
Abstract
Background:
Two strategies to interrogate the insulin growth factor 1 receptor (IGF-1R) pathway were investigated: vertical inhibition with dalotuzumab and MK-2206 or ridaforolimus to potentiate PI3K pathway targeting and horizontal cross-talk inhibition with dalotuzumab and MK-0752 to exert effects against cellular proliferation, angiogenesis, and stem cell propagation.
Methods:
A phase I, multi-cohort dose escalation study was conducted in patients with advanced solid tumours. Patients received dalotuzumab (10 mg kg–1) and escalating doses of MK-2206 (90–200 mg) or escalating doses of dalotuzumab (7.5–10 mg kg–1) and MK-0752 (1800 mg) weekly. Upon maximum tolerated dose determination, patients with low-RAS signature, high-IGF1 expression ovarian cancer were randomised to dalotuzumab/MK-2206 versus dalotuzumab/ridaforolimus, whereas patients with high IGF1/low IGF2 expression colorectal cancer received dalotuzumab/MK-0752.
Results:
A total of 47 patients were enrolled: 29 in part A (18 in the dalotuzumab/MK-2206 arm and 11 in the dalotuzumab/MK-0752 arm) and 18 in part B (6 in each arm). Dose-limiting toxicities (DLTs) for dalotuzumab/MK-2206 included grade 4 neutropenia and grade 3 serum sickness-like reaction, maculopapular rash, and gastrointestinal inflammation. For dalotuzumab/MK-0752, DLTs included grade 3 dehydration, rash, and diarrhoea. Seven patients remained on study for >4 cycles.
Conclusions:
Dalotuzumab/MK-2206 and dalotuzumab/MK-0752 combinations were tolerable. Further developments of prospectively validated predictive biomarkers to aid in patient selection for anti-IGF-1R therapies are needed.
Similar content being viewed by others


Main
The insulin-like growth factor pathway is commonly deregulated in cancer to maintain the malignant phenotype (Pollak, 2008; Gao et al, 2012). Two ligands, insulin growth factor 1 (IGF1) and IGF2, bind the IGF1 receptor (IGF-1R) to induce its auto-phosphorylation. The activated receptor transduces signals downstream through the MAPK and PI3K pathways (Baserga et al, 2003; Pollak, 2008; Gao et al, 2012; Pollak, 2012). High circulating IGF1 levels have been associated with increased risks of prostate (Chan et al, 1998; Rowlands et al, 2009) and colorectal (Ma et al, 1999) cancers, whereas high IGF2 levels have been associated with poor prognosis in patients with osteosarcoma (Avnet et al, 2009). Dalotuzumab (MK-0646) is a humanised IgG1 monoclonal antibody that binds IGF-1R with high affinity (Kd ∼1 nM) and selectively blocks IGF-1R (Pandini et al, 2007; Broussas et al, 2009; Atzori et al, 2011). In a phase I clinical trial of single-agent dalotuzumab, the most common adverse events were fatigue, hyperglycaemia, and back pain. Maximum tolerated dose (MTD) was not reached, and the recommended phase II dose (RP2D) was declared at 10 mg kg–1 per week (Atzori et al, 2011).
Anti-IGF-1R monoclonal antibodies have thus far shown limited success in clinical development (Jassem et al, 2010; Guha, 2013). This lack of significant antitumour activity has partly been attributed to the resistance (Pollak, 2012; Yee, 2012; Guha, 2013). The activation of the PI3K/AKT/mTOR pathway through IGF-1R-independent mechanisms has been described as a basis of resistance to anti-IGF-1R therapy (Hägerstrand et al, 2010; Guha, 2013). AKT and mTOR inhibitors can in turn upregulate IGF-1R via feedback loops, resulting in diminished efficacy of these compounds (Di Cosimo et al, 2005; O'Reilly et al, 2006; Wan et al, 2006; Tabernero et al, 2008). An unbiased RNAi screen for enhancers of dalotuzumab efficacy revealed that blockage of the PI3K/AKT/mTOR pathway achieved the highest synergy, and thus drugs targeting this pathway were the strongest candidates for combination therapy (Di Cosimo et al, 2010).
MK-2206 is an oral, potent allosteric inhibitor of AKT1, AKT2, and AKT3 (IC50 values of 5, 12, and 65 nM, respectively) (Yap et al, 2011). Its prolonged half-life (40–100 h) favoured the evaluation of several schedules in a phase I clinical trial (Biondo et al, 2011; Yap et al, 2011). The RP2Ds were 60 mg on alternating days (Yap et al, 2011) and 135–200 mg weekly (Biondo et al, 2011), with main toxicities of rash, nausea, vomiting, diarrhoea, and hyperglycaemia (Biondo et al, 2011; Yap et al, 2011).
Ridaforolimus is a non-pro-drug rapalog that has been evaluated both orally (Mita et al, 2013) and intravenously (IV; Mita et al, 2008) in phase I clinical trials. The RP2Ds were 40 mg daily (5 days on/2 days off) orally and 12.5 mg daily (5 days every 2 weeks) IV. Main adverse events included mucositis, fatigue, rash, nausea, diarrhoea, myelosuppression, and dyslipidaemia. A phase I clinical trial with dalotuzumab and ridaforolimus reported a RP2D of 10 mg kg–1 per week and 30 mg daily (5 days on/2 days off), respectively (Di Cosimo et al, 2010). Based on the tolerability established in a subsequent phase II study in breast cancer, the doses of dalotuzumab 10 mg kg–1 per week and ridaforolimus 20 mg daily (5 days on/2 day off) were recommended for further evaluation (Ebbinghaus et al, 2011).
The Notch pathway, implicated in the maintenance of tumour-initiating cells and enhancement of angiogenesis (Ranganathan et al, 2011; Aster and Blacklow, 2012), has demonstrated cross-talk with IGF-1R signalling (Eliasz et al, 2010; Medyouf et al, 2011; Gusscott et al, 2012). MK-0752 is an oral, potent, and specific gamma secretase inhibitor that has undergone phase I evaluation to define a RP2D of 1800 mg weekly. The main toxicities were fatigue, nausea, and diarrhoea (Krop et al, 2012). The concomitant activation of Notch and IGF-1R pathways is detected in certain cancer types, such as colorectal cancer (Naumov et al, 2012). In a panel of colorectal cancer cell lines, the combination of MK-0752 and dalotuzumab achieved greater growth inhibition than the individual single agents, with maximum activity in low IGF2 expressers (Naumov et al, 2012).
Based on the above, we conducted an open-label, parallel-dose escalation study evaluating the combinations of dalotuzumab with MK-0752 or MK-2206 in solid tumours with expansion cohorts in biomarker-positive ovarian and colon cancer patients of these phase I doublets at their RP2Ds, and an additional expansion cohort of ovarian cancer with dalotuzumab and ridaforolimus.
Patients and methods
Study design
This was a multicentre open-label phase I, parallel-arm study evaluating dalotuzumab in combination with MK-2206, MK-0752, or ridaforolimus. The primary objective of the study was to define the safety and the MTD of dalotuzumab/MK-2206 and dalotuzumab/MK-0752 doublets, whereas the secondary objective was to explore the preliminary antitumour activity in selected populations of these two regimens, as well as a third regimen of dalotuzumab and ridaforolimus for which a recommended dose for evaluation in part B of this study had been already established. Part A was dose escalation in all comers and part B was dose expansion.
Patient selection
In a retrospective analysis of a randomised phase II/III clinical trial that evaluated dalotuzumab in combination with cetuximab and irinotecan in patients with KRAS wild-type colorectal cancer, high IGF1 expression was associated with longer progression-free survival and overall survival, whereas high IGF2 expression was associated with shorter progression-free survival (Watkins et al, 2012). These findings supported the evaluation of dalotuzumab in combination with MK-0752 in patients with KRAS wild-type colorectal cancer with high IGF1 but low IGF2 expression in the expansion cohort.
A low score for a RAS signature generated in silico from three publicly available RAS pathway profiles (Sweet-Cordero et al, 2005; Bild et al, 2006; Blum et al, 2007) correlated with sensitivity to MK-2206 in a panel of breast cancer cell lines (Loboda et al, 2010). Tumour biopsies from patients with ovarian cancer have been shown to be enriched for co-expression of low RAS signature and high IGF1 expression, potential biomarkers of sensitivity to MK-2206/ridaforolimus and dalotuzumab, respectively (Haines et al, 2012), as shown with dalotuzumab combined with ridaforolimus in patient-derived xenografts (Haines et al, 2012). This preclinical work supported selection of patients with ovarian cancer with low RAS signature and high IGF1 co-expression in the expansion cohort of the arms evaluating dalotuzumab combined with MK-2206 or ridaforolimus.
Patient population
Eligible patients in part A had advanced solid tumours refractory to standard treatment. In part B, patients with KRAS wild-type colorectal cancer, high IGF1 expression but low IGF2 expression received dalotuzumab and MK-0752, whereas patients with platinum-resistant ovarian cancer, low RAS signature, and high IGF1 expression levels were randomised to receive dalotuzumab and MK-2206 arm or dalotuzumab and ridaforolimus arm. Patients must have had good performance status (ECOG 0 or 1) and adequate bone marrow, renal, and hepatic functions and coagulation parameters. Diabetic patients were allowed, provided that their fasting serum glucose was <160 mg dl–1 and HbA1c was ⩽8%. All patients must have had measurable disease per RECIST 1.1 (Eisenhauer et al, 2009), although patients with ovarian cancer in part B were eligible without RECIST measurable disease if they were evaluable by CA125 criteria as defined by the Gynecologic Cancer InterGroup (GCIG) (Rustin et al, 2011). Patients must have recovered from the toxicity from prior surgery or prior treatment-related toxicities. Patients with systemic treatment ⩽4 weeks prior, symptomatic ascites, pleural effusion, unstable brain metastasis, or any other concurrent medical condition that would expose them to a higher risk during the study were excluded. Females of childbearing potential must have had a negative pregnancy test. Patients of childbearing potential must have used adequate contraceptive methods.
Treatment
For the part A dalotuzumab/MK-2206 arm, patients enrolled in sequential cohorts assigned to escalating MK-2206 dose levels (DLs) with a fixed dose of dalotuzumab. Each cohort received a once-weekly dose of dalotuzumab 10 mg kg–1 IV and MK-2206 orally (PO) at one of the following DLs: 90 mg (DL1), 135 mg (DL2), 150 mg (DL2.5), or 200 mg (DL3). In the dalotuzumab/MK-0752 arm, patient cohorts received dalotuzumab 10 mg kg–1 IV and MK-0752 1800 mg PO once weekly (DL1). In both arms, DL1 (dalotuzumab 7.5 mg kg–1+MK-2206 90 mg once weekly or dalotuzumab 7.5 mg kg–1+MK-0752 1800 mg once weekly) could be explored if DL1 was deemed excessively toxic. Treatment cycles were 28 days long. A modified toxicity probability interval method guided the dose escalation (Ji et al, 2010; Ji and Wang, 2013). In part B, up to 12 patients were expected to receive dalotuzumab/MK-0752 at the provisional MTD, whereas up to 24 patients were planned to be randomised to either dalotuzumab/MK-2206 at the provisional MTD or to dalotuzumab 10 mg kg–1 IV once weekly and ridaforolimus 20 mg daily at 5 days per week PO (ratio 1 : 1).
Antitumour activity
Response was assessed by RECIST 1.1. Radiological evaluation was performed every two cycles. For patients with ovarian cancer participating in part B response was assessed by RECIST 1.1 and/or CA125 criteria. Response-evaluable patients had one baseline and at least one post-baseline CT scan that were reviewed independently by the central imaging vendor.
Dose-limiting toxicities
Dose-limiting toxicities (DLTs) were assessed according to National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events (CTCAE), version 4.0 (http://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03_2010-06-14_QuickReference_5x7.pdf). Patients were considered DLT evaluable if they have experienced a DLT during cycle 1 or have completed cycle 1 without a DLT. Any missed dose because of any reason other than toxicity would have made the patient DLT inevaluable.
The following treatment-related toxicities were considered as DLTs if occurring during the first cycle of treatment: grades 4 or 5 haematological toxicities (exception of neutropenia lasting <6 days), grades 3 or 4 neutropenia with fever (>38.5 °C), a persistent increase in QTc interval (>60 ms from baseline), any significant bradycardia, and any drug-related toxicity (regardless of grade) leading to a dose modification in the first cycle or causing a delay of >3 weeks from the next scheduled dose of study medication. Also, any grade 3 or worse non-haematological toxicity was a DLT, with the following exceptions: non-optimally treated grade 3 diarrhoea, vomiting, nausea, or dehydration; transient G3 transaminase elevation (<1 week); transient or non-optimally treated grades 3–4 hyperglycaemia; clinically nonsignificant, treatable, or reversible laboratory abnormalities; dalotuzumab infusion-related reactions; alopecia; inadequately treated hypersensitivity reactions. Treatment was held when patients experienced any toxicity meeting DLT criteria during any cycle of treatment. Treatment was resumed at a reduced dose if the toxicity recovered to grade ⩽1 within 3 weeks; otherwise, the patient was discontinued from the study. A patient who required >2 dose modifications for any single study drug was also discontinued.
Safety
Pretreatment patient evaluation included history and physical examination, ECOG performance status, laboratory analysis (haematology and biochemistry), electrocardiogram, audiometry, and ophthalmologic examination (only in MK-2206 arm). Physical examination and electrocardiogram were repeated at the beginning of each cycle and as clinically indicated. Audiometric assessment was repeated only if any grade ⩾2 ear disorder occurred. Ophthalmologic exam was repeated at treatment discontinuation. The other baseline assessments were repeated weekly before each treatment administration. Adverse events were graded according to the CTCAE 4.0.
Sites and recruitment
Four centres (two in the USA, one in Canada, and one in Israel) participated in part A, and nine centres (seven in the USA, one in Canada, and one in Israel) participated in part B. Three of the nine part B sites are members of Moffitt Cancer Center’s Total Cancer Care (TCC) consortium. TCC also includes a secure biorepository database containing molecular data from 17 hospitals and the Moffitt Cancer Center. Potential part B patients, who had consented to TCC for the purpose of donating to the biorepository, underwent a secure pre-selection process (following established standard operating procedures) to evaluate candidacy for the clinical trial. The study was conducted in accordance with the applicable regulatory requirements. The clinical trial registration number was NCT01243762. All patients provided written informed consent. The study was approved by each centre’s independent ethics committee.
Biomarker evaluation for patient selection
Archival samples from patients participating in the pre-screening study portion of the study were analysed by an external vendor (Almac Diagnostics, Craigavon, UK). After RNA extraction, samples from platinum-resistant ovarian cancer were analysed for IGF1 expression and RAS signature score. Samples from patients with colorectal cancer were analysed for IGF1 and IGF2 expression. The Rosetta/Merck Human RSTA Custom Affymetrix 1.0 microarray (Affymetrix, Santa Clara, CA, USA; GEO accession number GPL6793) was used to evaluate the RAS signature. Specific TaqMan-based assays were designed by Almac Diagnostics to determine the IGF1 and IGF2 mRNA levels by qRT-PCR.
For ovarian cancer samples, biomarker eligibility cut points were chosen conceptually, to broadly exclude potentially resistant tumours. Biomarker positivity was defined as a RAS score below the 50th percentile (⩽ −0.148) and IGF1 expression above the 75th percentile (ΔΔCt⩽1.44) of Almac validation samples. For colorectal cancer samples, biomarker eligibility cut points were based upon data from a retrospective analysis of a phase II/III Merck clinical study in metastatic colon cancer. Biomarker positivity was defined as IGF1 expression above the 75th percentile (ΔΔCt<2.03) and IGF2 expression below the top 10th percentile (ΔΔCt>−2.87). These thresholds could be adjusted during the pre-screening process to ensure that the positivity rate ranged between 15% and 50% of the analysed samples.
Pharmacokinetic analysis
Pre-dose samples were collected weekly during cycle 1 and on day 1 of subsequent cycles. Dalotuzumab concentration in serum was determined by an external lab via ELISA that captured dalotuzumab using the extracellular domain of recombinant human IGF-1R. Murine anti-human IgG FC antibodies conjugated to horseradish peroxidase were used to detect the captured dalotuzumab. The lower detection limit of dalotuzumab was 20 ng ml–1 (Atzori et al, 2011). MK-2206 and MK-0752 plasma concentrations were determined as previously described (Yap et al, 2011; Krop et al, 2012) by high-performance liquid chromatography with tandem mass spectroscopy (HPLC-MS/MS) at Merck Research Labs (West Point, PA, USA). Validation data documented adequate specificity, precision, and accuracy of the HPLC-MS/MS assay used for the study. Ridaforolimus and rapamycin concentrations were assessed in whole blood samples by an external lab using chromatography with tandem mass spectroscopy. Analyses for compartmental modelling were performed using WINNonlin Pro, Version 4.1 (Pharsight Corp, Mountain View, CA, USA) (Mita et al, 2008).
Statistical analysis
Standard descriptive statistics, such as the mean, median, range, and proportion, have been used to summarise the patient characteristics and frequency of adverse events. Categorical variables have been expressed as frequencies and percentages. Continuous variables have been summarised by their mean and range or their mean and s.d.
Results
Patient demographics
From December 2010 to February 2013, 47 patients participated in the clinical trial. Twenty-nine patients participated in part A (18 in the dalotuzumab/MK-2206 arm and 11 in the dalotuzumab/MK-0752 arm). In part B, 258 patients with ovarian cancer participated in the biomarker pre-screening; 32 patients (12.4%) were biomarker positive; among them, 13 signed consent and 12 (4.7%) were eligible and randomised (1 : 1) to receive treatment with dalotuzumab and MK-2206 or dalotuzumab and ridaforolimus. In the dalotuzumab/MK-0752 arm, 158 patients with colorectal cancer participated in the biomarker pre-screening; 37 (23.4%) were biomarker positive, 10 signed informed consent, and 6 (3.8%) finally participated in part B. Table 1 summarises the demographic and disease characteristics of the patients who participated in the study.
Dose escalation (part A)
Dalotuzumab+MK-2206 arm
No DLTs were observed in the three DLT-evaluable patients at DL1 (Table 2). Dose-limiting toxicities were observed in one out of six DLT-evaluable patients at DL2 (grade 3 serum sickness-like reaction) and in two of the three DLT-evaluable patients at DL3 (grade 3 maculopapular rash and grade 4 neutropenia/leukopenia). As DL3 was deemed too toxic, an intermediate DL (DL2.5) was evaluated. As none of the six DLT-evaluable patients at DL2.5 experienced a DLT, DL2.5 was considered the provisional MTD.
Dalotuzumab+MK-0752 arm
At DL1, two of the six DLT-evaluable patients (two additional patients were not DLT-evaluable) experienced DLTs (grade 3 dehydration and grade 3 rash). Based on the modified toxicity probability interval method, three patients were evaluated at DL-1. As none of the three DLT-evaluable patients experienced a DLT at this dose, DL1 was expanded in part B for confirmation of the RP2D as pre-established in the protocol according to the modified toxicity probability method.
Expansion cohorts (part B)
Ovarian cancer cohort
Twelve of 32 (37%) biomarker-eligible patients with recurrent platinum-resistant ovarian cancer participated in part B. One patient was a screening failure. The other 19 biomarker-eligible patients did not participate in the expansion cohort because of clinical deterioration before screening (5 patients), pursuit of other treatment options (4 patients), or inability to undergo screening before study accrual had closed (10 patients). Six biomarker-eligible patients received dalotuzumab/ridaforolimus; six received dalotuzumab/MK-2206. In the dalotuzumab/ridaforolimus arm, none of the three DLT-evaluable patients experienced a DLT; three additional patients were not DLT-evaluable. However, three of the four DLT-evaluable patients in the dalotuzumab/MK-2206 arm at DL2.5 experienced DLTs (grade 3 maculopapular rash in two patients and grade 3 gastrointestinal inflammation in one patient). It seemed that DL2.5 was too toxic; therefore, two additional patients were evaluated at DL2, neither of whom experienced a DLT.
Colorectal cancer cohort
Six of 37 (17%) biomarker-eligible patients with KRAS-wild-type colorectal cancer participated in the expansion cohort of dalotuzumab/MK-0752. Five patients were screening failures. The other 26 biomarker-eligible patients did not participate because of clinical deterioration before screening (2 patients), changed personal circumstances before screening (1 patient), or inability to undergo screening before study accrual had closed (23 patients). DL1 seemed too toxic, as one of the four DLT-evaluable patients experienced a DLT (grade 3 diarrhoea); an additional patient was not DLT-evaluable. Therefore, DL1 was evaluated in an additional patient that experienced a DLT (grade 3 diarrhoea). Based on the toxicity profile of the combination, the difficulty of identifying biomarker-positive patients and the lack of preliminary antitumour activity, this study arm was terminated.
Safety and tolerability
Dalotuzumab+MK-2206 arm
The most common treatment-related adverse event of any grade included fatigue (54%), hyperglycaemia (38%), diarrhoea (29%), dermatological adverse events, including rash (38%), maculopapular rash (29%), and dry skin (29%; Table 3). Hyperglycaemia (17%), maculopapular rash (13%), and fatigue (8%) were the most common grade 3-related adverse events. Twelve patients experienced serious adverse events (SAEs), but only three SAEs were related to study medication (Supplementary Table 1): grade 3 gastrointestinal inflammation and grade 2 anaemia (one patient), both of which resolved ad integrum, and persistent grade 4 neutropenia, for which the outcome could not be assessed (patient was lost to follow-up).
Dalotuzumab+ridaforolimus arm
Among the six patients treated, the most common treatment-related adverse events were stomatitis (three patients), mucosal inflammation (two patients), and infusion-related reaction (two patients) (Table 3). There were three grade 3 treatment-related adverse events: abdominal pain, fatigue, and ECG T-wave inversion (an SAE requiring hospitalisation). Another treatment-related SAE, grade 2 pneumonitis, was also reported. Both SAEs resolved ad integrum but required treatment discontinuation (Supplementary Table 1).
Dalotuzumab+MK-0752 arm
The most common treatment-related adverse events were nausea (65%), diarrhoea (59%), anorexia (59%), fatigue (53%), and vomiting (41%). The most common grade 3 treatment-related events included diarrhoea and dehydration (18% each) (Table 4). Six patients collectively experienced 10 treatment-related SAEs (Supplementary Table 1): infusion-related reaction (one patient); grade 3 diarrhoea, hypokalaemia, and hypophosphataemia (one patient); grade 3 dehydration (two patients); grade 4 vomiting, grade 3 nausea, grade 2 upper abdominal pain (one patient) and grade 3 diarrhoea, despite study drug reduction (one patient), which resolved with sequelae in 12 days. The remaining SAEs resolved ad integrum in ⩽15 days (study drug interrupted for six SAEs and discontinued for one).
Response evaluation
Dalotuzumab+MK-2206 arm
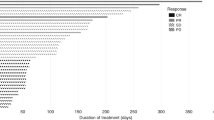
During part A, no partial or complete responses were observed among the 15 evaluable patients. In five patients, tumour remained stable for at least two cycles (Supplementary Figure 1A). Among those, four patients remained on study for four or more cycles (two colorectal cancer, one renal cell cancer, and one uterine leiomyosarcoma; Figure 1A). In part B, none of the four evaluable patients achieved a partial or complete response by RECIST 1.1 or GCIG (Supplementary Figures 1A and 2). Stable disease was reported for two patients, lasting four cycles for one of them. The tumour growth rate before participating in the study is not available; hence, conclusions on the efficacy of this combination cannot be drawn.

Time on treatment for the patients in the dalotuzumab+MK-2206 and dalotuzumab+ridaforolimus ( A ) and dalotuzumab+MK-0752 arm ( B).
Dalotuzumab+ridaforolimus arm
No patient achieved a complete or partial response by RECIST 1.1 or GCIG. Two patients were response evaluable; one experienced disease progression after one cycle of treatment. In the second patient, the disease remained stable for five cycles (Figure 1A). Similarly to the dalotuzumab/MK-2206 arm, the significance in terms of efficacy of this finding is unknown based on the lack of data on tumour growth rate before study participation.
Dalotuzumab+MK-0752 arm
All of the 12 evaluable patients (8 in part A and 4 in part B) experienced disease progression in the first radiological evaluation (Figure 1B and Supplementary Figure 1B).
Pharmacokinetics
Trough concentrations (Ctrough) after the first administration of dalotuzumab (7.5 and 10 mg kg–1) were comparable to those observed in earlier studies at these DLs and generally exceeded the target threshold level of 25 μg ml–1 (Table 5 and Figure 2). Accumulation upon repeated dosing at these DLs was also in line with earlier observations (Figure 2). From a comparison between the tested drug combinations in the study and comparison with earlier studies, no effect of MK-0752, MK-2206, and MK-8669 on the PK of dalotuzumab is apparent, although it should be noted that trough levels are a relatively insensitive measure for such effects (Figure 2).

Dalotuzumab (MK-0646) ( A ), MK-2206 ( B ), and MK-0752 ( C ) pharmacokinetic profile. Ridaforolimus pharmacokinetic profile was not evaluable as a large proportion of samples were obtained post-dose instead of pre-dose.
A dose proportional increase of MK-2206 Ctrough levels was not discernible in patients treated with MK-2206 doses of 90, 135, 150, and 200 mg QW (Table 5 and Figure 2). MK-2206 Ctrough levels were comparable with previous studies. Upon weekly dosing with 1800 mg MK-0752 plasma, MK-0752 trough levels were comparable between the patients receiving 7.5 mg kg–1 and those receiving 10 mg kg–1 dalotuzumab (Table 5 and Figure 2). Owing to notable differences between scheduled pre-dose and actual sampling times (up to 2.5 h post dose) for the patients treated with the MK-8669-dalotuzumab combination, several observations exceeded the range of trough levels expected based on earlier studies (Table 5).
Discussion
Existent IGF-1R antagonists have limited anti-tumour activity as single agents (Haluska et al, 2010; Olmos et al, 2010; Atzori et al, 2011; Becerra et al, 2011; Schmitz et al, 2012; Macaulay et al, 2013; Ray-Coquard et al, 2013); as such, there is a strong interest to combine them with other drugs in an attempt to achieve synergistic effects. Similar to other combination trials involving anti-IGF-1R monoclonal antibodies, the observed adverse events in this study comprised known class-effect toxicities and no new relevant toxicities were described (Calvo et al, 2010; Di Cosimo et al, 2010; Naing et al, 2011; Quek et al, 2011). Still, in part B, the provisional MTDs for both doublets were found to be excessively toxic, requiring further evaluation of lower doses. Dalotuzumab 10 mg kg–1 and MK-2206 135 mg were determined to be the RP2D for this treatment arm, but no RP2D could be defined for the dalotuzumab/MK-0752 arm. Many factors, including the challenges inherent in combining cross-class targeted agents, likely contributed to our observations. As previously described, the determination of MTD for MK-2206 and MK-0752 as single agents has been challenging (Yap et al, 2011; Krop et al, 2012), requiring the evaluation of different treatment schedules to optimise tolerability. The addition of dalotuzumab to each of these agents further complicated MTD delineation for these doublets. These serve to highlight the value of using the modified toxicity probability interval method to better define the MTD in comparison to the classic ‘3+3’ method and the relevant role of expansion cohorts in phase I trials to better characterise toxicity profiles of targeted drugs or combinations (Manji et al, 2013). The tumour types evaluated in the expansion cohorts represent another potential factor accounting for the increased toxicity observed; patients with ovarian and colorectal cancers may be more prone to experience gastrointestinal toxicities because of prior surgical treatments and the possible abdominal involvement by metastatic disease.
This is one of the first reported phase I clinical trials using an IGF-1R monoclonal antibody in combination in which molecular profiling has been used for patient selection. We have shown that the application of a gene expression signature in combination with qRT-PCR is technically and clinically feasible, with average turnaround times from sample submission to result communication of 7 and 8 days for the colorectal and ovarian cancer groups, respectively. With these efforts, the pre-screening positivity rates in both arms were 16% and 26% for ovarian and colorectal groups, respectively. Potential areas of improvement for biomarker selection approach should be reviewed. Contributing factors to the observed differences between the biomarker-eligible rates as reported from the prior studies, the TCC database, and the actual biomarker-positive rate in our study population are multifactorial and include variances in the tissue preparation (formalin-fixed paraffin-embedded vs fresh frozen; macrodissection) and assay platform (microarray vs qPCR).
Selection of the correct biomarker with validated cut points is crucial to the success of biomarker-driven trials. Many factors affect the yield of consistent, clinically applicable results. Recently presented gene expression analysis of patients with ovarian cancer treated with ganitumab (anti-IGF-1R monoclonal antibody) in a phase II study revealed that IGF1 is one of the upregulated genes in the cluster that predicts greater benefit from the treatment (Ray-Coquard et al, 2013). This further supports our selection approach; still, the IGF1/IGF2 expression biomarker used in this study was developed from the tumour samples of patients receiving dalotuzumab combined with cetuximab (Watkins et al, 2012). Thus, it is undetermined whether it has applications to other dalotuzumab-containing combinations. In the dalotuzumab plus MK-0752 arm, the IGF1 and IGF2 expression criteria do not take into account any dependency on the Notch pathway. No Notch pathway biomarker has been prospectively validated, but some recently published potential biomarkers based on protein and gene expression could be evaluated (Zhang et al, 2013). From the lessons learned, the authors charge that biomarker studies validating clinical utility are required. This is further supported by multiple groups, including the National Cancer Institute/Cancer Therapy Evaluation Program (Dancey et al, 2010) or European Organisation for Research and Treatment of Cancer (Daidone et al, 2012).
There are currently several combinations of IGF-1R and mTOR inhibitors under evaluation in patients with breast cancer–dalotuzumab/ridaforolimus (NCT01220570, NCT01234857 (Ebbinghaus et al, 2011) and cixutumumab/temsirolimus (NCT00699491)–as responses were seen in this patient population in the initial dalotuzumab and ridaforolimus studies (Di Cosimo et al, 2010). A study (NCT01708161) evaluating ganitumab and BYL719 (PI3K α-isoform-specific inhibitor) is recruiting patients with breast or ovarian cancer. For both tumour types, only patients with PIK3CA mutations or amplifications are selected (Cao et al, 2013). It remains unknown whether a signature derived from a gene expression profile, such as the one used for patient selection in this study, could better identify activation of the PI3K/AKT/mTOR pathway, in comparison to mutations or amplifications in a single point of the pathway.
Although the high attrition rates are not surprising for a phase I clinical trial population, the accrual rates limited the evaluation of efficacy in the expansion cohorts; no arm had >4 evaluable patients. Among them, no objective response by RECIST was observed, but two patients with ovarian cancer remained on study for at least 4 months. Based on the limited number of patients, ultimately no preliminary conclusion on efficacy or on the predictive value of the current selection strategy can be established.
In conclusion, this study utilises a molecular selection approach in expansion cohorts to obtain additional safety and preliminary efficacy data, in order to facilitate developmental decisions for multiple targeted combinations. The toxicity management has been challenging, especially in the MK-0752 and dalotuzumab arm, and further alterations in treatment schedule will be necessary to overcome this problem. The patient selection process represented another relevant challenge. As molecular selection is becoming more prevalent in the clinic, the development of locally accessible, multiplexed, validated molecular profiling programs is becoming instrumental to perform relevant molecular analyses while the patient is still receiving standard treatment. Although this strategy would miss secondary mutations or changes in the microenvironment occurring because of selective pressure under standard treatment, many of the key molecular alterations would still be present in these initial samples and the results will be readily available upon treatment progression. Thus, patients could be offered tailored treatment in a timely manner.
Accession codes
Change history
11 November 2014
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Aster JC, Blacklow SC (2012) Targeting the Notch pathway: twists and turns on the road to rational therapeutics. J Clin Oncol 30: 2418–2420.
Atzori F, Tabernero J, Cervantes A, Prudkin L, Andreu J, Rodriguez-Braun E, Domingo A, Guijarro J, Gamez C, Rodon J, Di Cosimo S, Brown H, Clark J, Hardwick JS, Beckman RA, Hanley WD, Hsu K, Calvo E, Rosello S, Langdon RB, Baselga J (2011) A phase I pharmacokinetic and pharmacodynamic study of dalotuzumab (MK-0646), an anti-insulin-like growth factor-1 receptor monoclonal antibody, in patients with advanced solid tumors. Clin Cancer Res 17: 6304–6312.
Avnet S, Sciacca L, Salerno M, Gancitano G, Cassarino MF, Longhi A, Zakikhani M, Carboni JM, Gottardis M, Giunti A, Pollak M, Vigneri R, Baldini N (2009) Insulin receptor isoform A and insulin-like growth factor II as additional treatment targets in human osteosarcoma. Cancer Res 69: 2443–2452.
Baserga R, Peruzzi F, Reiss K (2003) The IGF-1 receptor in cancer biology. Int J Cancer 107: 873–877.
Becerra C, Salazar R, Garcia-Carbonero R, Thomas AL, Vazquez-Mazon F, Cassidy J, Maughan T, Gallen M, Iveson T, Hixon M, Gualberto A, Yin D, Bergsland EK, Li D (2011) Phase II trial of figitumumab in patients with refractory, metastatic colorectal cancer (mCRC). 2011 ASCO Annual Meeting; 9 June 2011; Chicago, IL, USA, Abstract 3525.
Bild AH, Yao G, Chang JT, Wang Q, Potti A, Chasse D, Joshi MB, Harpole D, Lancaster JM, Berchuck A, Olson JA Jr, Marks JR, Dressman HK, West M, Nevins JR (2006) Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature 439: 353–357.
Biondo A, Yap TA, Yan L, Patnaik A, Fearen I, Baird RD, Papadopoulos KP, Delgado LM, Taylor A, Lupinacci L, Blackman SC, Decordova S, Tall M, Heaton S, Garrett MD, Sullivan D, De Bono JS, Tolcher AW (2011) Phase I clinical trial of an allosteric AKT inhibitor, MK-2206, using a once weekly (QW) dose regimen in patients with advanced solid tumors. 2011 ASCO Annual Meeting; 20 May 2011; Chicago, IL, USA, Abstract 3037.
Blum R, Elkon R, Yaari S, Zundelevich A, Jacob-Hirsch J, Rechavi G, Shamir R, Kloog Y (2007) Gene expression signature of human cancer cell lines treated with the ras inhibitor salirasib (S-farnesylthiosalicylic acid). Cancer Res 67: 3320–3328.
Broussas M, Dupont J, Gonzalez A, Blaecke A, Fournier M, Corvaia N, Goetsch L (2009) Molecular mechanisms involved in activity of h7C10, a humanized monoclonal antibody, to IGF-1 receptor. Int J Cancer 124: 2281–2293.
Calvo E, Ma W, Tolcher AW, Hidalgo M, Soria J, Bahleda R, Gualberto A, Denis LJ, Millham RD, Adjei AA (2010) Phase (P) I study of PF-00299804 (PF) combined with figitumumab (FI; CP-751,871) in patients (pts) with advanced solid tumors (ASTs). 2010 ASCO Annual Meeting; 14 June 2010; Chicago, IL, USA, Abstract 3026.
Cao ZA, Pinzon-Ortiz M, Chen Y, Li X, Beltran PJ, Gansert JL, Peters M, Schlegel R, Schumacher KM, Huang A (2013) Targeting PIK3CA mutant breast cancer with the combination of PIK3CA-specific inhibitor, BYL719, and IGF1-R antibody, ganitumab. 2013 ASCO Annual Meeting; 17 June 2013; Chicago, IL, USA, Abstract e13525.
Chan JM, Stampfer MJ, Giovannucci E, Gann PH, Ma J, Wilkinson P, Hennekens CH, Pollak M (1998) Plasma insulin-like growth factor-I and prostate cancer risk: a prospective study. Science 279: 563–566.
Daidone MG, Foekens JA, Harbeck N, Martens J, Brunner N, Thomssen C, Hall JA, Salgado R, Dittmer J, Geurts-Moespot A, Duffy MJ, Sweep FCGJ, Schmitt M (2012) Identification, validation and clinical implementation of cancer biomarkers: translational strategies of the EORTC PathoBiology Group. Eur J Cancer 10: 120–127.
Dancey JE, Dobbin KK, Groshen S, Jessup JM, Hruszkewycz AH, Koehler M, Parchment R, Ratain MJ, Shankar LK, Stadler WM, True LD, Gravell A, Grever MR (2010) Guidelines for the development and incorporation of biomarker studies in early clinical trials of novel agents. Clin Cancer Res 16: 1745–1755.
Di Cosimo S, Bendell JC, Cervantes-Ruiperez A, Roda D, Prudkin L, Stein MN, Leighton-Swayze A, Song Y, Ebbinghaus S, Baselga J (2010) A phase I study of the oral mTOR inhibitor ridaforolimus (RIDA) in combination with the IGF-1R antibody dalotozumab (DALO) in patients (pts) with advanced solid tumors. 2010 ASCO Annual Meeting; 14 June 2010, Chicago, IL, USA, Abstract 3008.
Di Cosimo S, Seoane J, Guzman M, Rojo F, Jimenez J, Anido J, Arribas J, Cognetti F, Baselga J (2005) Combination of the mammalian target of rapamycin (mTOR) inhibitor everolimus (E) with the insulin like growth factor-1-receptor (IGF-1-R) inhibitor NVP-AEW-541: a mechanistic based anti-tumor strategy. 2005 ASCO Annual Meeting; 28 May 2005, Orlando, FL, USA, Abstract 3112.
Ebbinghaus S, Blum JL, Cortes J, Rugo HS, Swanton C, Eaton L, Song Y, Zhang T, Baselga J (2011) A phase II study of ridaforolimus (RIDA) and dalotuzumab (DALO) in estrogen receptor-positive (ER+) breast cancer. 2011 ASCO Meeting; 9 June 2011, Chicago, IL, USA, Abstract: TPS110.
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45: 228–247.
Eliasz S, Liang S, Chen Y, De Marco MA, Machek O, Skucha S, Miele L, Bocchetta M (2010) Notch-1 stimulates survival of lung adenocarcinoma cells during hypoxia by activating the IGF-1R pathway. Oncogene 29: 2488–2498.
Gao J, Chang YS, Jallal B, Viner J (2012) Targeting the insulin-like growth factor axis for the development of novel therapeutics in oncology. Cancer Res 72: 3–12.
Guha M (2013) Anticancer IGF1R classes take more knocks. Nat Rev Drug Discov 12: 250–250.
Gusscott S, Kuchenbauer F, Humphries RK, Weng AP (2012) Notch-mediated repression of miR-223 contributes to IGF1R regulation in T-ALL. Leuk Res 36: 905–911.
Hägerstrand D, Lindh MB, Peña C, Garcia-Echeverria C, Nistér M, Hofmann F, Östman A (2010) PI3K/PTEN/Akt pathway status affects the sensitivity of high-grade glioma cell cultures to the insulin-like growth factor-1 receptor inhibitor NVP-AEW541. Neuro-Oncology 12: 967–975.
Haines BB, Wick MJ, Papadopoulos KP, Tolcher AW, Tammam JG, Chaney MF, Nebozhyn M, Zhang T, Ebbinghaus SW, Cheng JD, Zawel L, Sathyanarayanan S (2012) Response biomarkers to IGF1R and mTOR inhibitor combination therapy in ovarian carcinoma. AACR 103rd Annual Meeting; 31 March–4April, Chicago, IL, USA, Abstract 1868.
Haluska P, Worden F, Olmos D, Yin D, Schteingart D, Batzel GN, Paccagnella ML, de Bono JS, Gualberto A, Hammer GD (2010) Safety, tolerability, and pharmacokinetics of the anti-IGF-1R monoclonal antibody figitumumab in patients with refractory adrenocortical carcinoma. Cancer Chemother Pharmacol 65: 765–773.
Jassem J, Langer CJ, Karp DD, Mok T, Benner RJ, Green SJ, Park K, Novello S, Strausz J, Gualberto A (2010) Randomized, open label, phase III trial of figitumumab in combination with paclitaxel and carboplatin versus paclitaxel and carboplatin in patients with non-small cell lung cancer (NSCLC). 2010 ASCO Annual Meeting; 14 June 2010, Chicago, IL, USA, Abstract 7500.
Ji Y, Liu P, Li Y, Bekele BN (2010) A modified toxicity probability interval method for dose-finding trials. Clin Trials 7: 653–663.
Ji Y, Wang SJ (2013) Modified toxicity probability interval design: a safer and more reliable method than the 3+3 design for practical phase I trials. J Clin Oncol 31: 1785–1791.
Krop I, Demuth T, Guthrie T, Wen PY, Mason WP, Chinnaiyan P, Butowski N, Groves MD, Kesari S, Freedman SJ, Blackman S, Watters J, Loboda A, Podtelezhnikov A, Lunceford J, Chen C, Giannotti M, Hing J, Beckman R, LoRusso P (2012) Phase I pharmacologic and pharmacodynamic study of the gamma secretase (Notch) inhibitor MK-0752 in adult patients with advanced solid tumors. J Clin Oncol 30: 2307–2313.
Loboda A, Nebozhyn M, Klinghoffer R, Frazier J, Chastain M, Arthur W, Roberts B, Zhang T, Chenard M, Haines B, Andersen J, Nagashima K, Paweletz C, Lynch B, Feldman I, Dai H, Huang P, Watters J (2010) A gene expression signature of RAS pathway dependence predicts response to PI3K and RAS pathway inhibitors and expands the population of RAS pathway activated tumors. BMC Med Genomics 3: 26.
Ma J, Pollak MN, Giovannucci E, Chan JM, Tao Y, Hennekens CH, Stampfer MJ (1999) Prospective study of colorectal cancer risk in men and plasma levels of insulin-like growth factor (IGF)-I and IGF-binding protein-3. J Natl Cancer Inst 91: 620–625.
Macaulay VM, Middleton MR, Protheroe AS, Tolcher A, Dieras V, Sessa C, Bahleda R, Blay JY, LoRusso P, Mery-Mignard D, Soria JC (2013) Phase I study of humanized monoclonal antibody AVE1642 directed against the type 1 insulin-like growth factor receptor (IGF-1R), administered in combination with anticancer therapies to patients with advanced solid tumors. Ann Oncol 24: 784–791.
Manji A, Brana I, Amir E, Tomlinson G, Tannock IF, Bedard PL, Oza A, Siu LL, Abdul Razak AR (2013) Evolution of clinical trial design in early drug development: systematic review of expansion cohort use in single-agent phase I cancer trials. J Clin Oncol 31: 4260–4267.
Medyouf H, Gusscott S, Wang H, Tseng JC, Wai C, Nemirovsky O, Trumpp A, Pflumio F, Carboni J, Gottardis M, Pollak M, Kung AL, Aster JC, Holzenberger M, Weng AP (2011) High-level IGF1R expression is required for leukemia-initiating cell activity in T-ALL and is supported by Notch signaling. J Exp Med 208: 1809–1822.
Mita MM, Mita AC, Chu QS, Rowinsky EK, Fetterly GJ, Goldston M, Patnaik A, Mathews L, Ricart AD, Mays T, Knowles H, Rivera VM, Kreisberg J, Bedrosian CL, Tolcher AW (2008) Phase I trial of the novel mammalian target of rapamycin inhibitor deforolimus (AP23573; MK-8669) administered intravenously daily for 5 days every 2 weeks to patients with advanced malignancies. J Clin Oncol 26: 361–367.
Mita MM, Poplin E, Britten CD, Tap WD, Rubin EH, Scott BB, Berk L, Rivera VM, Loewy JW, Dodion P, Haluska F, Sarantopoulos J, Mita A, Tolcher A (2013) Phase I/IIa trial of the mammalian target of rapamycin inhibitor ridaforolimus (AP23573; MK-8669) administered orally in patients with refractory or advanced malignancies and sarcoma. Ann Oncol 24: 1104–1111.
Naing A, Kurzrock R, Burger A, Gupta S, Lei X, Busaidy N, Hong D, Chen HX, Doyle LA, Heilbrun LK, Rohren E, Ng C, Chandhasin C, LoRusso P (2011) Phase I trial of cixutumumab combined with temsirolimus in patients with advanced cancer. Clin Cancer Res 17: 6052–6060.
Naumov G, Howard S, Seeley A, Ware C, Reilly J, Cheng J, Sathyanarayanan S (2012) Development of novel combination therapy with dalotuzumab and GSI for the treatment of cetuximab refractory colorectal cancer. 24th EORTC-NCI-AACR Symposium on Molecular Targets and Cancer Therapeutics; November, Dublin, Ireland, Abstract 299.
O'Reilly KE, Rojo F, She Q-B, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, Baselga J, Rosen N (2006) mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res 66: 1500–1508.
Olmos D, Postel-Vinay S, Molife LR, Okuno SH, Schuetze SM, Paccagnella ML, Batzel GN, Yin D, Pritchard-Jones K, Judson I, Worden FP, Gualberto A, Scurr M, de Bono JS, Haluska P (2010) Safety, pharmacokinetics, and preliminary activity of the anti-IGF-1R antibody figitumumab (CP-751,871) in patients with sarcoma and Ewing's sarcoma: a phase 1 expansion cohort study. Lancet Oncol 11: 129–135.
Pandini G, Wurch T, Akla B, Corvaia N, Belfiore A, Goetsch L (2007) Functional responses and in vivo anti-tumour activity of h7C10: a humanised monoclonal antibody with neutralising activity against the insulin-like growth factor-1 (IGF-1) receptor and insulin/IGF-1 hybrid receptors. Eur J Cancer 43: 1318–1327.
Pollak M (2008) Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer 8: 915–928.
Pollak M (2012) The insulin receptor/insulin-like growth factor receptor family as a therapeutic target in oncology. Clin Cancer Res 18: 40–50.
Quek R, Wang Q, Morgan JA, Shapiro GI, Butrynski JE, Ramaiya N, Huftalen T, Jederlinic N, Manola J, Wagner AJ, Demetri GD, George S (2011) Combination mTOR and IGF-1R inhibition: phase I trial of everolimus and figitumumab in patients with advanced sarcomas and other solid tumors. Clin Cancer Res 17: 871–879.
Ranganathan P, Weaver KL, Capobianco AJ (2011) Notch signalling in solid tumours: a little bit of everything but not all the time. Nat Rev Cancer 11: 338–351.
Ray-Coquard I, Haluska P, O'Reilly S, Cottu PH, Elit L, Provencher DM, Beckmann MW, Bosserman LD, Jacod S, Houe V, Loberg RD, Glaspy JA, Karlan B, Slamon DJ, Konecny GE (2013) A multicenter open-label phase II study of the efficacy and safety of ganitumab (AMG 479), a fully human monoclonal antibody against insulin-like growth factor type 1 receptor (IGF-1R) as second-line therapy in patients with recurrent platinum-sensitive ovarian cancer. 2013 ASCO Annual Meeting; 17 June 2013; Chicago, IL, USA, Abstract 5515.
Rowlands M-A, Gunnell D, Harris R, Vatten LJ, Holly JMP, Martin RM (2009) Circulating insulin-like growth factor peptides and prostate cancer risk: a systematic review and meta-analysis. Int J Cancer 124: 2416–2429.
Rustin GJ, Vergote I, Eisenhauer E, Pujade-Lauraine E, Quinn M, Thigpen T, du Bois A, Kristensen G, Jakobsen A, Sagae S, Greven K, Parmar M, Friedlander M, Cervantes A, Vermorken J (2011) Definitions for response and progression in ovarian cancer clinical trials incorporating RECIST 1.1 and CA 125 agreed by the Gynecological Cancer Intergroup (GCIG). Int J Gynecol Cancer 21: 419–423.
Schmitz S, Kaminsky-Forrett MC, Henry S, Zanetta S, Geoffrois L, Bompas E, Moxhon A, Mignion L, Guigay J, Knoops L, Hamoir M, Machiels JP (2012) Phase II study of figitumumab in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck: clinical activity and molecular response (GORTEC 2008-02). Ann Oncol 23: 2153–2161.
Sweet-Cordero A, Mukherjee S, Subramanian A, You H, Roix JJ, Ladd-Acosta C, Mesirov J, Golub TR, Jacks T (2005) An oncogenic KRAS2 expression signature identified by cross-species gene-expression analysis. Nat Genet 37: 48–55.
Tabernero J, Rojo F, Calvo E, Burris H, Judson I, Hazell K, Martinelli E, SRy Cajal, Jones S, Vidal L, Shand N, Macarulla T, Ramos FJ, Dimitrijevic S, Zoellner U, Tang P, Stumm M, Lane HA, Lebwohl D, Baselga J (2008) Dose- and schedule-dependent inhibition of the mammalian target of rapamycin pathway with everolimus: a phase I tumor pharmacodynamic study in patients with advanced solid tumors. J Clin Oncol 26: 1603–1610.
Wan X, Harkavy B, Shen N, Grohar P, Helman LJ (2006) Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene 26: 1932–1940.
Watkins DJ, Ayers M, Cunningham D, Tabernero J, Tejpar S, Kim TY, Kim TW, Kim SY, Roh JK, Beale PJ, Park YS, Guren TK, Loboda A, Nebozhyn M, Mogg R, Lunceford J, Schmidt EV, Brown HM, Sathyanarayanan S, Mauro DJ (2012) Molecular analysis of the randomized phase II/III study of the anti-IGF-1R antibody dalotuzumab (MK-0646) in combination with cetuximab (Cx) and irinotecan (Ir) in the treatment of chemorefractory KRAS wild-type metastatic colorectal cancer (mCRC). 2012 ASCO Annual Meeting; 20 May 2012; Chicago, IL, USA, Abstract 3531.
Yap TA, Yan L, Patnaik A, Fearen I, Olmos D, Papadopoulos K, Baird RD, Delgado L, Taylor A, Lupinacci L, Riisnaes R, Pope LL, Heaton SP, Thomas G, Garrett MD, Sullivan DM, de Bono JS, Tolcher AW (2011) First-in-man clinical trial of the oral pan-AKT inhibitor MK-2206 in patients with advanced solid tumors. J Clin Oncol 29: 4688–4695.
Yee D (2012) Insulin-like growth factor receptor inhibitors: baby or the bathwater? J Natl Cancer Inst 104: 975–981.
Zhang CC, Yan Z, Zong Q, Fang DD, Painter C, Zhang Q, Chen E, Lira ME, John-Baptiste A, Christensen JG (2013) Synergistic effect of the gamma-secretase inhibitor PF-03084014 and docetaxel in breast cancer models. Stem Cells Transl Med 2: 233–242.
Acknowledgements
Total Cancer Care is enabled, in part, by the generous support of the DeBartolo Family, and we thank the many patients who so graciously provided data and tissue to the Total Cancer Care Consortium. We thank Rasa Hamilton, Moffitt Cancer Center, for her editing assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
MBJ and JDC are employees of Merck. AG was an employee of Merck when the study was conducted. PH, JF, and LLS had received research funding from Merck. The remaining authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Brana, I., Berger, R., Golan, T. et al. A parallel-arm phase I trial of the humanised anti-IGF-1R antibody dalotuzumab in combination with the AKT inhibitor MK-2206, the mTOR inhibitor ridaforolimus, or the NOTCH inhibitor MK-0752, in patients with advanced solid tumours. Br J Cancer 111, 1932–1944 (2014). https://doi.org/10.1038/bjc.2014.497
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2014.497
Keywords
This article is cited by
-
Is more better? An analysis of toxicity and response outcomes from dose-finding clinical trials in cancer
BMC Cancer (2021)
-
FoxP3-miR-150-5p/3p suppresses ovarian tumorigenesis via an IGF1R/IRS1 pathway feedback loop
Cell Death & Disease (2021)
-
Targeting cancer stem cell pathways for cancer therapy
Signal Transduction and Targeted Therapy (2020)
-
Dual targeting of IGF-1R and ErbB3 as a potential therapeutic regimen for ovarian cancer
Scientific Reports (2019)
-
A phase I trial of MK-2206 and hydroxychloroquine in patients with advanced solid tumors
Cancer Chemotherapy and Pharmacology (2019)
