
Abstract
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) induces apoptosis and preferentially kills tumor cells by engaging specific glycosylated death receptors, resulting in the internalization of ligand/receptor complexes and recruitment of the initiator caspase-8 to an activation platform known as the death-inducing signaling complex (DISC). However, emergence of TRAIL-resistant sub-populations may contribute to therapeutic failure. To investigate resistance mechanisms, we isolated a stable TRAIL-resistant sub-population of the metastatic colon cancer cell line LS-LIM6, designated LIM6-TR. LIM6-TR cells are impaired in endocytosis of TRAIL/death receptors complexes and failed to recruit/activate caspase-8 to the DISC upon TRAIL stimulation. Differential activation of Wnt and JNK pathways is not responsible for acquisition of TRAIL resistance. LIM6-TR cells display a marked increase in cell-surface expression of galectin-3, an endogenous lectin, which co-localizes with and binds death receptors. Silencing of galectin-3 restores TRAIL sensitivity and promotes TRAIL-mediated endocytosis of TRAIL/death receptors complexes. Inhibitors of galectin-3 and glycosylation also re-sensitize LIM6-TR to TRAIL and restore internalization of ligand/receptors complexes. These studies identify a novel TRAIL-resistance mechanism in which galectin-3 impedes trafficking of death receptor by anchoring them in glycan nano-clusters, blocking the execution of the apoptosis signal.
Similar content being viewed by others


Main
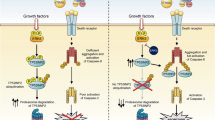
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), a member of the tumor necrosis factor family, can induce apoptotic cell death by engaging specific death receptors, DR4 and DR5.1 The observation that TRAIL can preferentially eliminate tumor cells has resulted in several potential therapeutic agents, including recombinant TRAIL protein and agonist monoclonal antibodies that target death receptors. Many of these agents are currently being evaluated in clinical trials.2 However, the emergence of TRAIL-surviving sub-populations may contribute to therapeutic failure. Homotrimeric TRAIL triggers the extrinsic pathway of apoptosis by binding to the heavily glycosylated DR4 and DR5.3 O-linked and N-linked glycosylation promotes oligomerization, clustering and rapid endocytosis of the receptors accompanied by recruitment of the adaptor, Fas-associated death domain (FADD), and the initiator caspase, procaspase-8, to assemble a death-inducing signaling complex (DISC).4 However, the exact role of TRAIL/receptors in endocytosis is unclear. It has been reported that an efficient cell death signal requires internalization of TRAIL/receptors complexes,5, 6 but other studies have suggested that receptor-mediated endocytosis is not required for TRAIL-induced apoptosis.7, 8
Two types of signaling pathway can be transmitted from the DISC to execute the apoptotic program. In the first type, the signal is independent of mitochondrial activation and cannot be blocked by Bcl-2 and Bcl-XL. In this pathway, TRAIL initiates signaling by the formation of the DISC that recruits and activates pro-caspase-8. Activated caspase-8 then processes directly the executioner, caspase-3. In the second type, the signal requires amplification through activation of the effector, caspase-9, by the mitochondria and can be blocked by Bcl-2 and Bcl-XL. In this pathway, activation of caspase-8 results in Bid cleavage, and subsequent mitochondrial depolarization and release of apoptogenic factors. One such factor, cytochrome c, binds Apaf1 and promotes oligomerization and recruitment of apical caspase-9 into a high molecular weight complex termed the apoptosome. Assembly of the apoptosome induces processing of caspase-9 and subsequent activation of caspase-3.9 In addition to its direct apoptosis-inducing effect, TRAIL may simultaneously induce multiple cell proliferative signaling pathways that involve proteins such as nuclear NF-kappa-B, MAPK's, JNK's, PI3 K, and Akt.10 Depending on the cell type and the relative strength and duration of the ligand signal, treatment with TRAIL may stimulate either apoptosis or cell proliferation and the disruption of the balance between these mechanisms may influence cell fate.11 TRAIL has been implicated in immune surveillance against tumor initiation, development, and metastasis, suggesting a potential application to cancer therapy. However, resistance to TRAIL-mediated apoptosis in cancer cells may limit the successes of TRAIL therapy. Approximately 50% of tested tumor cell lines are resistant to TRAIL, suggesting that the molecular regulation of TRAIL-induced apoptosis is complex.12 Resistance to TRAIL can occur at different points in the signaling pathways triggered by TRAIL, such as dysfunction of the death receptors DR4 and DR5,13 defects in the FADD14 and caspase-8,15 and activation of the survival PI3K/Akt pathway.16 In addition, galectin-3 has recently emerged as a modulator of TRAIL. On one hand, it sensitizes Bt-549 human breast cancer cells to TRAIL by upregulating PTEN and inactivating Akt.17 On the other hand, over-expression of galectin-3 in J82 human bladder carcinoma cells activates Akt and confers resistance to TRAIL.18
Galectin-3 is a unique member of a family of highly conserved animal lectins characterized by their ability to recognize multiple N-acetyl-lactosamine sequences that can be displayed on both N- and O-linked glycans on cell surface glycoconjugates. It consists of three structural domains: (a) an amino-terminal domain, which is essential for galectin-3 homo-dimerization, (b) a COOH-terminal domain containing a single carbohydrate-recognition domain (CRD), and (c) a collagen-like sequence linking the amino-terminal domain to the CRD.19 Galectin-3 is found in the cytoplasm, on the cell surface, in the nucleus, and is secreted by tumor and inflammatory cells. Most studies have found a positive correlation between total galectin-3 and colon cancer progression. Galectin-3 concentrations have been found to be increased in sera from colorectal cancer patients and to be higher in those with metastatic disease than in patients with localized tumors.20 The unique structure of this protein enables it to interact with a plethora of ligands in a carbohydrate-dependent or -independent manner. Although galectin-3 possesses only one CRD, it exhibits bi/multivalent binding properties, which are enabled by homo-oligomerization through its amino-terminal domain.21 It has been postulated that oligomeric galectin-3 modulates functions critical to the development and maintenance of the tumor phenotype, including cell adhesion, migration, invasion, angiogenesis, immune function, and apoptosis.22 Furthermore, multivalent galectin-3 acts as a scaffolding molecule by simultaneously binding glycan ligands on multiple glycoproteins on the cell surface, such as growth factor receptors (EGFR, K-Ras, VEGF and bFGF, and TCR) and extracellular matrix (ECM) proteins like fibronectin (heterotypic clustering).23 Alternatively, it can bind and segregate the same receptors into different membrane domains (homotypic clustering).24 Clustering of surface glycoprotein receptors can significantly modulate their function and their influence on cellular responses. Galectin/glycan lattices may control the dialog between tumor and tumor-associated stromal and immune cells.25 Circulating dimeric or multimeric soluble galectin-3 induces homotypic aggregation, immune evasion, and enhanced survival, and favors homing of blood-borne cells to secondary sites.26 Galectin-3 secreted from tumor stromal cells and/or cancer-initiating cells exhibits immunosuppressive properties and modulates cytokine release.27 Here we report that cell surface galectin-3 immobilizes death receptors on human colon cancer cells by trapping them in a nano-cluster lattice, blocking DISC formation, and recruitment of the apoptosis-initiating protease, procaspase-8. Our results identify a novel mechanism for the acquisition of TRAIL resistance.
Results
Generation of stable TRAIL-resistant human colon cancer cells
TRAIL has cytotoxic effects against most tumor cells. However, a fraction of a given tumor cell population cannot be killed even at high doses of TRAIL. To explore how these resistant survivors escape from TRAIL-induced death, we obtained TRAIL-resistant stable cells by subjecting LS-LiM6 metastatic human colon cancer cells to repeated exposure to TRAIL. Exposure of LS-LiM6 cells to TRAIL resulted in a dose- (Figure 1a) and time-dependant (Figure 1b) reduction in cell viability, which plateaued at 35% viable cells at TRAIL concentrations of 100 ng/ml within 5 h post exposure. Residual surviving cells were propagated with periodic exposure to TRAIL to yield a TRAIL-resistant stable cell line, LiM6-TR. As shown in Figures 1a and b, less than 5% loss of viable cells could be detected in LIM6-TR cells after exposure to 200 ng/ml TRAIL for 24 h, in contrast to about 65% loss of viability observed in parental LS-LiM6 cells. Extended exposure to higher doses or longer duration of TRAIL treatment did not increase cell death. The TRAIL-resistant LiM6-TR cells did not display cross-resistance to other cytotoxic agents, such as oxaliplatin or irinotecan (data not shown). To determine the nature of cell death induced by TRAIL we monitored the accumulation of cell populations in Sub-G1 by DNA-content staining with propidium iodide (PI) and the caspase-dependent cleavage of PARP. PI staining (Figure 1c) showed that TRAIL (100 ng/ml) induced significant apoptotic cell death (63.9% of cells in sub-G1) within 24 h post exposure, whereas LIM6-TR cells were resistant to TRAIL (5.3%). The PARP assay (Figure 2d) supports these findings.

Generation of TRAIL-resistant LiM6-TR colon cancer cells. The TRAIL-resistant LiM6-TR stable cell line was obtained as described under ‘Materials and Methods’. The parental LS-LiM6 and resistant LiM6-TR were periodically tested for TRAIL response. (a) TRAIL-dependent loss of viable cells, measured by the MTT assay. Each data point represents the mean and standard deviation of experiments performed in triplicate. (b) Apoptotic cell death was confirmed by the accumulation of cells at sub-diploid fraction following exposure to 100 ng/ml TRAIL for 4 h and 24 h. Bars denote percentage of sub-diploid cells. (c) Immunoblots of PARP show the full length 116- and 89-kDa apoptosis related cleaved fragments only in the parental LS-LiM6 exposed to 100 μg/ml TRAIL, confirming apoptotic cell death induced by TRAIL

Differential regulation of pro-survival signals is not responsible for the acquisition of TRAIL resistance by LiM6-TR. (a) Parental LS-LiM6 and TRAIL-resistant LiM6-TR were treated with 100 ng/ml TRAIL for 24 h and modulation of pro-survival signaling was compared by western blot analysis. (b) Effects of inhibition of apoptosis by the caspase-3 inhibitor Ac-DEVD-FMK (10 and 100 μ M) on the activation status of JNK, PTEN, and beta-catenin in parental LS-LiM6 cells. (c) JNK inhibitor SP600125 (25 μ M) failed to confer resistance to TRAIL as determined by western blots of PARP cleavage. (d) TRAIL-dependent accumulation of apoptotic cells in sub-diploid faction is not prevented by JNK inhibitor. Bars denote percentage of sub-diploid cells
Pro-survival and apoptosis signaling in TRAIL-sensitive (LS-LiM6) versus TRAIL-resistant (LiM6-TR) colon cancer cells
TRAIL activates JNK and shuts off Wnt pathways only in LS-LiM6
TRAIL can trigger both pro-survival and apoptosis signaling, and disruption of the balance between these mechanisms may determine cell fate. TRAIL deactivates PI3K/Akt in both parental sensitive LS-LiM6 and the resistant variant LiM6-TR (Figure 2a). It activates JNK and shuts off Wnt signal transduction pathways only in responsive LS-LiM6 cells (Figure 2a). The differential activation of Wnt is not responsible for the acquisition of TRAIL resistance, because downregulation of beta-catenin could be blocked by the caspase-3 specific inhibitor Ac-DEVD-FMK (Figure 2b), indicating that blocking of Wnt signaling accompanies the apoptosis signaling activated by TRAIL. The differential activation of JNK is also not responsible for the acquisition of TRAIL resistance, because specific inhibition of JNK activation by SP600125 failed to confer resistance to LS-LiM6 cells (Figures 2c and d). Furthermore, activation of JNK is independent of the apoptosis signaling cascade elicited by TRAIL, because the caspase-3 inhibitor (Ac-DEVD-FMK) failed to block JNK phosphorylation, but completely abrogated PARP cleavage and apoptotic cell death (Figure 2b).
TRAIL fails to activate caspase-8 in TRAIL-resistant colon cancer cells
We evaluated protein levels and processing of endogenous caspases in TRAIL-sensitive LS-LiM6 and TRAIL-resistant LiM6-TR cells in response to TRAIL. Exposure of parental LS-LiM6 cells to TRAIL activates the initiator caspase-8 (Figure 3a) that triggers a mitochondria-dependent apoptotic amplification loop by activating Bid, inducing the release of cytochrome C (Figure 3b). The resulting activation of the effector caspase-9 subsequently leads to the activation of the executioner caspase-3 and apoptotic cell death as determined by PARP cleavage. In contrast, TRAIL fails to activate caspase-8 and the subsequent caspase cascade in resistant LiM6-TR cells, suggesting that the acquisition of TRAIL resistance in LiM6-TR cells occurs upstream to caspase-8 at the level of the DISC assembly.

TRAIL fails to activate caspase-8 and the subsequent apoptosis cascade in TRAIL-resistant colon cancer cells. (a) Western blot analysis of apoptosis cascade in parental LS-LiM6 cells and TRAIL-resistant LiM6-TR cells with or without TRAIL treatment. (b) Secretion of cytochrome C from the mitochondria (M) to cytosol (C) after TRAIL treatment of LS-LiM6, but not LiM6-TR cells. COX4 is the mitochondrial marker
TRAIL fails to induce the recruitment of caspase-8 to the DISC in LIM6-TR cells
The initial step in TRAIL-mediated apoptosis is the assembly of the DISC involving the formation of TRAIL/death receptor trimers, which recruit the adaptor molecule FADD and caspase-8 to propagate the apoptotic signal.4 To investigate whether the acquisition of TRAIL resistance in LiM6-TR is because of a defect in DISC formation, we compared expression levels of DISC components in LS-LiM6 and LiM6-TR. Protein expression levels of the death receptors DR4 and DR5, and death-associated proteins FADD, FAF1, and FLIP are similar in both the responsive LS-LiM6 and the unresponsive LiM6-TR cells (Figure 4a). Expression of the decoy receptors, DcR1 and DcR2, were not detected (data not shown) in either LS-LiM6 or LiM6-TR. We also compared surface expression of DR4 and DR5 in parental LS-LiM6 and resistant LiM6-TR cells by flow cytometry. Surface expression of DR4 and DR5 are almost identical in TRAIL-sensitive LS-LiM6 and TRAIL-resistant LiM6-TR cells (Figures 4b and c). Next, we investigated whether TRAIL induces the assembly of a functional DISC in LIM6-TR cells. Cells were treated with a complex of Flag-tagged TRAIL and anti-Flag to precipitate the DISC. Analysis of the DISC components revealed that although TRAIL binds DR4 and DR5 in LIM6-TR cells, it fails to induce the recruitment and activation of caspase-8 to the DISC (Figure 4d).

The expression of the DISC components is not changed in LiM6-TR. (a) Total protein expression of members of the DISC was compared between LS-LiM6 and LiM6-TR by western blot analysis. (b) Comparison of cell surface expression of the death receptor DR4 between LS-LiM6 (blue) and LiM6-TR (red) was performed by FACS analysis, using Alexa-Fluor 647 fluorophore and Fortessa FACS cytometer. (c) Comparison of cell surface expression of the death receptor DR5 between LS-LiM6 (blue) and LiM6-TR (red) was performed by FACS analysis, using Alexa-Fluor 488 fluorophore and XL-MCL FACS cytometer. Control staining (gray) differs between DR4 and DR5 labeling. (d) Cells were treated with a complex of Flag-tagged TRAIL and anti-Flag antibody, and cell lysates were subjected to DISC immunoprecipitation. Associated proteins were detected by immunoblotting with the indicated antibodies
Galectin-3 cell surface expression is elevated in TRAIL-resistant colon cancer cells
Galectin-3 is a modulator of apoptosis that may exert anti- or pro-apoptotic activity depending on the nature of the stimulus and cell type. Galectin-3 sensitizes Bt-549 human breast cancer cells to TRAIL,17 but confers resistance to TRAIL to J82 human bladder carcinoma cells.18 To investigate whether galectin-3 has a role in the acquisition of TRAIL resistance by LiM6-TR, we compared expression levels of galectin-3 in total cell lysates of LS-LiM6 and LiM6-TR by western blot analysis. As shown in Figure 5a, expression of galectin-3 is markedly increased in LiM6-TR. Next, we examined surface expression of galectin-3 in LS-LiM6 and LiM6-TR. Confocal microscopy and FACS analysis revealed that surface expression of galectin-3 is 3-fold higher in LiM6-TR than in LS-LiM6 (Figures 5b and c), suggesting that surface galectin-3 may interfere with the initial phase of TRAIL-induced signaling, clustering of death receptors, and the assembly of the DISC. Confocal images suggested that LIM6-TR cells form larger cell clusters than the parental LIM6 cells. This may be because of the fact that LIM6-TR cells express higher levels of surface galectin-3, which can mediate homotypic cell aggregation.28

Cell surface galectin-3 confers TRAIL resistance. (a) Comparison of total galectin-3 protein expression between LS-LiM6 and LiM6-TR by western blot analysis. (b) Comparison of surface expression of galectin-3 between parental LS-LiM6 and TRAIL-resistant LiM6-TR cells. Cells were fixed and processed for confocal microscopy using anti-galectin-3 (mAb TIB166, red) and visualized using Alexa Fluor-conjugated secondary antibody. (c) Comparison of cell surface expression of galectin-3 between LS-LiM6 (blue) and LiM6-TR (red) was performed by FACS analysis. Control staining, gray. (d) LiM6-TR cells were treated with galectin-3 sh-RNA (TR-SH-G) or non-targeting control sh-RNA (TR-C-SH) lentiviral particles. Depletion of galectin-3 expression was confirmed by western blot analysis. (e) Cell surface expression of galectin-3 was monitored by FACS analysis of LiM6-TR-G-SH (red) and LiM6-TR-C-SH (blue). Control staining, gray. (f) LiM6-TR,TR-G-SH, and TR-C-SH cells were treated with 100 ng/ml TRAIL for 5 h and loss of viable cells was determined by the MTT assay. Data are reported as mean±S.E.M. (g) Apoptotic cell death was quantified by flow cytometry analysis of the PI-stained cells. Bars denote percentage of sub-diploid cells
Galectin-3 depletion restores TRAIL-responsiveness to TRAIL-resistant colon cancer cells
To determine whether galectin-3 is indeed responsible for the acquisition of TRAIL-resistance in LiM6-TR, we infected the cells with lentiviral particles harboring different galectin-3 sh-RNAs, which specifically depleted the galectin-3 protein (Figure 5d) with a concomitant reduction in surface expression levels of galectin-3 (Figure 5e). Galectin-3 knockdown did not alter the amount of surface death receptors, DR4 and DR5, and unmodified caspase-8 (data not shown). However, it re-sensitized LiM6-TR to TRAIL-induced apoptotic cell death to the same levels as the parental TRAIL-sensitive LS-LiM6 cells (Figures 5f and g), providing independent evidence for the functional importance of galectin-3.
Surface galectin-3 impedes trafficking of death receptors in TRAIL-resistant colon cancer cells
Binding of homotrimeric TRAIL to death receptors DR4 and DR5 induces oligomerization and rapid endocytosis of the receptors, leading to regulation of apoptosis execution.4, 5 We investigated whether galectin-3 interferes with this process, which may result in the acquisition of TRAIL resistance in LiM6-TR. First, we compared the pattern of surface distribution of DR4, DR5, and galectin-3 in unstimulated LS-LiM6 and LiM6-TR. Confocal microscopy revealed a mostly diffuse pattern with some definite, small punctuate surface labeling of DR4 and DR5 in parental TRAIL-sensitive LS-LiM6 cells (Figure 6a). This is in agreement with previous reports of self-association of DR4 and DR5 into trimeric assemblies in unstimulated cells.29, 30 Galectin-3 labeling of LS-LiM6 was predominantly of a diffuse pattern and not homogenous. This heterogeneity is consistent with the non-clonal origin of LS-LiM6.31 In contrast, a robust punctate, patchy pattern of the death receptors and of galectin-3 is seen in LiM6-TR cells (Figure 6a), indicating the oligomerization of DR4 and DR5 into high order complexes. Confocal image analysis revealed co-localization of galectin-3 with DR4 and DR5 on the surface of LiM6-TR, as indicated by Pearson's coefficient values of 0.7 and 0.6, respectively, but not LS-LiM6 (−0.1 and −0.1) (Figure 6a). Co-immunoprecipitation with either DR4 or DR5 antibodies was able to pull down galectin-3 (Figure 6b), suggesting direct association between the death receptors and galectin-3. These findings suggest either homotypic clustering and segregation of death receptors or heterotypic clustering of death receptors to ECM proteins by galectin-3. To test whether DR4 and DR5 are anchored and trapped within a lattice formed by surface galectin-3, we applied two independent methods. First, we monitored loss of surface expression of DR4 and DR5 following a short exposure of LS-LiM6 and LiM6-TR to TRAIL. TRAIL caused loss of both DR4 and DR5 receptors in LS-LiM6, but not in LiM6-TR (Figure 6d). Exposure of LiM6-TR galectin-3 knockout cells (TR-G-SH) to TRAIL resulted in loss of DR4 (Supplementary Figure S1) and DR5 (not shown) receptors resembling the effects on the parental TRAIL-responsive LS-LiM6 cells. In a second validation, we monitored DR4 and DR5 membrane transport and endocytosis by confocal microscopy. TRAIL treatment of LS-LiM6 caused internalization and uptake of both DR4 and DR5 (Figure 6c). In contrast, neither DR4 nor DR5 uptake was detected in LiM6-TR cells under the same conditions (Figure 6c). TRAIL-untreated cells showed no uptake of either DR4 or DR5 (Figure 6c, upper panels). Uptake of DR4 (Supplementary Figure S2) and DR5 (not shown) similar to that in LS-LiM6 was observed in the LiM6-TR-derived galectin-3 knockout cells (TR-G-SH) treated with TRAIL, consistent with the hypothesized role of galectin-3 in immobilizing DR4 and DR5 on the surface of LiM6-TR.

Surface galectin-3 anchors DR4 and DR5 in nano-clusters and impedes endocytosis. (a) Cells were labeled with anti-DR4 (green), anti-DR5 (gray), anti-galectin-3 (red) and nuclear staining by DAPI (blue), and surface labeling was visualized with Alexa Fluor-conjugated secondary antibodies by confocal microscopy. The white boxes of merged images are magnified to show detailed morphology (detail). Morphometric analysis performed by the 3I's Slide book 5.0, computing Pearson's coefficient for degree of overlapping. (b) Lysates from LS-LIM6 or LIM6-TR cells were immunoprecipitated with either anti-DR4, anti-DR5, or control IgG antibodies. The precipitates were immunoblotted with anti-galectin-3 antibody (TIB 166). (c) TRAIL-dependent internalization of DR4 and DR5. Cells were pre-labeled with anti-DR4 (green) and anti-DR5 (gray) antibodies, and exposed to 100 ng/ml TRAIL on ice for 1 h. The temperature was raised to 37°C for 30 min to allow receptor internalization or kept at 0°C for controls. Surface labeling was removed by treatment with 2 mM acetic acid, then the cells were fixed and permeabilized. Internalization of death receptor immunocomplexes was visualized with Alexa Fluor-conjugated secondary antibodies by confocal microscopy. (d) TRAIL-dependent loss of cell surface DR4 and DR5. Cells were treated with 100 ng/ml TRAIL for 30 min at 37°C followed by labeling with either anti-DR4 or anti-DR-5 antibodies and cell surface death receptor expression was determined by flow cytometry analysis
Glycosylation inhibitors restore TRAIL sensitivity to TRAIL-resistant colon cancer cells
Although galectin-3 possesses only one CRD, it exhibits bi/multivalent binding properties, which are enabled by homo-oligomerization through its amino-terminal domain.21It has been reported that multivalent surface galectin-3 acts as a scaffolding molecule by binding N-linked and O-linked glycan ligands on multiple glycoproteins of the cell surface.23 The death receptors DR4 and DR5 possess several ectodomain O-linked oligosaccharides.4 Amino acid sequences predict a consensus extracellular N-glycosylation site (Asn156) only in DR4. To examine whether the formation of galectin-3-DR4/DR5 glycan lattices is essential for the acquisition of TRAIL resistance by LiM6-TR, we pretreated LiM6-TR with a galectin-3 carbohydrate-binding inhibitor N-Acetyl-lactosamine, with the N-glycosylation inhibitors, swainsonine and tunicamycin, or with the O-glycosylation inhibitor benzyl-alpha-N-Acetyl-galactosamine. Disruption of galectin-3-DR4/DR5 nano-cluster lattice formation by 24 h pretreatment with each of the four inhibitors efficiently re-sensitized the resistant LiM6-TR to TRAIL-induced apoptotic cell death (Figure 7a). This was accompanied by TRAIL-induced loss of surface expression of DR4 and DR5 as determined by flow cytometry analysis (Figure 7b).

Inhibitors of glycosylation and galectin-3 binding re-sensitize LiM6-TR to TRAIL. (a) LiM6-TR cells were pretreated with either 5 mM LacNAc, 2 μg/ml tunicamycin, 2 μg/ml swainsonine, or 1 mM benzyl-alpha-GalNAc for 24 h followed by exposure to 100 ng/ml TRAIL for 5 h. Apoptosis was determined by the accumulation of PI-labeled cells in the sub-diploid fractions by flow cytometry analysis. (b) LiM6-TR cells were treated with inhibitors of glycosylation and galectin-3 binding and analyzed for TRAIL-dependent loss of surface DR5 receptor by flow cytometry. In control experiments (red), cells were pre-treated but were not stimulated with TRAIL. Control staining, gray. Only data for DR5 is shown; similar results showed TRAIL-dependent loss of surface DR4 in tunicamycin-treated LiM6-TR
Discussion
Acquired resistance to apoptosis of tumor cells is a major obstacle for cancer chemotherapy, including TRAIL-based therapy. Elucidation of mechanisms underlying resistance of tumor cells to TRAIL-induced apoptosis has led to an improved understanding of regulation of the death receptor signaling pathway. Many ways by which tumor cells can acquire TRAIL resistance have been reported. The micro-RNAs, miR-221 and miR-222, interfere with TRAIL signaling through their effects on the cell cycle regulator protein p27kip1. Furthermore, by targeting PTEN and TIMP3, miR-221 and miR-222 induce TRAIL resistance and enhance cellular migration through the activation of the Akt pathway and metallopeptidase.32 Another group of resistance mechanisms involve characteristics that inhibit apoptosis in a general manner such as reduced caspase expression,15 increased expression of caspase inhibitors, such as XIAP, cIAP2,33 or expression of Bcl-2.34 A group of resistance mechanisms more specific for TRAIL signaling include defects in death receptors or increased expression of inhibitors that are selective for death receptors like FLIP or the decoy receptors.35 Recently, a novel method of TRAIL-resistance involving expression of the peptidyl O-glycosyl N-acetylgalactosaminyl transferase 14 (GALNT14) was identified.4 In this study we provide evidence for a new mechanism of how metastatic colon cancer cells may escape from the killing mediated by TRAIL.
In the present study, we established stable TRAIL-resistant colon cancer cells by repetitive exposure of LS-LiM6 human metastatic colon cancer cells to TRAIL. TRAIL-resistant cells showed no cross-resistance to other apoptosis-inducing agents, such as oxaliplatin, irinotecan, or 5-FU. Parental, TRAIL-sensitive LS-LiM6 cells behave like classical type II cells in which TRAIL promotes oligomerization, clustering, and rapid endocytosis of the death receptors, DR4 and DR5, followed by the assembly of the DISC. The DISC/caspase-8 signal requires an amplification loop to activate executioner caspases. This signal is mediated by the proapoptotic BCL-2 family protein Bid, which links the extrinsic apoptotic pathway to the intrinsic pathway by triggering mitochondrial membrane permeabilization and activation of the effector caspase-9, thereby amplifying caspase-3 activation.36 In contrast, TRAIL-resistant LiM6-TR cells are impaired in assembling a functional DISC, and recruitment and activation of the initiator caspase-8. The levels of death receptors DR4 and DR5 as well as decoy receptors DcR1 and DcR2, and other components of the DISC were not affected by TRAIL selection. However, the cell surface expression of the endogenous mammalian lectin, galectin-3, was significantly elevated in LiM6-TR as compared with parental LS-LiM6 cells.
Studies have indicated endocytosis as a means for coordinating the type, duration, and amplitude of signals transduced by surface receptors. In the case of TRAIL, there are conflicting reports about the requirement of internalization to an efficient cell death signal. Although most studies agree that TRAIL triggers a rapid endocytosis of death receptors in a time- and concentration-dependent manner, there is a debate about its contribution to the apoptotic signal. Several studies have shown that blockade of receptor internalization does not inhibit TRAIL-induced apoptosis but, rather, amplifies the apoptotic signaling of TRAIL.7, 8 In contrast, other studies have suggested that endocytosis is required to execute TRAIL-induced apoptosis.5, 6 Here we provide evidence that surface galectin-3 on LiM6-TR forms heterodimeric complexes with DR4 and DR5, thus blocking their endocytosis and preventing the execution of the apoptosis signal.
Previous studies have suggested that galectin-3 is capable of attenuating the output signal of a variety of other receptors such as K-ras, EGFR, TCR, VEGF, and bFGF by trapping them in nano-cluster lattices.23 Galectin-3 recognizes multiple N-acetyl-lactosamine sequences, which can be displayed on both N- and O-glycans on cell surface glycoconjugates. The branching of complex N-glycans attached to growth factor receptors can alter cell surface receptor functions, including their levels of cell surface retention, rates of internalization, and subsequent intracellular signaling.37 It has been reported that galectin-3 crosslinks Mgat5-modified N-glycans on EGFR and TGFbR at the cell surface and delays the removal by constitutive endocytosis, amplifying and sustaining the output signal.38 Similarly, O-glycosylation of death receptors DR4 and DR5 by GalNT14 promotes ligand-stimulated clustering and controls tumor cell sensitivity to TRAIL.4 TRAIL receptors are pre-ligand assembled into trimers through pre-ligand assembly domain, and TRAIL binding causes further death receptor oligomerization resulting in internalization of receptor/ligand complexes and recruitment of adaptor proteins to the caspase activation platform (DISC).29 Supporting this idea are recent findings that other members of the TNF family of death ligands such as TNF and CD95L induce apoptosis with an absolute requirement for internalization of their corresponding receptors and the formation of a DISC.39, 40 In this study, we show that surface galectin-3 is capable of blocking death receptor internalization by formation of carbohydrate-dependent nano-cluster lattices.
We have observed a similar resistance mechanism in a TRAIL-resistant subpopulation of the human colon cancer cell line DLD1 and the metastatic breast cancer cell line GI101AB. It was reported that accelerated degradation of caspase-8 is responsible for the acquisition of TRAIL resistance in DLD1/TRAIL-R cells.15 We observed elevated surface expression levels of galectin-3 in both DLD1/TRAIL-R and GI101-TR cells (Supplementary Figure S3). Furthermore, TRAIL failed to induce activation of caspase-8 and death receptors endocytosis in both cell lines (data not shown). These findings suggest that acquisition of TRAIL resistance by this mechanism is not unique to a single cell line or tumor type.
In addition to its direct apoptosis-inducing effect, TRAIL may simultaneously induce alternative pro-survival signaling pathways such as nuclear NF-kappa-B and MAPK's, including ERK, JNK, PI3 K, and Akt. However, it is not clear how activation of these pathways is related to the apoptosis signal. Previously, we reported that galectin-3 upregulates PTEN expression that results in the inhibition of Akt signaling and sensitizes breast cancer cells, Bt-549, to TRAIL.17 These observations are in agreement with other reports that inhibition of PI3 K/Akt pathway sensitizes tumor cells to TRAIL treatment or even reverses TRAIL resistance. TRAIL inhibited Akt signaling in TRAIL-resistant LiM6-TR cells, as well as in the parental TRAIL-sensitive LS-LiM6 cells, suggesting that inhibition of the PI3/Akt pathway is not a general prerequisite for acquiring TRAIL resistance. In contrast, TRAIL phosphorylated JNK and downregulated beta-catenin expression only in LS-LiM6 cells. Loss of JNK activation in LiM6-TR is not responsible for the acquisition of TRAIL resistance, but, however, is independent/parallel to the caspase cascade, aiding in the execution of the apoptotic cascade. Our findings also suggest that shutting off Wnt signaling is an integral part of the apoptosis signaling elicited by TRAIL, as it is downstream of caspase-3 activation.
These findings provide evidence that galectin-3-dependent clustering of DR4 and DR5 on the surface of colon cancer cells can confer resistance to death receptor-mediated apoptosis. They suggest a novel mechanism by which tumor cells may become resistant to TRAIL and provide a mechanism-based strategy to restore TRAIL responsiveness.
Materials and Methods
Cell culture, reagents, subcellular fractionation and western blot
See Supplementary Information.
Generation of stable TRAIL-resistant LiM6-TR cells
Parental LS-LiM6 cells31 were treated with 100 ng/ml human recombinant TRAIL (R&D Systems, Danvers, MA, USA, #375-TEC) for 24 h, resulting in 65% cell death. The apoptotic cells (cells floating in the medium) were then removed and the surviving cells were propagated in the presence of 10 ng/ml TRAIL. After five cycles of exposure to TRAIL, the stable TRAIL-resistant cell line LiM6-TR was obtained. The sensitivity of LiM6-TR to TRAIL was periodically examined by PI (Sigma, St. Louis, MO, USA) staining and flow cytometry, and never exceeded 5% cell death.
Cytotoxicity and apoptosis assays
Cell viability was assessed by the MTT (3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide) assay (Promega, Madison, WI, USA, G109) according to manufacturers protocol. Colorimetric reading at 570 nm was performed using an MRX Revelation microplate reader (Dynex, Richfield, MN, USA). The results are expressed as mean percentage of viable cells from triplicate determinations. Apoptosis was assessed by PARP degradation and was quantified by using PI staining (BD Biosciences, San Diego, CA, USA, #556547) and flow cytometry, and analyzed by CellQuant PRO software (BD Biosciences) as previously described.17
DISC isolation and co-immunoprecipitation experiments
1 × 107 cells were plated to achieve 80% confluence and treated with 500 ng/ml Flag-tagged TRAIL complexed with anti-Flag antibody (Enzo, Farmington, NY, USA) for 30 min at 37°C, and then trypsinized, spun down, washed twice with ice-cold PBS, and lysed for 30 min on ice in DISC immunoprecipitation (IP) lysis buffer (30 mM Tris, pH 7.5, 150 mM NaCl, 10% glycerol, 1% Triton X-100, with anti-protease and phosphatase inhibitors cocktail, from Pierce, Rockford, IL, USA). Cell lysates were incubated with Protein G Plus agarose (Santa Cruz Biotechnology, Santa Cruz, CA, USA) overnight at 4°C. In control experiments, the Flag-tagged TRAIL and anti-Flag complex were added after cell lysis. In co-immunoprecipitation experiments, untreated cell lysates were incubated with either anti-DR4 or anti-DR5 or normal IgG antibodies (in control experiments) overnight at 4°C. Complexes were precipitated by Protein G Plus agarose. The beads were subsequently washed four times with DISC IP lysis buffer and boiled in 2 × SDS sample buffer for 5 min. Samples were resolved on SDS-PAGE gels and subjected to western immunoblot analysis.
Flow cytometry assays
Assays of surface expression and internalization of galectin-3 and death receptors were performed as follows: Cells were collected in Versene solutions (Invitrogen, Carlsbad, CA, USA, #15040-066), washed, and blocked with flow cytometry buffers (Santa Cruz Biotechnology) followed by labeling with 5 μg of either rat anti-galectin-3 (TIB-166 ATCC, Manassas, VA, USA) or goat anti-DR4 or mouse anti-DR5 (R&D Systems) or with the corresponding control IgG antibodies (Abcam, Cambridge, MA, USA) for 1 h on ice. Secondary antibodies used were Alexa Fluor 594-labeled donkey anti-rat, Alexa Fluor 647-labeled donkey anti-goat, or Alexa Fluor 488-labeled donkey anti-mouse, respectively (Invitrogen). For the assessment of loss of cell surface death receptors, cells were pretreated with 100 ng/ml TRAIL for 30 min at 37°C followed by labeling with anti-DR4 or anti-DR5 as above. Cells were then fixed with FCM fixative buffer (Santa Cruz Biotechnology, #sc-3622) and analyzed with a XL-MCL FACS Cytometer (BD Biosciences) for samples labeled with Alexa Fluor 488, or Fortessa FACS cytometer (BD Biosciences) for samples stained with Alexa Fluor 647, and FlowJo version 8.8.6 acquisition/analysis software (BD Biosciences).
Confocal microscopy
Cells were cultured on glass chamber slides and treated as in FACS analysis. Nuclei were stained with ProLong Gold antifade reagent with DAPI (Invitrogen, #P36935). For monitoring death receptor endocytosis, cells were labeled with anti-DR4 and anti-DR5 antibodies and incubated with 100 ng/ml TRAIL on ice for 30 min. Internalization of death receptor immunocomplexes was initiated by raising the temperature to 37°C for 30 min. The cell surface-labeled DR4 and DR5 were removed by incubation with 0.2 M acetic acid in 0.2 M NaCl on ice for 5 min. Cells were then fixed and permeabilized with FCM buffer (Santa Cruz Biotechnology, #sc-3622) and incubated with the corresponding Alexa Fluor-conjugated antibodies as used for FACS analysis. Cells were visualized with a × 20, × 60, or × 100 oil immersion objective on an Olympus FluoView FV300 laser scanning confocal microscope (Olympus, Melville, NY, USA). Co-localization was quantified morphometrically using 3Is Slide Book 5.0 Computing Pearson's coefficients.
sh-RNA silencing of galectin-3
The galectin-3 sh-RNA (#sc-155994-V) and non-targeting control sh-RNA (#sc-108080) lentiviral particles were from Santa Cruz Biotechnology. Cells were infected with 20 μl of 1 × 106 infectious units of virus according to manufacturer's protocol. After transduction, stable cell lines expressing the galectin-3 (or control) sh-RNA were isolated by selection with puromycin. Following propagation for four passages, cells were harvested for measuring protein expression by western blot analysis or cell surface expression by FACS and confocal microscopy analysis, and tested for viability assays following exposure to TRAIL.
Abbreviations
- TRAIL:
-
tumor necrosis factor-related apoptosis-inducing ligand
- DISC:
-
death-inducing signaling complex
- FADD:
-
Fas-associated death domain
- CRD:
-
carbohydrate-recognition domain
- ECM:
-
extracellular matrix
- PI:
-
propidium iodide
- MTT:
-
(3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide
- IP:
-
immunoprecipitation
- sh-RNA:
-
short hair pin-ribonucleic acid
References
Ashkenazi A, Herbst RS . To kill a tumor cell: the potential of proapoptotic receptor agonists. J Clin Invest 2008; 118: 1979–1990.
Wiezorek J, Holland P, Graves J . Death receptor agonists as a targeted therapy for cancer. Clin Cancer Res 2010; 16: 1701–1708.
Abdulghani J, El-Deiry WS . TRAIL receptor signaling and therapeutics. Expert Opin Ther Targets 2010; 14: 1091–1108.
Wagner KW, Punnoose EA, Januario T, Lawrence DA, Pitti RM, Lancaster K et al. Death-receptor O-glycosylation controls tumor-cell sensitivity to the proapoptotic ligand Apo2L/TRAIL. Nat Med 2007; 13: 1070–1077.
Zhang Y, Yoshida T, Zhang B . TRAIL induces endocytosis of its death receptors in MDA-MB-231 breast cancer cells. Cancer Biol Ther 2009; 8: 917–922.
Akazawa Y, Mott JL, Bronk SF, Werneburg NW, Kahraman A, Guicciardi ME et al. Death receptor 5 internalization is required for lysosomal permeabilization by TRAIL in malignant liver cell lines. Gastroenterology 2009; 136: 2365–2376.
Kohlhaas SL, Craxton A, Sun XM, Pinkowski MJ, Cohen GM . Receptor-mediated endocytosis is not required for tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis. J Biol Chem 2007; 282: 12831–12841.
Austin CD, Lawrence DA, Peden AA, Varfolomeev EE, Totpal K, De Maziere AM et al. Death-receptor activation halts clathrin-dependent endocytosis. Proc Natl Acad Sci USA 2006; 103: 10283–10288.
Suliman A, Lam A, Datta R, Srivastava RK . Intracellular mechanisms of TRAIL: apoptosis through mitochondrial-dependent and -independent pathways. Oncogene 2001; 20: 2122–2133.
Ehrhardt H, Fulda S, Schmid I, Hiscott J, Debatin KM, Jeremias I . TRAIL induced survival and proliferation in cancer cells resistant towards TRAIL-induced apoptosis mediated by NF-kappaB. Oncogene 2003; 22: 3842–3852.
Falschlehner C, Emmerich CH, Gerlach B, Walczak H . TRAIL signalling: decisions between life and death. Int J Biochem Cell Biol 2007; 39: 1462–1475.
Walczak H, Miller RE, Ariail K, Gliniak B, Griffith TS, Kubin M et al. Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat Med 1999; 5: 157–163.
Jin Z, McDonald ER, Dicker DT, El-Deiry WS . Deficient tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) death receptor transport to the cell surface in human colon cancer cells selected for resistance to TRAIL-induced apoptosis. J Biol Chem 2004; 279: 35829–35839.
Kuang AA, Diehl GE, Zhang J, Winoto A . FADD is required for DR4- and DR5-mediated apoptosis: lack of trail-induced apoptosis in FADD-deficient mouse embryonic fibroblasts. J Biol Chem 2000; 275: 25065–25068.
Zhang L, Zhu H, Teraishi F, Davis JJ, Guo W, Fan Z et al. Accelerated degradation of caspase-8 protein correlates with TRAIL resistance in a DLD1 human colon cancer cell line. Neoplasia 2005; 7: 594–602.
Larribere L, Khaled M, Tartare-Deckert S, Busca R, Luciano F, Bille K et al. PI3 K mediates protection against TRAIL-induced apoptosis in primary human melanocytes. Cell Death Differ 2004; 11: 1084–1091.
Mazurek N, Sun YJ, Liu KF, Gilcrease MZ, Schober W, Nangia-Makker P et al. Phosphorylated galectin-3 mediates tumor necrosis factor-related apoptosis-inducing ligand signaling by regulating phosphatase and tensin homologue deleted on chromosome 10 in human breast carcinoma cells. J Biol Chem 2007; 282: 21337–21348.
Oka N, Nakahara S, Takenaka Y, Fukumori T, Hogan V, Kanayama HO et al. Galectin-3 inhibits tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by activating Akt in human bladder carcinoma cells. Cancer Res 2005; 65: 7546–7553.
Barondes SH, Castronovo V, Cooper DN, Cummings RD, Drickamer K, Feizi T et al. Galectins: a family of animal beta-galactoside-binding lectins. Cell 1994; 76: 597–598.
Iurisci I, Tinari N, Natoli C, Angelucci D, Cianchetti E, Iacobelli S . Concentrations of galectin-3 in the sera of normal controls and cancer patients. Clin Cancer Res 2000; 6: 1389–1393.
Gong HC, Honjo Y, Nangia-Makker P, Hogan V, Mazurak N, Bresalier RS et al. The NH2 terminus of galectin-3 governs cellular compartmentalization and functions in cancer cells. Cancer Res 1999; 59: 6239–6245.
Dumic J, Dabelic S, Flogel M . Galectin-3: an open-ended story. Biochim Biophys Acta 2006; 1760: 616–635.
Garner OB, Baum LG . Galectin-glycan lattices regulate cell-surface glycoprotein organization and signalling. Biochem Soc Trans 2008; 36: 1472–1477.
Thiemann S, Baum LG . The road less traveled: regulation of leukocyte migration across vascular and lymphatic endothelium by galectins. J Clin Immunol 2011; 31: 2–9.
Earl LA, Bi S, Baum LG . Galectin multimerization and lattice formation are regulated by linker region structure. Glycobiology 2011; 21: 6–12.
Nangia-Makker P, Balan V, Raz A . Regulation of tumor progression by extracellular galectin-3. Cancer Microenviron 2008; 1: 43–51.
Wei J, Barr J, Kong LY, Wang Y, Wu A, Sharma AK et al. Glioma-associated cancer-initiating cells induce immunosuppression. Clin Cancer Res 2010; 16: 461–473.
Inohara H, Raz A . Functional evidence that cell surface galectin-3 mediates homotypic cell adhesion. Cancer Res 1995; 55: 3267–3271.
Chan FK, Chun HJ, Zheng L, Siegel RM, Bui KL, Lenardo MJ . A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science 2000; 288: 2351–2354.
Clancy L, Mruk K, Archer K, Woelfel M, Mongkolsapaya J, Screaton G et al. Preligand assembly domain-mediated ligand-independent association between TRAIL receptor 4 (TR4) and TR2 regulates TRAIL-induced apoptosis. Proc Natl Acad Sci USA 2005; 102: 18099–18104.
Bresalier RS, Raper SE, Hujanen ES, Kim YS . A new animal model for human colon cancer metastasis. Int J Cancer 1987; 39: 625–630.
Garofalo M, Di Leva G, Romano G, Nuovo G, Suh SS, Ngankeu A et al. miR-221&222 regulate TRAIL resistance and enhance tumorigenicity through PTEN and TIMP3 downregulation. Cancer Cell 2009; 16: 498–509.
Cummins JM, Kohli M, Rago C, Kinzler KW, Vogelstein B, Bunz F . X-linked inhibitor of apoptosis protein (XIAP) is a nonredundant modulator of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis in human cancer cells. Cancer Res 2004; 64: 3006–3008.
Fulda S, Wick W, Weller M, Debatin KM . Smac agonists sensitize for Apo2L/TRAIL- or anticancer drug-induced apoptosis and induce regression of malignant glioma in vivo. Nat Med 2002; 8: 808–815.
Igney FH, Krammer PH . Death and anti-death: tumour resistance to apoptosis. Nat Rev Cancer 2002; 2: 277–288.
Gross A, Yin XM, Wang K, Wei MC, Jockel J, Milliman C et al. Caspase cleaved BID targets mitochondria and is required for cytochrome c release, while BCL-XL prevents this release but not tumor necrosis factor-R1/Fas death. J Biol Chem 1999; 274: 1156–1163.
Lau KS, Partridge EA, Grigorian A, Silvescu CI, Reinhold VN, Demetriou M et al. Complex N-glycan number and degree of branching cooperate to regulate cell proliferation and differentiation. Cell 2007; 129: 123–134.
Guo HB, Johnson H, Randolph M, Lee I, Pierce M . Knockdown of GnT-Va expression inhibits ligand-induced downregulation of the epidermal growth factor receptor and intracellular signaling by inhibiting receptor endocytosis. Glycobiology 2009; 19: 547–559.
Lee KH, Feig C, Tchikov V, Schickel R, Hallas C, Schütze S et al. The role of receptor internalization in CD95 signaling. EMBO J 2006; 25: 1009–1023.
Schneider-Brachert W, Tchikov V, Neumeyer J, Jakob M, Winoto-Morbach S et al. Compartmentalization of TNF receptor 1 signaling: internalized TNF receptosomes as death signaling vesicles. Immunity 2004; 21: 415–428.
Acknowledgements
Supported by National Cancer Institute grant RO1CA69480 (RSB). Cytometric analyses, confocal microsccopy, and STR DNA fingerprinting were done by the Flow Cytometry and Cellular Imaging Core Facility and the Characterized Cell Line Core, both supported by the Cancer Center Support grant, NCI # CA16672.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by JP Medema
Supplementary Information accompanies the paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article
Cite this article
Mazurek, N., Byrd, J., Sun, Y. et al. Cell-surface galectin-3 confers resistance to TRAIL by impeding trafficking of death receptors in metastatic colon adenocarcinoma cells. Cell Death Differ 19, 523–533 (2012). https://doi.org/10.1038/cdd.2011.123
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2011.123
Keywords
This article is cited by
-
Galectin-3 promotes secretion of proteases that decrease epithelium integrity in human colon cancer cells
Cell Death & Disease (2023)
-
Mucin 21 confers resistance to apoptosis in an O-glycosylation-dependent manner
Cell Death Discovery (2022)
-
Glycosylation and raft endocytosis in cancer
Cancer and Metastasis Reviews (2020)
-
Decoding the sweet regulation of apoptosis: the role of glycosylation and galectins in apoptotic signaling pathways
Cell Death & Differentiation (2019)
-
N-glycosylation of mouse TRAIL-R restrains TRAIL-induced apoptosis
Cell Death & Disease (2018)
