
Abstract
Background:
Ligands of transmembrane receptor tyrosine kinases have important roles in cell proliferation, survival, migration and differentiation in solid tumours. We conducted this study to evaluate the relationship between concentration of serum ligands and prognosis of patients with metastatic colorectal cancer (mCRC) treated with anti-epidermal growth factor receptor (EGFR) antibodies.
Methods:
Between August 2008 and August 2011, serum samples were obtained from KRAS wild-type patients who met the inclusion criteria and received an anti-EGFR antibody treatment. Serum concentration of ligands was measured by an enzyme-linked immunosorbent assay, and somatic mutations of KRAS, BRAF, PIK3CA and BRAF were analysed by direct sequencing.
Results:
A total of 103 patients were enrolled in the present study. At the pretreatment serum levels, patients with high levels of hepatocyte growth factor (HGF) had shorter progression-free survival (PFS) and overall survival (OS) compared with those with low levels of HGF (median PFS: 6.4 months vs 4.4 months; P<0.001, median OS: 15.3 months vs 8.0 months; P<0.001, respectively). Patients with high levels of epiregulin (EREG) also had shorter PFS and OS compared with those with low levels of EREG (median PFS: 6.6 months vs 4.9 months; P=0.016, median OS: 13.8 months vs 7.4 months; P=0.048, respectively). In addition, patients whose serum levels of ligands were elevated at progressive disease had shorter PFS and OS compared with other patients.
Conclusions:
Our study indicated that high levels of HGF and EREG were associated with resistance to treatment with anti-EGFR antibodies in KRAS wild-type patients with mCRC. Our findings will contribute to the newly combination therapy on the treatment of anti-EGFR antibodies.
Similar content being viewed by others

Main
Colorectal cancer (CRC) is the second most common cause of cancer in women, the third most common in men and the fourth leading cause of cancer deaths worldwide (Jemal et al, 2011). Anti-EGFR monoclonal antibodies are active drugs, and gene mutations of KRAS codons 12 and 13 are recognised as strong predictive factors for no clinical benefit of anti-EGFR antibody treatment in mCRC (Jonker et al, 2007; Van Cutsem et al, 2009; Douillard et al, 2010). Several biomarkers for response to treatment with anti-EGFR antibodies have been investigated in mCRC. In particular, genomic changes in the EGFR downstream signal pathway, such as KRAS, BRAF, PIK3CA and NRAS, have been demonstrated to be associated with poor response and prognosis in mCRC patients who received anti-EGFR antibody treatment (Loupakis et al, 2010; De Roock W et al, 2010; Mao et al, 2012). However, these gene mutations are present at low frequency in mCRC, so further search for novel biomarkers is required.
EGFR is a receptor tyrosine kinase, activated by the binding of ligand to its extracellular domain, which triggers intracellular signalling through the RAS/RAF/MAPK and PI3K/AKT pathways that subsequently may modulate cell proliferation, adhesion, angiogenesis, migration and survival (Mendelsohn and Baselga, 2006; Scaltriti and Baselga, 2006). EGFR is a member of a family of related growth factor receptor tyrosine kinases that in addition to EGFR (ErbB1) include HER2/neu (ErbB2), HER3 (ErbB3) and HER4 (ErbB4). Eleven ligands have been identified in the ErbB family in humans: EGF, transforming growth factor-α (TGF-α), HB-EGF, betacellulin, amphiregulin (AREG), epiregulin (EREG), epigen and the neuregulins (NRG 1–4) (Yarden, 2001). Ligands binding to ErbB receptors induce the formation of receptor homodimers and heterodimers and activation of the intrinsic kinase domain, resulting in phosphorylation of specific tyrosine residues, which serve as docking sites for a range of proteins, the recruitment of which leads to the activation of intracellular signal pathways (Hynes and Lane, 2005). EREG is known to bind more weakly to EGFR and ErbB4 than EGF but is much more potent than EGF and leads to a prolonged state of receptor activation (Shelly et al, 1998), but the molecular roles of EREG in cancer cells are not well known. Recently, some studies indicated that EREG and AREG expression of mRNA or immunohistochemistry (IHC) in tumour tissues was associated with response to and prognosis for the treatment with cetuximab in mCRC patients (Khambata-Ford et al, 2007; Jacobs et al, 2009). On the other hand, the clinical significance of serum levels of these ligands is still unclear with regard to the treatment with anti-EGFR antibodies in mCRC.
Hepatocyte growth factor/scatter factor (HGF/SF) is a mesenchymal cytokine with a number of biological activities, including mitogenic, motogenic and/or morphogenic properties in epithelial tissues. HGF is also known as an angiogenesis factor by its ability to promote endothelial cell growth, survival and migration (Bussolino et al, 1992). HGF is a significant ligand of MET, which elicits multiple cellular responses regulating cell survival, morphogenesis, adhesion, migration, breakdown of extracellular matrix and angiogenesis (Birchmeier et al, 2003; Lesko and Majka, 2008). Recently, molecular target therapy directed at the HGF/MET signal pathway has been developed in solid tumours, and biomarkers for effectiveness of HGF/MET monoclonal antibody and tyrosine kinase inhibitor therapy are evaluated (Cecchi et al, 2012).
Use of anti-VEGF antibody (bevacizumab) as front-line therapy is the standard treatment for mCRC patients (Hurwitz et al, 2004; Saltz et al, 2008). A treatment that targets ligands of transmembrane receptor tyrosine kinases may be a promising approach in mCRC. We conducted this study to evaluate the relationship between serum concentrations of ligands and efficacy of anti-EGFR antibody treatment.
Materials and Methods
Patients and sample collection
Between August 2008 and August 2011, specimens collected by endoscopic biopsy or surgical resection from 337 patients with advanced CRC were screened for the genomic status of KRAS codons 12 and 13 at the Gastrointestinal Oncology Division in National Cancer Center Hospital. We selected the mCRC patients who received anti-EGFR antibody treatment and whose tumours were KRAS wild-type (codons 12 and 13).
Blood samples in our study were obtained from residual blood samples of previous laboratory tests. Separated serum was stocked at −20 °C at the Biobank at the division of clinical laboratories in National Cancer Center Hospital until use. We selected samples from these patients at two points as follows: (1) within 2 weeks before initiation of the treatment with anti-EGFR antibodies, and (2) within 2 weeks after diagnosis of progressive disease (PD) of anti-EGFR antibodies. In addition, we collected formalin-fixed paraffin-embedded (FFPE) tumour samples and performed genomic analyses of KRAS (codons 61, 146), BRAF (V600E), PIK3CA (exons 9, 20) and NRAS (codons 12/13, 61).
We enrolled the KRAS wild-type patients who met the inclusion criteria as follows: pathologically proven adenocarcinoma, metastatic or recurrent CRC, KRAS wild-type patients who had previously received one or more regimens of systemic chemotherapy, an Eastern Cooperative Oncology Group (ECOG) Performance status (PS) of 0–2, no significant abnormality of liver and renal function, patients who received combined chemotherapy or monotherapy with anti-EGFR antibodies, and who showed disease deterioration by computed tomography (CT) after anti-EGFR antibody treatment. Main exclusion criteria included the following: previous chemotherapy targeting the EGF pathway, other duplicated advanced cancer, and metastasis to central nervous system. Patients continued to receive chemotherapy until PD or intolerable toxicity from chemotherapy intervened. The response was evaluated by contrast-enhanced CT every 2–3 months. Patients’ consent for the use of clinical materials was obtained, and this study was undertaken after approval by the institutional review boards.
Elisa
We chose ligands such as EGF, TGF-α, AREG, EREG, NRG, HGF and IGF-1, which was previously associated with resistance to target therapy for EGFR or HER2 in solid tumours.
HGF concentrations in serum were measured using a commercially available enzyme test (Human HGF Quantikine ELISA Kit, DHG00, R&D Systems, Minneapolis, MN, USA), and procedures were as follows: (1) we prepared all reagents, standard dilutions and samples as directed in the product insert, (2) added 150 μl of assay diluent to each well and then 50 μl of standard and sample to each well, (3) covered with a plate sealer and incubated at room temperature for 2 h, (4) aspirated each well and washed, repeating the process three times for a total of four washes, (5) we added 200 μl of HRP conjugate to each well, (6) covered with a new plate sealer, incubated at room temperature for 2 h and aspirated and washed four times, (7) we added 200 μl TMB substrate solution to each well and incubated at room temperature for 30 min with protection from light, (8) we added 50 μl of stop solution to each well, and (9) read at 450 nm within 30 min and set wavelength correction to 540 nm or 570 nm.
Concentrations of EREG in serum were measured using Human epiregulin ELISA kit (CSB-EL007779HU, CUSABIO, Wuhan, China). Procedures were as follows: (1) we prepared all reagents, standards and samples (5 × diluted with sample diluent), (2) added 100 μl of standard and sample per well and covered with a plate sealer and then incubated for 2 h at 37 °C, (3) removed the liquid from each well, (4) added 100 μl of Biotin-antibody (1 × ) to each well and covered with a plate sealer and then incubated for 1 h at 37 °C, (5) aspirated each well and washed, repeating the process two times for a total of three washes, (6) added 100 μl of HRP-avidin (1 × ) to each well and covered with a plate sealer and incubated for 1 h at 37 °C, (7) repeated the aspiration/wash process (for) five times, (8) added 90 μl of TMB substrate to each well and incubated for 15–30 min at 37 °C with protection from light, and (9) added 50 μl of Stop Solution to each well, read at 450 nm within 30 min and set wavelength correction to 540 nm or 570 nm.
We used ELISA kits to measure serum levels of ligands as follows: Uscn Life Science Inc. (Wuhan, China) (E90006Hu) for AREG, R&D (DEG00) for EGF, R&D (DTGA00) for TGF-α, CUSABIO (CSB-E17153h) for NRG and R&D (DG00) for IGF-1.
Direct sequencing of KRAS, BRAF, PIK3CA and NRAS
DNA samples were extracted from FFPE tumour tissue sections. Tumour cell-rich area in the H/E section was marked under a microscope, and tissue was scraped from the corresponding area of another deparaffinised unstained section. DNA from the scraped-off tissue sample was isolated using the QIAamp DNA FFPE Tissue Kit (QIAGEN KK, Tokyo, Japan). Exon 2 (codons 12, 13), exon 3 (codon 61), exon 4 (codon 146) of KRAS gene and exon 15 (codon 600) of BRAF gene and exon 9 (codons 542, 545), exon 20 (codon 1047) of PIK3CA gene and exon 2 (codons 12, 13) and exon 3 (codon 61) of NRAS gene were amplified by PCR (the GeneAmp PCR System 9700 thermal cycler, Applied Biosystems, Foster City, CA, USA). The PCR products were visualised using agarose gel electrophoresis with ethidium bromide staining and directly sequenced using an ABI 3130x/Genetic Analyzer (Life Technologies Japan (Applied Biosystems), Tokyo, Japan) according to the manufacturer’s instructions.
Assessment and statistical analysis
To assess the associations of ligand protein with the objective response rate (ORR), disease control rate (DCR), progression-free survival (PFS) and overall survival (OS), the values for each ligand were categorised into low and high values with respect to the median. The efficacy consisted of RR, DCR, PFS and OS. Assessment of therapeutic response consisted of complete response (CR), partial response (PR), stable disease (SD), PD and not evaluated (NE), according to the Response Evaluation Criteria in Solid Tumors criteria ver. 1.0. ORR was defined as the proportion of patients whose best response was a CR or PR among all patients. DCR was defined as the proportion of patients whose best response was a CR, a PR or SD. PFS was defined as the interval from initiation of anti-EGFR therapy to the occurrence of PD or death without evidence of progression. OS was defined as the interval from initiation of anti-EGFR therapy to death or last follow-up.
Differences in the distribution of variables were evaluated using the Fisher’s exact test or χ2-test, as appropriate. PFS and OS were estimated by the Kaplan–Meier method, and survival curves were compared by the log-rank test. All tests were two-sided, and a P-value <0.05 was defined as statistically significant. We estimated hazard ratios (HRs) and the corresponding 95% confidence intervals (CIs) for PFS and OS using univariate and multivariate analysis by Cox proportional hazards model. Variables of patients’ background included age, gender, ECOG PS, histological type, primary site, stage and metastatic sites. Receiver operating characteristics (ROC) curve analysis was performed to determine the optimal cutoff values for serum ligands as continuous variables.
We performed statistical analyses by the SPSS statistical software, version 19 (IBM, Tokyo, Japan).
Results
A total of 113 KRAS wild-type patients met the selection criteria between August 2008 and August 2011 in our hospital. Background characteristics are summarised in Table 1. As previous chemotherapy, all patients received the standard fluoropyrimidine-based chemotherapy regimen, 112 of the 113 patients (99.0%) received oxaliplatin, and 94 of the 113 patients (91.3%) received irinotecan. Thirty-two patients (31.1%) received subsequent treatment after treatment with anti-EGFR antibodies. Nine patients were enrolled in phase I trials, and five patients received hepatic arterial infusion chemotherapy of 5-fluorouracil for liver metastases. Three patients received a combination of irinotecan plus mitomycin C, and one patient received immunotherapy. Fourteen patients received previous chemotherapy, which had been withdrawn due to intolerable adverse events.
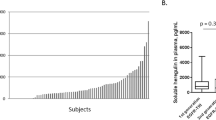
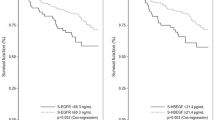
Serum samples from 103 patients were used to measure the concentrations of ligands. Ten patients were excluded, because serum samples at two points were insufficient to analyse by ELISA. Results of serum levels of ligands are shown in Figure 1 (graphs) and Supplementary Data 1 (Table). We evaluated the prognostic role of these ligands by log-rank test. When the cutoff values set the median of serum levels of ligands, serum HGF and EREG were associated with prognosis on the treatment of anti-EGFR antibodies. Survival curves estimated by the Kaplan–Meier method are shown in Figure 2. We also evaluated the prognostic roles of other ligands, but there was no significant correlation between the serum concentration of each ligand (cutoff value: median) and clinical outcome such as PFS (Supplementary Data 2).

Graphical representations of serum concentrations of ligands measured by ELISA. Individual data of serum EREG, HGF, EGF, AREG, NRG, IGF-1 and TGF-α are summarized by graphs. Blue bars show serum levels at pre-treatment and red bars show those at progression disease.

Survival curves of pretreatment hepatocyte growth factor (HGF) and epiregulin (EREG) levels among KRAS wild-type patients and all wild-type of KRAS, BRAF, PIK3CA and NRAS. Survival curves of PFS in terms of HGF levels are shown among (A) KRAS wild-type patients and (B) among all wild-type patients. Survival curves of PFS in terms of EREG levels are shown among (C) KRAS wild-type patients and (D) among all wild-type patients. Among KRAS wild-type patients, survival curves of OS in (E) HGF levels and (F) EREG levels are shown. Patients with low levels of ligands had longer OS compared with patients with high levels.
We divided 103 patients into two subgroups of high and low levels, respectively, of the pretreatment serum concentrations of HGF and EREG. There were no significant differences in patients’ characteristics between subgroups of each HGF and EREG levels (Supplementary Data 3).
Serum levels of HGF and EREG, genomic change of KRAS, BRAF, PIK3CA and NRAS
Serum concentrations of HGF and EREG were compared between subgroups of high/low levels and genomic mutations of KRAS, BRAF, PIK3CA and NRAS are shown in Table 2. The median serum concentration of HGF in patients whose pretreatment HGF levels were high was elevated at PD (pretreatment: 1672.5 pg ml−1, PD: 1990.2 pg ml−1). In contrast, in patients whose pretreatment HGF levels were low, the median HGF levels at PD were similar to pretreatment levels (pretreatment: 1154.8 pg ml−1, PD: 1166.0 pg ml−1). The median serum concentration of EREG was elevated at PD compared with pretreatment levels in patients whose pretreatment EREG levels were high (pretreatment: 1662.5 pg ml−1, PD: 2188.4 pg ml−1). The median of EREG levels at PD were similar to pretreatment levels in patients whose pretreatment EREG levels were low (pretreatment: 1,175.0 pg ml−1, PD: 1,274.9 pg ml−1). A total of 103 KRAS wild-type patients were investigated for genomic changes in KRAS, BRAF, PIK3CA and NRAS. Mutations of KRAS codon 61 (N=2, 1.9%), KRAS codon 146 (N=5, 4.9%), BRAF V600E (N=2, 1.9%), PIK3CA exon9 (N=4, 3.9%), NRAS codon 12/13 (N=2, 1.9%), and NRAS codon 61 (N=5, 4.9%) were detected. In two patients, there were concomitant mutations of KRAS codon 146 and PIK3CA exon 9 and NRAS codon 61and PIK3CA exon 9. Patients with genomic mutations achieved no response to anti-EGFR antibodies (SD: N=6, PD: N=12).
PFS and OS by pretreatment levels of HGF and EREG in serum
Survival curves of PFS according to pretreatment levels of HGF and EREG are shown in Figures 1A–D. Among the 103 KRAS wild-type patients, patients with low levels of serum HGF had significantly longer PFS compared with those with high levels of serum HGF (median: 6.4 vs 4.4 months, HR: 0.479, 95% CI: 0.313–0.732, P=0.00049). Patients with low levels of serum EREG also had significantly longer PFS compared with those with high levels of serum EREG (median: 6.6 vs 4.9 months, HR: 0.618, 95% CI: 0.416–0.917, P=0.016). Among the 57 all-wild-type patients, patients with low levels of serum HGF had significantly longer PFS compared with those with high levels of serum HGF (median: 10.0 months vs 5.5 months, HR: 0.362, 95% CI: 0.199–0.659, P=0.00053). Patients with low levels of serum EREG also had significantly longer PFS compared with those with high levels of serum EREG (median: 8.0 vs 5.0 months, HR: 0.584, 95%CI: 0.337–0.991, P=0.047).
Survival curves of OS according to pretreatment serum levels of HGF and EREG are shown in Figures 1E and F. Among KRAS wild-type patients, those with low levels of serum HGF had significantly longer PFS compared with those with high levels of serum HGF (median: 15.3 vs 8.0 months, HR: 0.425, 95% CI: 0.261–0.690, P=0.00065). Patients with low levels of serum EREG also had significantly longer PFS compared with those with high levels of serum EREG (median: 13.8 vs 7.4 months, HR: 0.621, 95%CI: 0.364–0.962, P=0.035).
ORR and DCR by subgroups of HGF and EREG levels in serum
ORR and DCR of the treatment with anti-EGFR antibodies according to serum levels of HGF and EREG are shown in Table 3. Among KRAS wild-type patients who were enrolled in this study, 34 patients and 37 patients achieved PR and SD, respectively, and 32 patients had no response to anti-EGFR antibodies. ORR and DCR were 33.0% (95% CI: 24.1–43.0) and 68.9% (59.1–77.7), respectively. There was no significant difference in ORR between subgroups of high HGF levels and low HGF levels (30.8% vs 35.3%, P=0.678). There also was no significant difference, but a better response rate was observed in patients with low EREG compared with those in the high EREG group (40.4% vs 23.5%, P=0.091).
Among all-wild-type patients, ORR and DCR were 42.1% (95% CI: 29.1–55.9) and 80.7% (68.1–90.0%), respectively, which were better than the results for KRAS wild-type patients. There were significant better ORR and DCR in patients with low EREG compared with those with high EREG (ORR: 56.6 vs 25.9%, P=0.035; DCR: 93.3 vs 66.6%, P=0.017).
Prognosis by pretreatment levels of HGF and EREG and change of serum levels after treatment
Survival curves by pretreatment levels of HGF and EREG (high/low) and change of serum levels at PD compared with pretreatment (elevation/no elevation) levels are shown in Figure 3. In common for HGF and EREG, patients whose pretreatment levels were low and whose serum levels were not elevated at PD tended to have longer PFS and OS compared with other subgroups. Even if serum concentration at pretreatment was low, patients with elevated serum levels at PD tended to have shorter prognosis compared with patients whose serum levels were not elevated.

Survival curves by the change of serum levels of ligands after treatment. Survival curves of PFS by pretreatment levels of HGF and EREG (high/low) and change of serum levels at PD compared with pretreatment (elevation/no elevation) levels are shown. (A) PFS curves divided by change of serum HGF levels. (B) PFS curves divided by change of serum EREG levels. (C) OS curves divided by change of serum HGF levels. (D) OS curves divided by change of serum EREG levels.
Appropriate cutoff values of serum EREG and HGF that were evaluated by ROC curve analysis
Among KRAS wild-type patients, we evaluated the appropriate cutoff values of serum ligands to set it so that it became a DCR best. As a result, cutoff values of serum EREG and HGF were 1412.65 pg ml−1 (sensitivity: 0.719, specificity: 0.549) and 1393.55 pg ml−1 (sensitivity: 0.620, specificity: 0.656), respectively. At this cutoff values of serum EREG and HGF, ORR and DCR are summarised in Table 4. There were significant better ORR and DCR in patients with low EREG compared with those with high EREG (ORR: 43.8 vs 23.6%, P=0.03; DCR: 83.3 vs 56.4%, P=0.003). There was no significant difference in ORR, but significant better DCR in patients with low HGF compared with those with high HGF (DCR: 78.6 vs 57.4%, P=0.021). Survival curves of PFS are shown in Supplementary Data 4. Patients with low levels of serum ligands had significantly longer PFS compared with those with high levels of serum ligands.
Univariate and multivariate analyses in terms of PFS and OS in KRAS-WT patients
Results of univariate and multivariate analyses of prognosis in terms of PFS and OS are shown in Table 5. Multivariate analyses of our study showed that HGF and EREG levels in serum were prognostic factors in PFS (HR: 0.562, 95% CI: 0.591–0.878, P<0.001; HR: 0.646, 95% CI: 0.426–0.976, P=0.039, respectively). ECOG PS and HGF levels in serum were prognostic factors in OS (HR: 5.476, 95% CI: 2.029–14.776, P<0.001; HR: 0.447, 95% CI: 0.267–0.747, P=0.002, respectively).
Discussion
Among the several ligands that stimulate EGFR, c-met and IGF-1 R, our study revealed that pretreatment serum concentrations of HGF and EREG had prognostic roles in the treatment with anti-EGFR antibodies in KRAS wild-type patients with mCRC. High levels of serum HGF and EREG at pretreatment were associated with shorter PFS and OS, whereas low levels of HGF and EREG were associated with better prognosis in the treatment with anti-EGFR antibodies. The response to anti-EGFR antibodies in terms of ORR and DCR showed no significant difference between serum levels of HGF and EREG but tended to be better in patients with low levels of serum HGF and EREG.
In the present study, we investigated the genomic mutations of KRAS, BRAF, PIK3CA and NRAS, because these mutations cause permanent activation of further downstream signal pathways and were known to be negative biomarkers for response to anti-EGFR antibody treatment (De Roock et al, 2010). In recent clinical trial of anti-EGFR antibodies in mCRC patients, RAS wild-type patients (KRAS, NRAS) are known to obtain clinical benefit of anti-EGFR antibodies compared with only KRAS wild-type patients. Among patients with all wild type of these mutations, we could separately evaluate the clinical significance of the predictive and prognostic roles of serum ligands. Actually, our results showed that high concentrations of serum HGF and EREG had significant prognostic value in predicting shorter PFS and OS in all wild-type patients. ORRs and DCR in patients with low levels of EREG were better than in patients with high levels of EREG.
To our knowledge, no previous studies in mCRC have evaluated the prognostic role of serum ligands in the treatment with anti-EGFR antibodies by monitoring changes in serum concentrations from initiation of treatment to diagnosis of PD. Our study revealed that elevation of serum concentrations of HGF and EREG at the time of PD were likely to associate with a poor prognosis. This may indicate that the serum levels of these ligands were associated with both early PD and delayed resistance to anti-EGFR antibodies. Previous reports show that the HGF/Met pathway was a significant factor in the development of resistance to EGFR-target therapy. For example, Wheeler et al (2008) reported that cetuximab-resistant cells manifested strong activation of ErbB2, ErbB3 and c-Met. EGFR upregulation promoted increased dimerisation with erbB-2 and erbB-3, leading to their transactivation. Yano et al (2008) reported that HGF induces EGFR-TKI resistance in EGFR mutant lung cancer. Engelman et al (2007) indicated that MET amplification causes gefitinib resistance by driving ErbB3-dependent activation of PI3K in non-small cell lung cancer (NSCLC). Xu et al (2010) indicated that EGFR regulates Met levels and invasiveness through hypoxia-inducible factor-1alpha in NSCLC. In CRC, Bardelli et al (2013) reported that the amplification of the MET receptor drives resistance to anti-EGFR therapies in CRC. These reports support that there is a cross-talk of signal pathways between members of the ErbB family and HGF/Met pathway.
Recently, phase I and II trials of HGF-targeted monoclonal antibodies, such as TAK701, AMG102 (rilotumumab) and SCH900105 (AV299), have been carried out in solid tumours (Meetze et al, 2009). A phase II randomised trial that compared rilotumumab or ganitumab (AMG 479) with panitumumab vs panitumumab alone in KRAS wild-type patients with mCRC revealed that the arm with rilotumumab achieved better response and PFS compared with panitumumab alone (Eng et al, 2011). In this trial, biomarker analyses of Met expression by IHC revealed that there was no association between efficacy and intensity of Met by IHC. Our data showing that high HGF levels in serum were associated with poor prognosis in the treatment with anti-EGFR antibody also support the ongoing clinical trials of HGF target therapy with a combination of anti-EGFR antibodies, and high HGF concentration in serum might be a promising biomarker for predicting prognosis.
EREG is mainly known as a ligand of EGFR and ErbB-4 and induces tyrosine phosphorylation of EGFR, ErbB-2, ErbB-3 and ErbB-4. Bio-pathological features and prognostic roles of EREG were evaluated in some solid tumours, such as oral squamous cell carcinoma, breast cancer, CRC and malignant fibrous histiocytoma (Yamamoto et al, 2004; Shigeishi et al, 2008; Révillion et al, 2008). In CRC, high levels of mRNA for EREG and AREG in tumour tissues at pretreatment are more likely to have anti-tumour activity resulting from cetuximab therapy (Khambata-Ford et al, 2007). This indicates EREG has a high dependency to EGFR and significant ligand for anti-EGFR antibodies treatment. In this report, there was no significant correlation between EREG protein in blood and mRNA levels in tumour tissues. The authors suggested that this might indicate the existence of posttranscriptional regulation of these genes. We thought that serum protein may also be affected by not only tumour tissues but also the normal tissues, and posttranslational modifications such as microRNAs or ubiquitination may be associated with serum levels of ligands (Lu et al, 2007; Mlcochova et al, 2013). From these considerations, mRNA in tumour tissue and levels of serous protein of these ligands may have the different meaning as biomarkers. Both tumour mRNA and serum protein are important to evaluate the mechanism of resistance of chemotherapy in any tumours. Next step, we need to validate the prognostic roles of both mRNA levels in tumour tissues and protein levels in serum on the anti-EGFR antibodies treatment.
Our study revealed that only EREG in ligands of ErbB family was significantly associated with prognosis in mCRC patients who received anti-EGFR antibodies. Anti-EGFR antibodies function as a competitive antagonist that inhibits the binding of ligands to EGFR. Roepstorff et al (2009) previously reported that EGFR ligands differentially affect EGFR endocytosis and recycling. The degree of competitive inhibition by anti-EGFR antibodies is considered to be different by ligand type. We would like to investigate the degree or change of competitive inhibition of EREG compared with other ligands by administrating the anti-EGFR antibodies in CRC cells. In addition, LGR5 – which is a Wnt target gene that has been reported to be a marker for normal stem cells in the intestine – may be expressed in colon cancer cells and may be an important molecule to identify colon cancer stem cells (Vermeulen et al, 2010). Kobayashi et al (2012) indicated that CRC cells with expression of LGR5 interconvert to drug-resistant LGR5-negative cells after chemotherapy. EREG was expressed in these LGR5-positive and -negative cells, and the numbers of tumour cells were decreased by anti-EREG antibody treatment in metastatic models. These reports and our study support the notion that EREG has significant molecular roles and may be a promising molecular therapeutic target in mCRC patients.
The present study has limitations. At first, our study was retrospective, and patients whose blood samples were not stocked were excluded. Second, power is limited with 103 cases of available KRAS wild-type patients. The results in this study may be just due to lack of power. Further validation of the findings is needed through other prospective studies with inhibitors of these ligands.
In conclusion, the present study revealed that serum HGF and EREG levels were associated with resistance to anti-EGFR antibodies in KRAS wild-type patients with mCRC. Target therapy studies regarding the HGF/Met pathway are now ongoing and under evaluation. According to our data, the concentration of serum HGF might be a potential biomarker for prediction of response and prognosis in dual target therapy with anti-EGFR antibodies and HGF/Met inhibitors. The clinical significance of serum EREG in mCRC has been unclear so far, but our data suggest a novel therapeutic strategy of administration of dual monoclonal anti-EGFR antibodies plus EREG antibodies to mCRC patients with high EREG levels in serum or elevation of serum EREG levels after treatment.
References
Bardelli A, Corso S, Bertotti A, Hobor S, Valtorta E, Siravegna G, Sartore-Bianchi A, Scala E, Cassingena A, Zecchin D, Apicella M, Migliardi G, Galimi F, Lauricella C, Zanon C, Perera T, Veronese S, Corti G, Amatu A, Gambacorta M, Diaz LA Jr, Sausen M, Velculescu VE, Comoglio P, Trusolino L, Di Nicolantonio F, Giordano S, Siena S (2013) Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov 3: 658–673.
Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF (2003) Met, metastasis, motility and more. Nat Rev Mol Cell Biol 4: 915–925.
Bussolino F, Di Renzo MF, Ziche M, Bocchietto E, Olivero M, Naldini L, Gaudino G, Tamagnone L, Coffer A, Comoglio PM (1992) Hepatocyte growth factor is a potent angiogenic factor which stimulates endothelial cell motility and growth. J Cell Biol 119: 629–641.
De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V, Papamichael D, Laurent-Puig P, Penault-Llorca F, Rougier P, Vincenzi B, Santini D, Tonini G, Cappuzzo F, Frattini M, Molinari F, Saletti P, De Dosso S, Martini M, Bardelli A, Siena S, Sartore-Bianchi A, Tabernero J, Macarulla T, Di Fiore F, Gangloff AO, Ciardiello F, Pfeiffer P, Qvortrup C, Hansen TP, Van Cutsem E, Piessevaux H, Lambrechts D, Delorenzi M, Tejpar S (2010) Effects of KRAS, BRAF, NRAS and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy–refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol 11: 753–762.
Douillard JY, Siena S, Cassidy J, Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J, Rivera F, Kocákova I, Ruff P, Błasińska-Morawiec M, Šmakal M, Canon JL, Rother M, Oliner KS, Wolf M, Gansert J (2010) Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: the PRIME study. J Clin Oncol 28: 4697–4705.
Eng C, Van Cutsem E, Noware E, Swieboda-Sadlej A, Tebbutt NC, Mitchell EP, Davidenko I, Oliner K, Chen L, Huang J, McCaffery I, Loh E, Smethurst D, Tabernero J (2011) A randomized, phase Ib/II trial of rilotumumab (AMG 102) or ganitumumab (AMG 479) with panitumumab (pmab) versus pmab alone in patients (pts) with wild-type (WT) KRAS metastatic colorectal cancer (mCRC): Primary and biomarker analyses. American Society of Clinical Oncology (ASCO) Annual Meeting: Chicago, IL, USA.
Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, Cappuzzo F, Mok T, Lee C, Johnson BE, Cantley LC, Jänne PA (2007) MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 316: 1039–1043.
Cecchi F, Rabe DC, Bottaro DP (2012) Targeting the HGF/Met signaling pathway in cancer therapy. Expert Opin Ther Targets 16: 553–572.
Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E, Ferrara N, Fyfe G, Rogers B, Ross R, Kabbinavar F (2004) Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 350: 2335–2342.
Hynes NE, Lane HA (2005) ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer 5: 341–354.
Ivenson T, Donehower RC, Davidenko I, Tjulandin S, Deptala A, Harrison M, Loh E, Jiang Y, Oliner K, Dubey S (2011) Safety and efficacy of epirubicin, cisplastin and capecitabine (ECX) plus rilutumab(R) as first-line treatment for unresectable locally advanced (LA) or metastatic (M) gastric or esophagogastric junction (EGJ) adenocarcinoma. European Multidisciplinary Cancer Congress: Stockholm, Sweden.
Jacobs B, De Roock W, Piessevaux H, Van Oirbeek R, Biesmans B, De Schutter J, Fieuws S, Vandesompele J, Peeters M, Van Laethem JL, Humblet Y, Pénault-Llorca F, De Hertogh G, Laurent-Puig P, Van Cutsem E, Tejpar S (2009) Amphiregulin and epiregulin mRNA expression in primary tumors predicts outcome in metastatic colorectal cancer treated with cetuximab. J Clin Oncol 27: 5068–5074.
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D (2011) Global cancer statistics. CA Cancer J Clin 61 (69–90): 2011.
Jonker DJ, O'Callaghan CJ, Karapetis CS, Zalcberg JR, Tu D, Au HJ, Berry SR, Krahn M, Price T, Simes RJ, Tebbutt NC, van Hazel G, Wierzbicki R, Langer C, Moore MJ (2007) Cetuximab for the treatment of colorectal cancer. N Engl J Med 357: 2040–2048.
Khambata-Ford S, Garrett CR, Meropol NJ, Basik M, Harbison CT, Wu S, Wong TW, Huang X, Takimoto CH, Godwin AK, Tan BR, Krishnamurthi SS, Burris HA 3rd, Poplin EA, Hidalgo M, Baselga J, Clark EA, Mauro DJ (2007) Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J Clin Oncol 25: 3230–3237.
Kobayashi S, Yamada-Okabe H, Suzuki M, Natori O, Kato A, Matsubara K, Jau Chen Y, Yamazaki M, Funahashi S, Yoshida K, Hashimoto E, Watanabe Y, Mutoh H, Ashihara M, Kato C, Watanabe T, Yoshikubo T, Tamaoki N, Ochiya T, Kuroda M, Levine AJ, Yamazaki T (2012) LGR5-positive colon cancer stem cells interconvert with drug resistant LGR5-negative cells and are capable of tumor reconstitution. Stem Cells 30: 2631–2644.
Lesko E, Majka M (2008) The biological role of HGF-MET axis in tumor growth and development of metastasis. Front Biosci 13: 1271–1280.
Loupakis F, Ruzzo A, Cremolini C, Vincenzi B, Salvatore L, Santini D, Masi G, Stasi I, Canestrari E, Rulli E, Floriani I, Bencardino K, Galluccio N, Catalano V, Tonini G, Magnani M, Fontanini G, Basolo F, Falcone A, Graziano F (2010) KRAS codon 61, 146 and BRAF mutations predict resistance to cetuximab plus irinotecan in KRAS codon 12 and 13 wild-type metastatic colorectal cancer. Br J Cancer 101: 715–721.
Lu Y, Li X, Liang K, Luwor R, Siddik ZH, Mills GB, Mendelsohn J, Fan Z (2007) Epidermal growth factor receptor (EGFR) ubiquitination as a mechanism of acquired resistance escaping treatment by the anti-EGFR monoclonal antibody cetuximab. Cancer Res 67: 8240–8247.
Mao C, Yang ZY, Hu XF, Chen Q, Tang JL (2012) PIK3CA exon 20 mutations as a potential biomarker for resistance to anti-EGFR monoclonal antibodies in KRAS wild-type metastatic colorectal cancer: a systematic review and meta-analysis. Ann Oncol 23: 1518–1525.
Meetze AK, Boudrow A, Venkataraman S, Medicherla S, Gyuris J, Han M (2009) Preclinical efficacy and pharmacodynamics of SCH 900105 (AV-299) and anti-HGF antibody in an intracranial glioblastoma. AACR-NCI-EORTC International Conference Molecular Target and Cancer Therapeutics: Boston, MA, USA.
Mendelsohn J, Baselga J (2006) Epidermal growth factor receptor targeting in cancer. Semin Oncol 33: 369–385.
Mlcochova J, Faltejskova P, Nemecek R, Svoboda M, Slaby O (2013) MicroRNAs targeting EGFR signalling pathway in colorectal cancer. J Cancer Res Clin Oncol 39: 1615–1624.
Révillion F, Lhotellier V, Hornez L, Bonneterre J, Peyrat JP (2008) ErbB/HER ligands inhuman breast cancer, and relationships with their receptors, the bio-pathological features and prognosis. Ann Oncol 19: 73–80.
Roepstorff K, Grandal MV, Henriksen L, Knudsen SL, Lerdrup M, Grøvdal L, Willumsen BM, van Deurs B (2009) Differential effects of EGFR ligands on endocytic sorting of the receptor. Traffic 10: 1115–1127.
Saltz LB, Clarke S, Díaz-Rubio E, Scheithauer W, Figer A, Wong R, Koski S, Lichinitser M, Yang TS, Rivera F, Couture F, Sirzén F, Cassidy J (2008) Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol 26: 2013–2019.
Scaltriti M, Baselga J (2006) The epidermal growth factor receptor pathway: a model for targeted therapy. Clin Cancer Res 12: 5268–5272.
Shelly M, Pinkas-Kramarski R, Guarino BC, Waterman H, Wang LM, Lyass L, Alimandi M, Kuo A, Bacus SS, Pierce JH, Andrews GC, Yarden Y (1998) Epiregulin is a potent pan-ErbB ligand that preferentially activates heterodimeric receptor complexes. J Biol Chem 273: 10496–10505.
Shigeishi H, Higashikawa K, Hiraoka M, Fujimoto S, Mitani Y, Ohta K, Takechi M, Kamata N (2008) Expression of epiregulin, a novel epidermal growth factor ligand associated with prognosis in human oral squamous cell carcinomas. Oncol Rep 19: 1557–1564.
Van Cutsem E, Köhne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A, D'Haens G, Pintér T, Lim R, Bodoky G, Roh JK, Folprecht G, Ruff P, Stroh C, Tejpar S, Schlichting M, Nippgen J, Rougier P (2009) Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med 360: 1408–1417.
Vermeulen L, De Sousa E, Melo F, van der Heijden M, Cameron K, de Jong JH, Borovski T, Tuynman JB, Todaro M, Merz C, Rodermond H, Sprick MR, Kemper K, Richel DJ, Stassi G, Medema JP (2010) Wnt activity defines colorectal cancer stem cells and is regulated by the microenviroment. Nat Cell Biol 12: 468–476.
Wheeler DL, Huang S, Kruser TJ, Nechrebecki MM, Armstrong EA, Benavente S, Gondi V, Hsu KT, Harari PM (2008) Mechanisms of acquired resistance to cetuximab: role of HER (ErbB) family members. Oncogene 27: 3944–3956.
Xu L, Nilsson MB, Saintigny P, Cascone T, Herynk MH, Du Z, Nikolinakos PG, Yang Y, Prudkin L, Liu D, Lee JJ, Johnson FM, Wong KK, Girard L, Gazdar AF, Minna JD, Kurie JM, Wistuba II, Heymach JV (2010) Epidermal growth factor receptor regulates MET levels and invasiveness through hypoxia-inducible factor-1alpha in non-small cell lung cancer cells. Oncogene 29: 2616–2627.
Yamamoto T, Akisue T, Marui T, Nakatani T, Kawamoto T, Hitora T, Nagira K, Fujita I, Matsumoto K, Kurosaka M (2004) Expression of betacellulin, heparin-binding epidermal growth factor and epiregulin in human malignant fibrous histiocytoma. Anticancer Res 24: 2007–2010.
Yano S, Wang W, Li Q, Matsumoto K, Sakurama H, Nakamura T, Ogino H, Kakiuchi S, Hanibuchi M, Nishioka Y, Uehara H, Mitsudomi T (2008) Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor–activating mutations. Cancer Res 68: 9479–9487.
Yarden Y (2001) The EGFR family and its ligands in human cancer: signalling mechanisms and therapeutic opportunities. Eur J Cancer 37: S3–S8.
Acknowledgements
We appreciate very much the participation of patients and their families in this study and thank all co-investigators for their contributions: Ms Mari Mizoguchi, Ms Hideko Morita (analyses by ELISA), Mrs Eri Onishi (support to clinical research), Ms Karin Yokozawa, and Ms Shoko Nakamura (for preparing serum).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on British Journal of Cancer website
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Takahashi, N., Yamada, Y., Furuta, K. et al. Serum levels of hepatocyte growth factor and epiregulin are associated with the prognosis on anti-EGFR antibody treatment in KRAS wild-type metastatic colorectal cancer. Br J Cancer 110, 2716–2727 (2014). https://doi.org/10.1038/bjc.2014.230
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2014.230
Keywords
This article is cited by
-
Evaluation of the additional prophylactic effect of topical steroid ointment to systemic minocycline against anti-epidermal growth factor antibody-induced skin toxicities in metastatic colorectal cancer treatment
Supportive Care in Cancer (2024)
-
Prognostic and predictive biomarkers for anti-EGFR monoclonal antibody therapy in RAS wild-type metastatic colorectal cancer: a systematic review and meta-analysis
BMC Cancer (2023)
-
Risk factor analysis for anti-epidermal growth factor receptor monoclonal antibody–induced skin toxicities in real-world metastatic colorectal cancer treatment
Supportive Care in Cancer (2023)
-
Cetuximab resistance induced by hepatocyte growth factor is overcome by MET inhibition in KRAS, NRAS, and BRAF wild-type colorectal cancers
Journal of Cancer Research and Clinical Oncology (2022)
-
Mechanistically detailed systems biology modeling of the HGF/Met pathway in hepatocellular carcinoma
npj Systems Biology and Applications (2019)
