
Abstract
Background:
The Par complex – comprising partition-defective 6 (Par6), Par3, and atypical protein kinase C (aPKC) – is crucial for cell polarisation, the loss of which contributes to cancer progression. Transforming growth factor β (TGFβ)-induced phosphorylation of Par6 on the conserved serine 345 is implicated in epithelial-to-mesenchymal transition (EMT) in breast cancer. Here we investigated the importance of phosphorylated Par6 in prostate cancer.
Methods:
We generated a p-Par6345-specific antibody and verified its specificity in vitro. Endogenous p-Par6345 was analysed by immunoblotting in normal human prostate RWPE1 and prostate cancer (PC-3U) cells. Subcellular localisation of p-Par6345 in migrating TGFβ-treated PC-3U cells was analysed by confocal imaging. Invasion assays of TGFβ-treated PC-3U cells were performed. p-Par6 expression was immunohistochemically analysed in prostate cancer tissues.
Results:
TGFβ induced Par6 phosphorylation on Ser345 and its recruitment to the leading edge of the membrane ruffle in migrating PC-3U cells, where it colocalised with aPKCζ. The p-Par6–aPKCζ complex is important for cell migration and invasion, as interference with this complex prevented prostate cancer cell invasion. High levels of activated Par6 correlated with aggressive prostate cancer.
Conclusions:
Increased p-Par6Ser345 levels in aggressive prostate cancer tissues and cells suggest that it could be a useful novel biomarker for predicting prostate cancer progression.
Similar content being viewed by others


Main
Prostate cancer is the most prevalent cancer and the second leading cause of cancer death among males. In the United States in 2012, an estimated 241 740 new cases of prostate cancer and 28 170 deaths from prostate cancer were reported (Siegel et al, 2012). Normal prostate tissue comprises gland units, in which epithelial cells polarise in the apical–basal direction and align into lumen or tubular structures to facilitate secretion (McNeal, 1988; Ware, 1994). This polarised cell organisation maintains proper organ function and regulates cell proliferation. Disturbance of this normal tissue organisation and cell polarity is a hallmark of malignancy (Aranda et al, 2008; Etienne-Manneville, 2008).
Cell polarisation is also essential for cell migration, with a front–rear axis driving cell movement in response to extracellular signals. In the leading edge of a migrating cell (facing the direction of movement), the actin cytoskeleton is reorganised to form membrane ruffling or spike-like filopodia. At the back of the migrating cell, the nucleus localises oriented towards the leading edge, with the microtubule-organising centre (MTOC) and Golgi apparatus positioned in front of the nucleus (Ridley et al, 2003; Etienne-Manneville, 2008).
In epithelial polarity regulation, there are three crucial polarity protein complexes: Par/aPKC, Crumbs, and Scribble. The Par complex – including Par3, Par6, Cdc42, and atypical protein kinase C (aPKC) – is a master regulator of apical–basal polarity, which is involved in almost all metazoan cells. Par6 exists in three mammalian Par6 isoforms (A, B, and C) with conserved protein-interaction domains. The Phox/Bem1 (PB1) domain localises at the N-terminus of Par6 and binds to other PB1 domain-containing proteins (e.g., aPKC), while the C-terminus of Par6 harbours the PSD-95/Dlg/ZO-1 (PDZ) domain that binds Par3. The middle of Par6 contains the Cdc42–Rac interaction binding (CRIB) motif, which binds the Cdc42 or Rac GTPases. Par3 localises in tight junctions at the apical–lateral border, providing anchorage for the Par complex (Joberty et al, 2000; Lin et al, 2000). Par6 functions as a scaffolding protein for Par complex assembly by direct interaction with Par3, aPKC, and Cdc42/Rac1 at the apical–lateral border. Under normal conditions, Par complexes localise in cell junctions to maintain epithelial cell–cell contacts and cell polarity. In neuronal cells, the polarity complex is required for initial axon formation and is recruited to the axon tip in a manner dependent on phosphoinositide 3-kinase (PI3K) (Shi et al, 2003). In cancer cells, mislocalisation and high expression of polarity proteins are reportedly related to cancer progression (Roh and Margolis, 2003; Etienne-Manneville and Hall, 2003b; Aranda et al, 2008).
TGFβ regulates different cellular processes – including cell migration, proliferation, differentiation, apoptosis, and epithelial–mesenchymal transition (EMT) (Massague, 2008; Heldin et al, 2009) – and activates downstream signalling pathways via its type I and type II serine/threonine kinase receptors. For the canonical Smad signalling pathway, TGFβ first binds the constitutively active TGFβ type II receptor (TβRII) and then recruits and phosphorylates the GS domain of the TGFβ type I receptor (TβRI). This activates the TβRI kinase domain, which in turn phosphorylates Smad2/Smad3 (Feng and Derynck, 2005; Massague et al, 2005). TGFβ also activates non-Smad pathways, including the p38/JNK MAPK pathway, Rho-like GTPase signalling pathway, PI3K–AKT–mTOR pathway, and Ras–Erk–MAPK pathway (Moustakas and Heldin, 2005; Mu et al, 2012).
Par6 interacts with the TGFβ receptor complex and is phosphorylated upon TGFβ stimuli (Wang et al, 2003; Ozdamar et al, 2005; Bose and Wrana, 2006). Phosphorylated Par6 has a crucial role in TGFβ-induced EMT, which is reportedly related to breast cancer progression (Viloria-Petit et al, 2009). Sustained TGFβ receptor signalling has also been observed in prostate cancer (Mu et al, 2011), but its role remains unclear.
In the present study, we investigated whether p-Par6 is involved in TGFβ-induced migratory responses in prostate cancer cells. Our results showed that TGFβ induced Par6 phosphorylation, and in turn, p-Par6 formed a complex with aPKC at the leading edge of membrane ruffles, which was important for migration and invasion of prostate cancer cells. Interestingly, prostate cancer cell invasion could be prevented by interfering with polarity complex formation. Moreover, analysis of Par6 signalling in prostate cancer cells and tissues revealed that high levels of p-Par6 correlated to prostate cancer progression.
Materials and methods
Cell culture
The human prostate cancer cell line PC-3U originated from PC-3 (Franzen et al, 1993), grown in RPMI-1640 with 10% foetal bovine serum (FBS) and L-glutamine. The human immortalised normal prostate cell line RPWE1 were purchased from ATCC (Manassas, VA, USA), grown according to the manufacturer’s recommendation in Keratinocyte Serum-Free Medium with bovine pituitary extract and human recombinant epidermal growth factor (GIBCO).
Antibodies and reagents
The rabbit polyclonal p-Par6 antibody was produced in our laboratory and its specificity was tested. For immunoblotting or immunofluorescence staining, we purchased antibodies against Par6 from Abcam (Cambridge, UK), p-PKCζ(H2) and N-cadherin from Santa Cruz Biotechnology (Dallas, TX, USA), E-cadherin from BD Biosciences (Franklin Lakes, NJ, USA), and antibodies against PARD6B(HPA), PKCζ(HPA), β-actin, and β-tubulin from Sigma Aldrich (St Louis, MO, USA). Rabbit anti-phospho-Smad2 antiserum was generated in-house and described previously (Persson et al, 1998). We purchased 4,6-diamidino-2-phenylindole dihydrochloride (DAPI) from Merk (Rahway, NJ, USA), TRITC-labelled phalloidin from Sigma Aldrich; Alexa Fluor 488 and Alexa Fluor 555 from Invitrogen (Carlsbad, CA, USA); LumiLight western blotting substrate and Pefabloc from Roche Diagnostics (Indianapolis, IN, USA); the PageRuler prestained protein ladder from Fermentas (Waltham, MA, USA), and PKCζ pseudosubstrate from Tocris Bioscience (Ellisville, MO, USA).
Western blotting
Cells were grown on 10-cm dishes. After 12–18 h of starvation in medium supplemented with 1% FBS, the indicated inhibitors were added into the media. One hour later, the cells were stimulated with TGFβ1 10 ng ml−1 (R&D System, Minneapolis, MN, USA) for 30 min. Next, the cells were washed once with ice-cold PBS and lysed in ice-cold lysis buffer containing 150 mM NaCl, 50 mM Tris (pH 8.0), 0.5% (v/v) DOC, 1% (v/v) NP40, 10% (v/v) glycerol, 1 mM aprotinin, 1 mM Pefabloc, and 2 mM sodium orthovanadate. After centrifugation, the supernatants were collected and protein concentrations were determined using the BCA Protein Measurement Kit (Nordic Biolabs, Täby, Sweden). Equal amounts of protein from the total cell lysate were run in SDS–PAGE and transferred onto polyvinylidine difluoride membranes. The membranes were blocked for 1 h with 5% BSA in TBS-Tween, followed by overnight incubation at 4 °C with primary antibody. Then the membranes were washed three times with TBS-Tween, incubated for 1 h with a secondary horseradish peroxidase-conjugated antibody, and developed with LumiLight PLUS Western Blot Substrate (Roche Diagnostics).
Western blotting using the fluorescence-based LI-COR detection system
The proteins were transferred to polyvinylidine difluoride membranes, which were then blocked for 1 h with 20 ml of Odyssey blocking solution, followed by overnight incubation at 4 °C with specific antibodies. The next day, the membranes were washed three times with TBS-Tween, followed by a 1-h incubation with IRDye 680RD or 800CW secondary antibodies (LI-COR Bioscience, Lincoln NE, USA) at room temperature. Finally, the membranes were rinsed three times with TBS-Tween and scanned using the infrared imaging scanner (Odyssey CLx Imager; LI-COR Bioscience).
Immunofluorescence staining for cultured cells
Wound-healing assays were performed on serum-starved confluent cells growing on sterile glass microscopy cover slides in six-well plates without coating, by using a 1000-μl pipette tip for scratching. Control cells and migrating cells facing the wound were subjected to immunofluorescence analyses of cytoskeletal rearrangements by actin stainings (phalloidin, which stains F-actin), or for analysis of subcellular localisation of proteins, as indicated in the figure legends. The slides were washed four times with PBS and then fixed in 4% paraformaldehyde for 30 min at room temperature. Next, they were washed four times in PBS and subsequently permeabilised in 0.1% Triton X-100 in PBS for 5 min. After four more washes in PBS, the slides were blocked overnight in 10 mM glycine. The next day, the slides were incubated at room temperature for 1 h in a humid chamber with the primary antibodies. After washing with PBS, the slides were incubated with fluorescent dye-labelled secondary antibodies for 45 min at room temperature, followed by 5-min incubation with DAPI. Thereafter, the mount medium was added on the slides for imaging. The samples were analysed at room temperature using a fluorescence microscope (Axioplan 2; Carl Zeiss Microimaging, Inc., San Diego, CA, USA) with a digital camera (C4742-95; Hamamatsu, Shizuoka, Japan), using a Plan-neofluar × 63 objective lens (Carl Zeiss MicroImaging, Inc.). The primary images were acquired using the camera’s QED software. The analyses of F-actin reorganisation in membrane ruffles and analyses of p-Par6 localisation in membrane ruffles were performed three times and approximately 200–300 cells facing the wound were counted under the microscope in each experiment.
Invasion assay
An invasion assay was performed using the CytoSelect Cell Invasion Assay kit (Cell Biolabs, Inc., San Diego, CA, USA). The collagen layer of the cell culture inserts was rehydrated in 300 μl serum-free RPMI-1640 media. Then 1 × 106 cells were seeded on the upper side of the chambers in 1% serum RPMI-1640 with or without TGFβ and in the absence or presence of inhibitors. The lower wells of the invasion plates were filled with 500 μl RPMI with 10% FBS. Non-invasive cells were removed from the upper chamber, and the invasive cells were photographed with a Leica (Wetzlar, Germany) DMR light microscope after staining with crystal violet cell stain solution. Colorimetric quantification was performed by transferring the inserts into 200 μl of extraction solution for 10 min. Optical density (OD) at 560 nm was measured in a 96-well plate using a Multiskan FC Microplate Photometer (Thermo Scientific, Waltham, MA, USA). Statistical analyses were performed using the SPSS software (Chicago, IL, USA), and P-values were calculated by one-way ANOVA.
Immunofluorescent staining of paraffin-embedded sections
Paraffin-embedded sections of prostate cancer tissue were kindly provided by the Department of Immunology, Genetics and Pathology of the Uppsala University Hospital. Ethical permission to use decoded tumour tissues to generate tissue slides was granted by the Uppsala ethical review board in full agreement with the Swedish Ethical Review Act. The sections were rehydrated twice in xylene for 10 min each; then in 100% ethanol for 10 min; followed by 5-min incubations in 95, 80, and 70% ethanol and deionised H2O; and finally in PBS for 10 min. Thereafter, the sections were treated with Antigen Retrieval Reagent (R&D) at 95 °C for 5 min, rinsed with PBS, blocked in 1% horse serum in PBS for 1 h at room temperature, and then incubated with primary antibodies overnight at 4 °C. After three 5-min washes with PBS, the sections were incubated with NorthernLights secondary antibody (R&D) for 45 min at room temperature. Finally, the sections were subjected to three more 5-min washes with PBS and then mounted with mounting media with DAPI and visualised using a fluorescence microscope (Axioplan 2; Carl Zeiss Microimaging, Inc.).
Tissue microarray (TMA) preparation from whole mounts of prostatectomy specimens and staining procedure
Prostatectomy specimens were obtained from a cohort of 127 patients operated between 1993 and 2002 at the Department of Urology and collected at the Department of Pathology, University Hospital, Uppsala, Sweden. Ethical permission to use decoded tumour tissues for the generation of tissue slides was granted by the Uppsala ethical review board in full agreement with the Swedish Ethical Review Act. From each specimen, the transverse section including the cancerous area with the most representative global Gleason score (GS) was used for TMA preparation. From each case, we took three 1-mm punches from such areas, along with one punch from normal prostate tissue. All cases were carefully reviewed regarding the extent and grade and the representativeness of the punches.
The sections were deparaffinised and retrieved in the above-described manner. Thereafter, the sections were incubated in 3% H2O2/methanol for 10 min, washed twice with PBS, and blocked in 5% normal goat serum for 1 h at room temperature. The sections were then incubated overnight at 4 °C with p-Par6 antibody and with normal rabbit IgG as a negative control. After three 5-min washes with PBS, the sections were incubated with secondary antibody (K4065; DAKO envision system, Glostrup, Denmark) for 45 min at room temperature. After three more washes in PBS, the sections were developed with AEC (SK-4200; Vector Laboratories, Burlingame, CA, USA) and mounted in aqueous mounting medium (H-5501; Vector Laboratories).
Quantitative assessment
Digital images of TMA sections were acquired by scanning with a Panoramic 250 Flash II (3DHistech, Ramsey, NJ, USA) and analysed using the ImageJ software (Lexington, KY, USA). Grey-scale values (pixel intensities) within the regions of interest were measured, and the minimum, maximum, and mean pixel intensity values were calculated to compare the intensities of immunoperoxidase reaction. From each group, we calculated the mean±s.e.m., and Student’s t tests were used to evaluate the between-group differences.
Results
TGFβ-induced p-Par6Ser345 in the leading edge of polarised prostate cancer cells
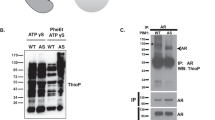
Ozdamar et al (2005) reported that the TGFβ type II receptor (TβRII) phosphorylates the C-terminus of Par6 at residue Ser345, which is important for TGFβ-induced dissolution of tight junctions during EMT. As there is no commercially available phospho-specific antibody for detection of p-Par6, we generated rabbit polyclonal antibodies against p-Par6Ser345 in-house. To verify the specificity of this antibody, Flag-tagged wild-type Par6 or the non-phosphorylated Par6 mutant Flag-tagged Par6Ser345Ala was expressed in human prostate cancer cells (PC-3U), followed by immunoblotting of p-Par6, total Par6, and Flag (Figure 1A). As expected, we observed a TGFβ-induced phosphorylation of the ectopically expressed WT Par6 but not of the Ser345Ala mutant Par6, thus confirming the specificity of the p-Par6Ser345 antibody. Use of the fluorescence-based LI-COR detection system enabled detection of p-Par6Ser345 together with Par6 and Flag antibodies. Similar results were obtained in human embryonic kidney (HEK) 293T cells (Supplementary Figure S1).

TGF β -induced phosphorylation of Par6 at serine 345. (A) PC-3U cells were transiently transfected with wild-type Flag-Par6 or mutant Flag-S345A, followed by treatment with TGFβ, lysis, and immunoblotting for p-Par6Ser345, Par6, Flag, and β-tubulin. NT means the group of cells that were not transfected with plasmids as a negative control. Bar graphs showing relative levels of p-Par6Ser345 present the mean±s.d. from three independent experiments. *P<0.05, Student’s t-test. (B) In a cell culture wound-healing assay, PC-3U cells were treated with TGFβ followed by fixation in 4% paraformaldehyde and immunofluorescence staining for p-Par6Ser345 and phalloidin. Arrows indicate membrane ruffles that formed towards the wound. Nuclei are stained with DAPI. Scale bar=20 μm. (C) Analysis for TGFβ-induced formation of membrane ruffles. Each column presents the mean±s.d. from three independent experiments in which around 200–300 cells were counted. *P<0.05, Student’s t-test.
Cell migration requires formation of cell protrusion called membrane ruffling and the formation of a polarised structure characterised by actin and microtubule polymerisation (Ridley et al, 2003; Etienne-Manneville, 2004; Ekman et al, 2012). Par6 is the major scaffolding protein enabling Par complex assembly and thus cell polarity regulation. Hence, it is likely that Par6 phosphorylation may affect the subcellular localisation and function of the polarity complex. Therefore, we next investigated the subcellular localisation of p-Par6Ser345 in a wound-healing assay, in which TGFβ was used to stimulate PC-3U cells to migrate to cover a wounded area (Figure 1B). Immunofluorescence imaging showed that TGFβ induced p-Par6Ser345 localisation in the membrane ruffles of the leading edge of migrating cells and bound to the cytoskeleton to direct cell migration towards the wound (Figure 1B). We also quantified the effects of TGFβ-induced membrane ruffling in PC-3U cells subjected to wound-healing assays, as described above. A significant increase in membrane ruffling was noted in cells treated with TGFβ, when compared with control cells (Figure 1C). These data are in line with our and other previous observations of TβRI localisation in the leading edge of migrating cells (To et al, 2008; Ekman et al, 2012). This finding suggests that TGFβ regulates the localisation of p-Par6 to the leading edge of migrating prostate cancer cells. Together, our data indicate that phosphorylation of Par6 at Ser345 and its recruitment to the leading edge of migrating cells is dependent on TGFβ.
TGFβ-induced formation of the p-Par6–aPKC complex in migrating prostate cancer cells
Par6 is a key adaptor protein for Par complex assembly through interactions with Par3, atypical PKC, and Cdc42/Rac1. Therefore, we further investigated whether a Par6–PKC complex could be formed in the frontier of migrating cells. Co-immunofluorescence staining in the wound-healing assay revealed TGFβ-induced co-localisation of p-Par6 and p-PKCζ in the membrane ruffles of the leading edge of migrating PC-3U cells (Figure 2A).

TGF β induces formation of a p-Par6Ser345–p-PKC ζ complex that connects to polarised microtubules. In a cell culture wound-healing assay, PC-3U cells were treated with TGFβ, fixed in 4% paraformaldehyde, and subjected to co-immunofluorescence staining (A) for p-Par6Ser345 and p-PKCζ and (B) for p-Par6Ser345 and β-tubulin. Arrows indicates membrane ruffles that formed towards the wound. Nuclei are stained with DAPI. Scale bar=20 μm.
The Par complex further recruits Cdc42 to form a large protein complex that orients the MTOC and regulates cell polarity (Etienne-Manneville, 2008); therefore, we further investigated the interaction between Par complexes and microtubules. Co-immunofluorescence analysis showed that p-Par6 localised exactly at the front tip of the polarised microtubule, with a tight connection to the polarised microtubules (Figure 2B). From these data, we concluded that TGFβ treatment led to Par6 phosphorylation and formation of the p-Par6–aPKC complex in the frontier of membrane ruffling, which is connected to the polarised microtubules to orient the MTOC and direct cell migration. However, the molecular mechanism for the involvement of the Par6 complex in orienting the MTOC must be further investigated.
Disturbance of the p-Par6–aPKCζ complex prevents invasion of prostate cancer cells
The p-Par6–aPKC complex is important in regulating cell polarity and cell migration, inspiring us to investigate whether interfering with the p-Par6–aPKC complex could prevent cell migration. An invasion assay demonstrated that PC-3U cells invaded the lower chamber upon 36 h of TGFβ treatment. As expected, PC-3U cell invasion was blocked by treatment with the PKCζ-specific inhibitor, PKCζ pseudosubstrate (PKCζ PS), or the PI3K inhibitor LY294002 (Figure 3A). Colorimetric quantification followed by statistical analysis revealed that the invasive capability of the PC-3U cells dramatically decreased following the use of these inhibitors (Figure 3B). This suggests that inhibitors of the p-Par6Ser345 pathway could prevent invasion of aggressive prostate cancer cells.

The p-Par6Ser345–p-PKC ζ complex is related to TGF β -induced invasion of PC-3U cells. (A) In an invasion assay, PC-3U cells were treated with TGFβ in the absence or presence of the PKCζ-specific inhibitor PKCζ pseudosubstrate (PKCζ PS) or the PI3K inhibitor LY2940002. Cells were visualised by staining with crystal violet cell stain solution. (B) Mean values for optical density (OD) of invasive cells. Error bars represent s.d. from three independent experiments. *P<0.05, one-way ANOVA.
Enhanced expression of p-Par6 and PKC in prostate cancer
To explore the role of p-Par6 in human prostate cancer, we analysed the expression levels and localisations of p-Par6 and PKCζ in prostate cancer tissue. A control experiment using only secondary antibodies verified the specificity of the antibodies in our study (Supplementary Figure S2). Normal prostate tissue showed low levels of p-Par6 and high levels of PKCζ (Figure 4A). Both proteins localised in the apical plasma membrane toward the secretary lumen, as well as in the cell tight junction in the lateral and basolateral plasma membrane. This localisation is important for maintaining the cell–cell junction and thus regulating apical–basal polarity under physiological conditions. Interestingly, prostate cancer tissue showed dramatically increased p-Par6 expression and slightly decreased PKCζ expression (Figure 4B). These polarity proteins were highly expressed in the invasive front where prostate cancer cells aggressively invaded into the adjacent mesenchymal tissue (Figure 4C). This finding indicates that enhanced expression and localisation of p-Par6 and PKCζ is related to prostate cancer invasiveness.

Increased expression levels of p-Par6Ser345 and PKC ζ in human prostate cancer and at the tumour invasion front. Representative images of p-Par6Ser345 and PKCζ expression in (A) human normal prostate tissue and (B) prostate cancer tissues (C) invasive front, obtained by immunofluorescence staining. Nuclei are stained with DAPI.
p-Par6 expression levels in prostate tissue and clinical correlation
To confirm the potential role of p-Par6 as a predictive biomarker, we analysed p-Par6 expression by using immunohistochemical staining in TMA sections from 127 prostate cancer samples, including 50 samples of adjacent normal prostatic tissue, 21 samples with GS<7, 26 samples with GS=7, and 30 samples with GS⩾7. Figure 5A shows representative images of p-Par6 expression. In normal prostate tissue, low levels of p-Par6 were detected in the glandular epithelium and strictly localised in the apical membrane towards the lumen and the basolateral border (Figure 5A). In contrast, samples of intermediate-grade (GS<7) and high-grade prostate cancer (GS⩾7) showed increased p-Par6 expression throughout the malignant epithelium. Interestingly, the p-Par6 level corresponded with the volume of malignant epithelium. Furthermore, p-Par6 expression was dramatically increased in poorly differentiated carcinoma where cancer cells proliferated and invaded into glandular lumens and the surrounding mesenchymal tissue (GS⩾7) (Figure 5A). Figure 5B presents the average grey-scale values for intensity of immunohistochemical staining for p-Par6, showing a significant greater p-Par6 expression in tumour samples compared with normal tissue samples (P<0.05).

p-Par6Ser345 expression in prostate cells and tissues and its clinical correlation. p-Par6Ser345 expression was evaluated by immunohistochemical staining in tissue microarray cores (TMA) of human prostate cancer tissues. (A) Representative images of p-Par6Ser345 staining in normal prostate tissue, moderately differentiated prostatic adenocarcinoma with a Gleason score (GS) of 3+3 (GS<7), prostatic adenocarcinoma with pseudoglandular morphology and GS of 4+3 (GS=7), and poorly differentiated prostatic adenocarcinoma with GS of 4+4 (GS>7). (B) p-Par6Ser345 staining was quantified using ImageJ. Mean±s.e.m. of estimated grey-scale values obtained from the following groups: normal tissue (n=50), GS<7 (n=21), GS=7 (n=26), and GS>7 (n=30). *P<0.05 for between-group differences, Student’s t-tests. (C) The prostate cancer cell line PC-3U and the noncancerous prostate epithelial cell line RWPE1 were treated with TGFβ as indicated, lysed, and immunoblotted as indicated. (D) Bar graphs showing relative levels of p-Par6Ser345 present the mean±s.d. from three independent experiments. *P<0.05, Student’s t-test. (E) PC-3U and RPWE1 cells were treated with TGFβ followed by fixation in 4% paraformaldehyde and immunofluorescence staining for E-cadherin and N-cadherin. Nuclei were stained with DAPI. Scale bar=20 μm.
As we observed increased expression levels of p-Par6 in aggressive prostate cancer tissues, we further investigated the amounts of p-Par6 in androgen-independent and aggressive PC-3U cells compared with immortalised normal prostate RWPE1 cells. Immunoblotting of total cell lysates from TGFβ-stimulated cells showed higher basal activity of p-Par6 and a TGFβ-induced response only in PC-3U cells and not in RWPE1 cells (Figures 5C and D). E-cadherin is a crucial component to maintain epithelial intracellular adhesion, and its expression is downregulated during TGFβ-induced EMT (Heldin et al, 2009). Loss of E-cadherin expression and increased N-cadherin expression is observed during prostate cancer progression (Gravdal et al, 2007) and EMT in mammary cells (Mani et al, 2008). Therefore, we also examined the expression of these adhesion molecules in the two cell lines. The expression of E-cadherin was reduced in PC-3U cells when compared with RWPE1 cells, and a TGFβ-induced increase of N-cadherin was observed only in PC-3U cells (Figure 5C). TGFβ-induced phosphorylation of Smad2 was observed in both the cell lines (Figure 5C). Immunofluorescence imaging showed a decrease in staining of E-cadherin in PC-3U cells, while in RWPE1 cells E-cadherin was found to be localised in cell–cell contacts (Figure 5E). Moreover, only in PC-3U cells, there was an increase of immunofluorescence staining of N-cadherin, in response to TGFβ (Figure 5E), suggesting that PC-3U cells have a mesenchymal phenotype. These findings suggested that p-Par6 is correlated to the switch in E-cadherin and N-cadherin expression in prostate cancer cells. From these data, we conclude that p-Par6 could be a useful biomarker for aggressive prostate cancer.
Discussion
In this study, we investigated whether TGFβ-induced p-Par6 signalling promoted migration and invasion in prostate cancer cells. We observed that TGFβ-induced Par6 activation was crucial for its localisation in the leading edge of membrane ruffles in the migrating cells. In turn, such localisation was important for formation of the p-Par6–aPKCζ complex that connects to the microtubule system and directs cell migration. We detected high levels of p-Par6 signalling in prostate cancer tissue, which was associated with high GS as well as in aggressive PC-3U cells but not in immortalised normal prostate (RWPE1) cells. We also found that PKCζ inhibition prevented prostate cancer cell invasion, indicating that the p-Par6–aPKC complex may be a novel target for therapeutic intervention in prostate cancer.
These findings provide functional evidence for the role of p-Par6 in tumour progression of prostate cancer cells.
We previously reported that Smad7 (a direct target gene for TGFβ) and adenomatous polyposis coli (APC) mediate TGFβ-induced migration of prostate cancer cells. We also found that Smad7 facilitates TGFβ-induced phosphorylation of p38 and GSK-3β, resulting in GSK-3β inactivation and β-catenin accumulation and, in turn, APC recruitment to the microtubule plus end in the leading edge to direct cell migration (Etienne-Manneville and Hall, 2003a; Ekman et al, 2012). Furthermore, GSK-3β is a substrate for aPKC, and inhibitory phosphorylation of GSK-3β by aPKC induces APC recruitment to the microtubule plus end of migrating cells (Etienne-Manneville and Hall, 2003a). TGFβ activates PKCζ in prostate cancer cells, which is important for the cleavage and nuclear translocation of TβRI (Mu et al, 2011). Here we observed Par6 phosphorylation in response to TGFβ stimuli, followed by recruitment to the leading edge of migrating prostate cancer cells. The localisation of p-Par6 is important for formation of the p-Par6–p-PKCζ complex in the membrane ruffles, where it connects to the microtubule system in migrating cells, potentially aiding in cell directional migration. It remains unclear whether p-Par6 is implicated in PKCζ activation or has a role in GSK-3β inactivation in prostate cancer cell migration. Further studies are needed to answer these questions.
Phosphorylation of Par6 on serine345 has an important role in TGFβ-induced EMT. In mammary gland epithelial cells, Par6 phosphorylation by TGFβ type II receptor recruits Smurf1 (an E3 ubiquitin ligase) to target RhoA for degradation, which causes tight junction dissolution and loss of cell polarity (Wang et al, 2003; Ozdamar et al, 2005). Moreover, TGFβ-induced p-Par6Ser345 has been implicated in cell polarity regulation. Interference with Par6 signalling blocks TGFβ-induced loss of acini-like structure and protrusive invasion in mammary tumour cells, which has a crucial role in regulating EMT and metastasis in breast cancer progression (Viloria-Petit et al, 2009).
During preparation of this manuscript, it was reported that PKCζ interacts with TGFβ receptors through Par6 and that aPKC kinase activity is important for Par6 phosphorylation. Moreover, the association between p-Par6Ser345 and PKCζ has a role in TGFβ-induced EMT and cell migration in non-small cell lung cancer (Gunaratne et al, 2013). In our present study, TGFβ–p-Par6 signalling linked PKCζ and microtubules, thus regulating migration and invasion of prostate cancer cells. PC-3U cells origininate from a bone metastasis of prostate cancer, and these cells have already partially undergone EMT, consistent with the reduced expression of E-cadherin in this cell line (Figure 5C) (Ekman et al, 2012), but the cells undergo further mesenchymal transformation with TGFβ treatment as an increasing expression of N-cadherin was detected (Figure 5C and E). Thus, either TGFβ–p-Par6 signalling may function in targeting RhoA degradation and cell–cell junction dissolution in EMT or it could facilitate PKCζ and microtubules in directing the migration of different cell types in a contextual manner in response to TGFβ.
Polarity signalling contributes to cancer progression in vivo. High Par6 activity correlates with human breast cancer having basal subtype features (Viloria-Petit et al, 2009), and PKCζ overexpression promotes the aggressive phenotype of human prostate cancer cells (Yao et al, 2010). Interestingly, our present results showed that high levels of p-Par6Ser345 correlated with malignancy of human prostate cancer. Upregulation of polarity proteins and overactivation of polarity signalling could disrupt cell orientation and facilitate cell migration (Etienne-Manneville, 2008). Moreover, polarity protein mislocalisation is crucial for initiation of invasion. Using a 3-D culture model with Madin–Darby canine kidney cells, (Gassama-Diagne et al, 2006) verified that PI(4,5)P2 and PTEN strongly regulate the localisation of Cdc42 and aPKC to the apical plasma membrane to maintain the polarised lumen structure. Furthermore, reduced levels of PTEN, annexin 2, or Cdc42 lead to intracellular accumulation of Par6/aPKC and formation of abnormal lumens (Martin-Belmonte et al, 2007). Consistent with these findings, our present results also showed mislocalisation of p-Par6 and PKCζ in prostate cancer tissue. PTEN is frequently mutated and deleted in prostate cancer (Li et al, 1997; Majumder and Sellers, 2005), which could lead to enhanced PI3K activity. It will be very interesting to investigate whether PTEN can counteract TGFβ–p-Par6-mediated invasion in prostate cancer cells.
Overall, our present results indicate that TGFβ–p-Par6 signalling is important for migration and invasion in prostate cancer cells. Further analysis of TGFβ–p-Par6 signalling might lead to development of useful biological markers for prediction of prostate cancer progression.
Change history
31 March 2015
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Aranda V, Nolan ME, Muthuswamy SK (2008) Par complex in cancer: a regulator of normal cell polarity joins the dark side. Oncogene 27 (55): 6878–6887.
Bose R, Wrana JL (2006) Regulation of Par6 by extracellular signals. Curr Opin Cell Biol 18 (2): 206–212.
Ekman M, Mu Y, Lee SY, Edlund S, Kozakai T, Thakur N, Tran H, Qian J, Groeden J, Heldin CH, Landstrom M (2012) APC and Smad7 link TGFbeta type I receptors to the microtubule system to promote cell migration. Mol Biol Cell 23 (11): 2109–2121.
Etienne-Manneville S (2004) Actin and microtubules in cell motility: which one is in control? Traffic 5 (7): 470–477.
Etienne-Manneville S (2008) Polarity proteins in migration and invasion. Oncogene 27 (55): 6970–6980.
Etienne-Manneville S, Hall A (2003a) Cdc42 regulates GSK-3beta and adenomatous polyposis coli to control cell polarity. Nature 421 (6924): 753–756.
Etienne-Manneville S, Hall A (2003b) Cell polarity: Par6, aPKC and cytoskeletal crosstalk. Curr Opin Cell Biol 15 (1): 67–72.
Feng XH, Derynck R (2005) Specificity and versatility in tgf-beta signaling through Smads. Annu Rev Cell Dev Biol 21: 659–693.
Franzen P, Ichijo H, Miyazono K (1993) Different signals mediate transforming growth factor-beta 1-induced growth inhibition and extracellular matrix production in prostatic carcinoma cells. Exp Cell Res 207 (1): 1–7.
Gassama-Diagne A, Yu W, ter Beest M, Martin-Belmonte F, Kierbel A, Engel J, Mostov K (2006) Phosphatidylinositol-3,4,5-trisphosphate regulates the formation of the basolateral plasma membrane in epithelial cells. Nat Cell Biol 8 (9): 963–970.
Gravdal K, Halvorsen OJ, Haukaas SA, Akslen LA (2007) A switch from E-cadherin to N-cadherin expression indicates epithelial to mesenchymal transition and is of strong and independent importance for the progress of prostate cancer. Clin Cancer Res 13 (23): 7003–7011.
Gunaratne A, Thai BL, Di Guglielmo GM (2013) Atypical protein kinase C phosphorylates Par6 and facilitates transforming growth factor beta-induced epithelial-to-mesenchymal transition. Mol Cell Biol 33 (5): 874–886.
Heldin CH, Landstrom M, Moustakas A (2009) Mechanism of TGF-beta signaling to growth arrest, apoptosis, and epithelial-mesenchymal transition. Curr Opin Cell Biol 21 (2): 166–176.
Joberty G, Petersen C, Gao L, Macara IG (2000) The cell-polarity protein Par6 links Par3 and atypical protein kinase C to Cdc42. Nat Cell Biol 2 (8): 531–539.
Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wigler MH, Parsons R (1997) PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 275 (5308): 1943–1947.
Lin D, Edwards AS, Fawcett JP, Mbamalu G, Scott JD, Pawson T (2000) A mammalian PAR-3-PAR-6 complex implicated in Cdc42/Rac1 and aPKC signalling and cell polarity. Nat Cell Biol 2 (8): 540–547.
Majumder PK, Sellers WR (2005) Akt-regulated pathways in prostate cancer. Oncogene 24 (50): 7465–7474.
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA (2008) The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133 (4): 704–715.
Martin-Belmonte F, Gassama A, Datta A, Yu W, Rescher U, Gerke V, Mostov K (2007) PTEN-mediated apical segregation of phosphoinositides controls epithelial morphogenesis through Cdc42. Cell 128 (2): 383–397.
Massague J (2008) TGFbeta in cancer. Cell 134 (2): 215–230.
Massague J, Seoane J, Wotton D (2005) Smad transcription factors. Genes Dev 19 (23): 2783–2810.
McNeal JE (1988) Normal histology of the prostate. Am J Surg Pathol 12 (8): 619–633.
Moustakas A, Heldin CH (2005) Non-Smad TGF-beta signals. J Cell Sci 118 (Pt 16): 3573–3584.
Mu Y, Gudey SK, Landstrom M (2012) Non-Smad signaling pathways. Cell Tissue Res 347 (1): 11–20.
Mu Y, Sundar R, Thakur N, Ekman M, Gudey SK, Yakymovych M, Hermansson A, Dimitriou H, Bengoechea-Alonso MT, Ericsson J, Heldin CH, Landstrom M (2011) TRAF6 ubiquitinates TGFbeta type I receptor to promote its cleavage and nuclear translocation in cancer. Nat Commun 2: 330.
Ozdamar B, Bose R, Barrios-Rodiles M, Wang HR, Zhang Y, Wrana JL (2005) Regulation of the polarity protein Par6 by TGFbeta receptors controls epithelial cell plasticity. Science 307 (5715): 1603–1609.
Persson U, Izumi H, Souchelnytskyi S, Itoh S, Grimsby S, Engstrom U, Heldin CH, Funa K, ten Dijke P (1998) The L45 loop in type I receptors for TGF-beta family members is a critical determinant in specifying Smad isoform activation. FEBS Lett 434 (1-2): 83–87.
Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR (2003) Cell migration: integrating signals from front to back. Science 302 (5651): 1704–1709.
Roh MH, Margolis B (2003) Composition and function of PDZ protein complexes during cell polarization. Am J Physiol Renal Physiol 285 (3): F377–F387.
Shi SH, Jan LY, Jan YN (2003) Hippocampal neuronal polarity specified by spatially localized mPar3/mPar6 and PI 3-kinase activity. Cell 112 (1): 63–75.
Siegel R, Naishadham D, Jemal A (2012) Cancer statistics, 2012. CA Cancer J Clin 62 (1): 10–29.
To C, Kulkarni S, Pawson T, Honda T, Gribble GW, Sporn MB, Wrana JL, Di Guglielmo GM (2008) The synthetic triterpenoid 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid-imidazolide alters transforming growth factor beta-dependent signaling and cell migration by affecting the cytoskeleton and the polarity complex. J Biol Chem 283 (17): 11700–11713.
Viloria-Petit AM, David L, Jia JY, Erdemir T, Bane AL, Pinnaduwage D, Roncari L, Narimatsu M, Bose R, Moffat J, Wong JW, Kerbel RS, O'Malley FP, Andrulis IL, Wrana JL (2009) A role for the TGFbeta-Par6 polarity pathway in breast cancer progression. Proc Natl Acad Sci USA 106 (33): 14028–14033.
Wang HR, Zhang Y, Ozdamar B, Ogunjimi AA, Alexandrova E, Thomsen GH, Wrana JL (2003) Regulation of cell polarity and protrusion formation by targeting RhoA for degradation. Science 302 (5651): 1775–1779.
Ware JL (1994) Prostate cancer progression. Implications of histopathology. Am J Pathol 145 (5): 983–993.
Yao S, Bee A, Brewer D, Dodson A, Beesley C, Ke Y, Ambroisine L, Fisher G, Moller H, Dickinson T, Gerard P, Lian LY, Risk J, Lane B, Smith P, Reuter V, Berney D, Gosden C, Scardino P, Cuzick J, Djamgoz MB, Cooper C, Foster CS (2010) PRKC-zeta expression promotes the aggressive phenotype of human prostate cancer cells and is a novel target for therapeutic intervention. Genes Cancer 1 (5): 444–464.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Mu, Y., Zang, G., Engström, U. et al. TGFβ-induced phosphorylation of Par6 promotes migration and invasion in prostate cancer cells. Br J Cancer 112, 1223–1231 (2015). https://doi.org/10.1038/bjc.2015.71
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2015.71
Keywords
This article is cited by
-
NF-κB signaling in neoplastic transition from epithelial to mesenchymal phenotype
Cell Communication and Signaling (2023)
-
Par6 Enhances Glioma Invasion by Activating MEK/ERK Pathway Through a LIN28/let-7d Positive Feedback Loop
Molecular Neurobiology (2023)
-
Partitioning defective 6 homolog alpha (PARD6A) promotes epithelial–mesenchymal transition via integrin β1-ILK-SNAIL1 pathway in ovarian cancer
Cell Death & Disease (2022)
-
Cell polarity and oncogenesis: common mutations contribute to altered cellular polarity and promote malignancy
The Nucleus (2020)
-
PKCζ facilitates lymphatic metastatic spread of prostate cancer cells in a mice xenograft model
Oncogene (2019)
