncogenic events contributing both to HCC development and progression (Calvisi et al, 2011). Thus, the ability of DNA-PKcs to induce lipid biosynthesis and activation of AKT further links DNA-PKcs to hepatocarcinogenesis. Nevertheless, our preliminary data indicate that the effect of DNA-PKcs inhibition is significantly more pronounced than suppression of AKT alone on the growth of various HCC cell lines (Calvisi DF et al, unpublished observation). This finding implies that DNA-PKcs exerts its biological activities in HCC cells both via AKT-dependent and -independent mechanisms. The latter mechanisms are currently under investigation.
Overall, our findings strongly suggest a crucial role of DNA-PKcs in human HCC development and progression, and the possibility that DNA-PKcs represents a prognostic marker for liver cancer. Of note, HCC cells are addicted to the activity of DNA-PKcs for their growth and survival, especially under conditions promoting genomic instability. This observation suggests that targeting DNA-PKcs might represent a promising therapeutic strategy against human HCC. Finally, we show that HSF1 induces DNA-PKcs upregulation through the activation of MAPK/JNK/AP-1 axis, which indicates that DNA-PKcs transduces HSF1 effects in HCC cells.
Change history
12 November 2013
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
An J, Huang YC, Xu QZ, Zhou LJ, Shang ZF, Huang B, Wang Y, Liu XD, Wu DC, Zhou PK (2010) DNA-PKcs plays a dominant role in the regulation of H2AX phosphorylation in response to DNA damage and cell cycle progression. BMC Mol Biol 11: 18.
An J, Yang DY, Xu QZ, Zhang SM, Huo Y, Shang ZF, Wang Y, Wu DC, Zhou PK (2008) DNA-dependent protein kinase catalytic subunit modulates the stability of c-Myc oncoprotein. Mol Cancer 7: 32.
Auckley DH, Crowell RE, Heaphy ER, Stidley CA, Lechner JF, Gilliland FD, Belinsky SA (2001) Reduced DNA-dependent protein kinase activity is associated with lung cancer. Carcinogenesis 22 (5): 723–727.
Bruix J, Boix L, Sala M, Llovet JM (2004) Focus on hepatocellular carcinoma. Cancer Cell 5 (3): 215–219.
Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T (2005) Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434 (7035): 913–917.
Calvisi DF, Factor VM, Ladu S, Conner EA, Thorgeirsson SS (2004) Disruption of beta-catenin pathway or genomic instability define two distinct categories of liver cancer in transgenic mice. Gastroenterology 126 (5): 1374–1386.
Calvisi DF, Ladu S, Pinna F, Frau M, Tomasi ML, Sini M, Simile MM, Bonelli P, Muroni MR, Seddaiu MA, Lim DS, Feo F, Pascale RM (2009) SKP2 and CKS1 promote degradation of cell cycle regulators and are associated with hepatocellular carcinoma prognosis. Gastroenterology 137 (5): 1816–1826.
Calvisi DF, Wang C, Ho C, Ladu S, Lee SA, Mattu S, Destefanis G, Delogu S, Zimmermann A, Ericsson JA, Brozzetti S, Staniscia T, Chen X, Dombrowski F, Evert M (2011) Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology 140 (3): 1071–1083.
Calvisi DF, Frau M, Tomasi ML, Feo F, Pascale RM (2012) Deregulation of signalling pathways in prognostic subtypes of hepatocellular carcinoma: Novel insights from interspecies comparison. Biochim Biophys Acta 1826 (1): 215–237.
Chen BP, Uematsu N, Kobayashi J, Lerenthal Y, Krempler A, Yajima H, Löbrich M, Shiloh Y, Chen DJ (2007) ATM is essential for DNA-PKcs phosphorylations at T2609 cluster upon DNA double strand break. J Biol Chem 282 (9): 6582–6587.
Dai C, Whitesell L, Rogers AB, Lindquist S (2007) Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell 130 (6): 1005–1018.
Ding Q, Reddy YV, Wang W, Woods T, Douglas P, Ramsden DA, Lees-Miller SP, Meek K (2003) Autophosphorylation of the catalytic subunit of the DNA-dependent protein kinase is required for efficient end processing during DNA double-strand break repair. Mol Cell Biol 23 (16): 5836–5848.
Dragoi AM, Fu X, Ivanov S, Zhang P, Sheng L, Wu D, Li GC, Chu WM (2005) DNA-PKcs, but not TLR9, is required for activation of Akt by CpG-DNA. EMBO J 24 (4): 779–789.
Feng J, Park J, Cron P, Hess D, Hemmings BA (2004) Identification of a PKB/Akt hydrophobic motif Ser-473 kinase as DNA-dependent protein kinase. J Biol Chem 279 (39): 41189–41196.
Fernandez-Capetillo O, Celeste A, Nussenzweig A (2003) Focusing on foci: H2AX and the recruitment of DNA-damage response factors. Cell Cycle 2 (5): 426–427.
Frau M, Ladu S, Calvisi DF, Simile MM, Bonelli P, Daino L, Tomasi ML, Seddaiu MA, Feo F, Pascale RM (2011) Mybl2 expression is under genetic control and contributes to determine a hepatocellular carcinoma susceptible phenotype. J Hepatol 55 (1): 111–119.
Garcia-Canton C, Anadón A, Meredith C (2012) γH2AX as a novel endpoint to detect DNA damage: applications for the assessment of the in vitro genotoxicity of cigarette smoke. Toxicol In Vitro 26 (7): 1075–1086.
Hosoi Y, Watanabe T, Nakagawa K, Matsumoto Y, Enomoto A, Morita A, Nagawa H, Suzuki N (2004) Upregulation of DNA-dependent protein kinase activity and Sp1 in colorectal cancer. Int J Oncol 25 (2): 461–468.
Hsu FM, Zhang S, Chen BP (2012) Role of DNA-dependent protein kinase catalytic subunit in cancer development and treatment. Transl Cancer Res 1 (1): 22–34.
Lee HS, Yang HK, Kim WH, Choe G (2005) Loss of DNA-dependent protein kinase catalytic subunit (DNA-PKcs) expression in gastric cancers. Cancer Res Treat 37 (2): 98–102.
Lee JS, Chu IS, Heo J, Calvisi DF, Sun Z, Roskams T, Durnez A, Demetris AJ, Thorgeirsson SS (2004) Classification and prediction of survival in hepatocellular carcinoma by gene expression profiling. Hepatology 40 (3): 667–676.
Lee KJ, Lin YF, Chou HY, Yajima H, Fattah KR, Lee SC, Chen BP (2011) Involvement of DNA-dependent protein kinase in normal cell cycle progression through mitosis. J Biol Chem 286 (14): 12796–12802.
Luceri C, De Filippo C, Caderni G, Gambacciani L, Salvadori M, Giannini A, Dolara P (2000) Detection of somatic DNA alterations in azoxymethane-induced F344 rat colon tumours by random amplified polymorphic DNA analysis. Carcinogenesis 21 (9): 1753–1756.
Mukherjee B, McEllin B, Camacho CV, Tomimatsu N, Sirasanagandala S, Nannepaga S, Hatanpaa KJ, Mickey B, Madden C, Maher E, Boothman DA, Furnari F, Cavenee WK, Bachoo RM, Burma S (2009) EGFRvIII and DNA double-strand break repair: a molecular mechanism for radioresistance in glioblastoma. Cancer Res 69 (10): 4252–4259.
Noguchi T, Shibata T, Fumoto S, Uchida Y, Mueller W, Takeno S (2002) DNA-PKcs expression in esophageal cancer as a predictor for chemoradiation therapeutic sensitivity. Ann Surg Oncol 9 (10): 1017–1022.
Nueda A, Hudson F, Mivechi NF, Dynan WS (1999) DNA-PK protects against heat-induced apoptosis. J Biol Chem 274 (21): 14988–14996.
Park EJ, Chan DW, Park JH, Oettinger MA, Kwon J (2003) DNA-PK is activated by nucleosomes and phosphorylates H2AX within the nucleosomes in an acetylation-dependent manner. Nucleic Acids Res 31 (23): 6819–6827.
Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A, McLendon R, Rasheed BA, Keir S, Nikolskaya T, Nikolsky Y, Busam DA, Tekleab H, Diaz LA, Hartigan J, Smith DR, Strausberg SRL, Marie K, Shinjo SM, Yan H, Riggins GJ, Bigner DD, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321 (5897): 1807–1812.
Pascale RM, Simile MM, De Miglio MR, Muroni MR, Calvisi DF, Asara G, Casabona D, Frau M, Seddaiu MA, Feo F (2002) Cell cycle deregulation in liver lesions of rats with and without genetic predisposition to hepatocarcinogenesis. Hepatology 35 (6): 1341–1350.
Peterson SR, Jesch SA, Chamberlin TN, Dvir A, Rabindran SK, Wu C, Dynan WS (1995) Stimulation of the DNA-dependent protein kinase by RNA polymerase II transcriptional activator proteins. J Biol Chem 270 (3): 1449–1454.
Shang ZF, Huang B, Xu QZ, Zhang SM, Fan R, Liu XD, Wang Y, Zhou PK (2010) Inactivation of DNA-dependent protein kinase leads to spindle disruption and mitotic catastrophe with attenuated checkpoint protein 2 phosphorylation in response to DNA damage. Cancer Res 70 (9): 3657–3666.
Shao SL, Cai Y, Wang QH, Yan LJ, Zhao XY, Wang LX (2007) Expression of GLUT-1, p63 and DNA-Pkcs in serous ovarian tumours and their significance. Zhonghua Zhong Liu Za Zhi 29 (9): 697–700.
Shintani S, Mihara M, Li C, Nakahara Y, Hino S, Nakashiro K, Hamakawa H (2003) Upregulation of DNA-dependent protein kinase correlates with radiation resistance in oral squamous cell carcinoma. Cancer Sci 94 (10): 894–900.
Söderlund Leifler K, Queseth S, Fornander T, Askmalm MS (2010) Low expression of Ku70/80, but high expression of DNA-PKcs, predict good response to radiotherapy in early breast cancer. Int J Oncol 37 (6): 1547–1554.
Tonotsuka N, Hosoi Y, Miyazaki S, Miyata G, Sugawara K, Mori T, Ouchi N, Satomi S, Matsumoto Y, Nakagawa K, Miyagawa K, Ono T (2006) Heterogeneous expression of DNA-dependent protein kinase in esophageal cancer and normal epithelium. Int J Mol Med 18 (3): 441–447.
Villanueva A, Newell P, Chiang DY, Friedman SL, Llovet JM (2007) Genomics and signaling pathways in hepatocellular carcinoma. Semin Liver Dis 27 (1): 55–76.
Weterings E, Chen DJ (2008) The endless tale of non-homologous end-joining. Cell Res 18 (1): 114–124.
Wong RH, Chang I, Hudak CS, Hyun S, Kwan HY, Sul HS (2009) A role of DNA-PK for the metabolic gene regulation in response to insulin. Cell 136 (6): 1056–1072.
Xing J, Wu X, Vaporciyan AA, Spitz MR, Gu J (2008) Prognostic significance of ataxia-telangiectasia mutated, DNA-dependent protein kinase catalytic subunit, and Ku heterodimeric regulatory complex 86-kD subunit expression in patients with non-small cell lung cancer. Cancer 112 (12): 2756–2764.
Yajima H, Lee KJ, Chen BP (2006) ATR-dependent phosphorylation of DNA-dependent protein kinase catalytic subunit in response to UV-induced replication stress. Mol Cell Biol 26 (20): 7520–7528.
Acknowledgements
This work was supported by Grants from Associazione Italiana Ricerche sul Cancro, Ministero dell’Istruzione Università e Ricerca and the Forschungsgemeinschaft DFG (German Research Foundation, Grant number Ev168/2-1 and Do622/2-1).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Evert, M., Frau, M., Tomasi, M. et al. Deregulation of DNA-dependent protein kinase catalytic subunit contributes to human hepatocarcinogenesis development and has a putative prognostic value. Br J Cancer 109, 2654–2664 (2013). https://doi.org/10.1038/bjc.2013.606
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2013.606
Keywords
This article is cited by
-
Role of PRKDC in cancer initiation, progression, and treatment
Cancer Cell International (2021)
-
A gene module identification algorithm and its applications to identify gene modules and key genes of hepatocellular carcinoma
Scientific Reports (2021)
-
From NASH to HCC: current concepts and future challenges
Nature Reviews Gastroenterology & Hepatology (2019)
-
Tumor DNA-dependent protein kinase catalytic subunit expression is associated with hepatitis B surface antigen status and tumor progression in patients with hepatocellular carcinoma
Scientific Reports (2018)
-
XLF-mediated NHEJ activity in hepatocellular carcinoma therapy resistance
BMC Cancer (2017)
