
Abstract
Fibrosis is a chronic process involving development and progression of multiple diseases in various organs and is responsible for almost half of all known deaths. Epithelial–mesenchymal transition (EMT) is the vital process in organ fibrosis. Lens is an elegant biological tool to investigate the fibrosis process because of its unique biological properties. Using gain- and loss-of-function assays, and different lens fibrosis models, here we demonstrated that microRNA (miR)-26a and miR-26b, members of the miR-26 family have key roles in EMT and fibrosis. They can significantly inhibit proliferation, migration, EMT of lens epithelial cells and lens fibrosis in vitro and in vivo. Interestingly, we revealed that the mechanisms of anti-EMT effects of miR-26a and -26b are via directly targeting Jagged-1 and suppressing Jagged-1/Notch signaling. Furthermore, we provided in vitro and in vivo evidence that Jagged-1/Notch signaling is activated in TGFβ2-stimulated EMT, and blockade of Notch signaling can reverse lens epithelial cells (LECs) EMT and lens fibrosis. Given the general involvement of EMT in most fibrotic diseases, cancer metastasis and recurrence, miR-26 family and Notch pathway may have therapeutic uses in treating fibrotic diseases and cancers.
Similar content being viewed by others


Main
Fibrosis is a chronic multiple organ disease and is responsible for almost half of all known deaths. Some severe fibrosis cases even have a lower survival rate than cancer.1 Pathological epithelial–mesenchymal transition (EMT) is a critical element in organ fibrosis, such as pulmonary fibrosis, renal fibrosis, hepatic fibrosis and ocular fibrosis. In fibrotic tissues, myofibroblasts accumulate and secrete excessive collagen, thereby damaging organ function and leading to its failure.2 There is currently no specific therapy that targets fibroblast-associated pathology. It is essential to better understand the cellular and molecular mechanism of fibrotic event. Former researches have proposed that lens is an elegant biological tool to investigate the fibrosis processes because of its unique biological properties.3 Lens fibrotic disorders, such as anterior subcapsular cataract (ASC) and posterior capsule opacification (PCO), are common causes of visual impairment all over the world. Therefore, using the lens as a model for fibrosis not only has direct relevance to lens fibrotic disorders, but also serves as a valuable experimental tool to understand fibrosis essentially.
ASC and PCO are two types of cataract but share many cellular and molecular features.4, 5 ASC is a primary cataract, which is characterized by dense light-scattering fibrotic regions underneath the anterior capsule of the lens. It is mainly caused by ocular trauma, inflammation or irritation.3 PCO is known as a secondary cataract, a major long-term complication of cataract surgery, occurring in 20 to 40% of patients after surgery, particularly, almost 100% in children and infants.6, 7 The cellular mechanism of ASC and PCO is the proliferation, migration and EMT of lens epithelial cells (LECs), leading to the transition from epithelium to fibroblasts, and the production of extracellular matrix (ECM) proteins (collagens I, IV and fibronectin), which finally contributes to the formation of subcapsular plaques beneath the lens anterior or posterior capsule.4, 8 TGFβ, especially TGFβ2, the major isoform in the aqueous humor of the eye, is the most important factor in these processes.9 Hence, inhibition of LECs proliferation, migration and EMT may be a potential strategy to prevent ASC and PCO.
Aberrant Notch signaling has been found in a range of different cancers and fibrotic diseases.10, 11, 12, 13 The inactivation of Notch signaling is important in overcoming drug resistance and the reversal of EMT phenotype, which may improve the overall survival of cancer and fibrosis patients. In mammals, there are four cell surface transmembrane receptors (Notch-1–4) and five ligands (Jagged-1, Jagged-2, Delta-like 1, Delta-like 3 and Delta-like 4).14 On ligand binding, the Notch receptors are cleaved by γ-secretase, releasing Notch intracellular domain (NICD), which subsequently translocates into the nucleus and regulates downstream target genes,15 including Hes and Hey.10 Previous studies have identified that Jagged-1/Notch signaling is required for TGFβ1-induced EMT in kidney and alveolar epithelial cells, using specific inhibitor of Notch signaling alleviates TGFβ1-EMT, suggesting that pharmacological targeting of Notch pathway may be beneficial in the treatment of kidney fibrosis and pulmonary fibrosis.16, 17, 18 Despite its definite effects on EMT inhibition, the function of Notch signaling in LEC–EMT is currently unstated.
MicroRNAs (miRNAs, miRs) are small noncoding RNAs that negatively regulate gene expression at the posttranscriptional level. Through binding to complementary sequences in the 3′-untranslated regions (3′-UTR) of their target mRNAs, miRNAs can induce mRNA degradation or translation suppression.19 Accumulating evidence has demonstrated that miRNAs have important roles in EMT, such as miR-200 family,20 miR-29b,21 miR-34a22 and miR-491-5p.23 MiR-26 family comprises two mature miRNAs: miR-26a and miR-26b. They have been both reported to be tumor suppressors in hepatocellular carcinoma,24 breast cancer25, 26 and glioma development.27 MiR-26b can inhibit tumor cell growth and induce cell apoptosis by targeting PTGS2 and SLC7A11,25, 26 and suppress tumor cell migration and invasion by directly regulating EphA2.27 Regard to the roles of miR-26 family in fibrosis, Liang et al.28 has demonstrated the antifibrotic role of miR-26a by directly targeting Smad4, inhibits the nuclear translocation of p-Smad3, and further represses TGFβ1-induced fibrogenesis in idiopathic pulmonary fibrosis. Wei et al.29 and Liang et al.30 also reported miR-26a can inhibit cardiac fibrosis and idiopathic pulmonary fibrosis by targeting collagen I, CTGF and HMGA2, respectively. The latest studies also demonstrated that miR-26b is significantly decreased in patient PCO tissues,31 and overexpression of it can inhibit LEC–EMT by targeting Smad4 and COX-2.32 However, the function of miR-26a in lens fibrosis is entirely unknown.
In the present study, we focused on miR-26a and -26b to further explore their functions in fibrosis using the lens as a model. We applied gain- and loss-of-function assays and different models to demonstrate miR-26a and miR-26b are EMT suppressors in lens fibrosis. Importantly, we revealed a novel mechanism underlying the functions of miR-26a and miR-26b by showing that they inhibit EMT via directly targeting Jagged-1 and suppressing Jagged-1/Notch signaling. Furthermore, using of Jagged-1 siRNA and Notch pathway specific inhibitor DAPT can also reverse LEC–EMT and ASC development. Our data suggest that miR-26 family and pharmacological targeting of Notch pathway may be of therapeutic value in the prevention and treatment of ASC, PCO and other organ fibrosis.
Results
Downregulation of miR-26a and -26b in TGFβ2-induced EMT in LECs and the injury-induced ASC mouse model
To determine which miRNA species may be involved in regulating LEC–EMT and the development of ASC, we first compared the miRNA expression profiles in TGFβ2-induced LEC–EMT. Microarray analysis revealed that 101 miRNAs were elevated, and 223 were downregulated in TGFβ2-induced LEC–EMT (Figures 1a and 1b). These miRNAs included miR-26b, which has been reported to be downregulated in human PCO tissues.31 Using real-time PCR analysis, we verified that miR-26b was reduced in response to TGFβ2 exposure at 24 and 48 h by 35% and 39%, respectively. As miR-26a also belongs to the miR-26 family, we examined the miR-26a expression meanwhile. Real-time PCR analysis demonstrated that miR-26a was simultaneously downregulated in TGFβ2-induced LEC–EMT (Figure 1c). In addition, we also validated the levels of miR-26a and -26b in injury-induced ASC model in mice. As expected, miR-26a and -26b were both rapidly decreased at day 1 and day 3 after injury, whereas at day 7, their expression returned to the baseline (Figure 1d). Collectively, these results indicate that miR-26a and -26b may have important roles in LEC–EMT and the development of ASC.

MiR-26a and miR-26b are downregulated in TGFβ2-induced EMT in LECs and injury-induced ASC model in mice. (a) MiRNA array analysis shows the representative downregulated miRNAs in TGFβ2-stimulated LECs for 48 h. The pseudocolor represents the intensity scale of the TGFβ2 (5 ng/ml) treatment group versus the control group (n=3 per group). (b) MiRNA array analysis shows the representative upregulated miRNAs in TGFβ2-stimulated LECs for 48 h. (c) Real-time PCR analysis of the expression of miR-26a and -26b in response to TGFβ2 (5 ng/ml) in LECs at different time points. (d) Real-time PCR analysis of miR-26a and -26b in injury-induced ASC in mice at day 1, day 3 and day 7 after injury. *P<0.05
MiR-26a and -26b inhibit LEC proliferation, migration and TGFβ2-induced EMT
To understand the biological functions of miR-26a and -26b in LECs, we then transfected LEC cell line SRA01/04 with miR-26a and -26b mimic and inhibitor oligonucleotides, and examined LECs proliferation, migration and TGFβ2-induced EMT by using gain- and loss-of-function experiments. Real-time PCR analysis confirmed that high and low levels of miR-26a and -26b expression were achieved in mimic- and inhibitor-transfected cells after 48 h compared with the negative control (NC)-transfected cells (Supplementary Figure S1a). Interestingly, EdU staining showed that the proliferations of LECs were markedly suppressed after transfection with miR-26a and -26b mimics, while increased in inhibitor-transfected cells (Figures 2a and b). Moreover, wound-healing assay demonstrated that overexpression of miR-26a and -26b both dramatically inhibited LECs migration, while downregulation of them induced LECs migration conversely (Supplementary Figures S1b and S1c). Subsequently, TGFβ2 was applied to establish LEC–EMT model in vitro. As showed in Figure 2 and Supplementary Figure S2, the results of real-time RCR, western blot and immunofluorescence staining all showed that TGFβ2 treatment induced EMT markers α-SMA, collagen type I (Col I), collagen type IV (Col IV) and fibronectin (FN), and EMT transcription factors Snail and Slug expression in LECs. Strikingly, overexpression of miR-26a and -26b both exhibited a significant downregulation of α-SMA, Col I, Col IV, FN, Snail and Slug induced by TGFβ2. Conversely, knockdown of miR-26a and -26b upregulated α-SMA, FN, Snail and Slug without TGFβ2 (Supplementary Figure S2b). These data suggest that miR-26a and -26b can significantly suppress LECs proliferation, migration and attenuate TGFβ2-induced EMT in vitro.

MiR-26a and -26b inhibit LECs proliferation and TGFβ2-induced EMT in vitro. (a) EdU staining analysis of LECs proliferation after transfected with miRNA negative control mimic (NC-m), miR-26a mimic (26a-m), or miR-26b mimic (26b-m), miRNA negative control inhibitor (NC-i), miR-26a inhibitor (26a-i), or miR-26b inhibitor (26b-i) for 48 h, respectively. Scale bar, 40 μm. (b) Quantification of EdU-positive cells (n=24 randomized fields per group); *P<0.05. (c) Immunofluorescent staining analysis of EMT markers FN, Col I and Col IV in LECs transfected with miRNA negative control mimic, miR-26a mimic or miR-26b mimic, and treated with TGFβ2 (5 ng/ml) for 48 h. Scale bar, 20 μm. (d) Western blot analysis of FN, α-SMA, Col IV, Snail and Slug protein levels in LECs transfected and treated as indicated in (c)
MiR-26a and -26b prevent injury-induced ASC in the mouse lens anterior capsular injury model
On the basis of the results in vitro, we next validated the anti-EMT roles of miR-26a and -26b in vivo. The mouse lens anterior capsular injury model is a well-established way for studying LECs proliferation, EMT, ECM components deposition and subcapsular plaque formation, as typically seen in human ASC and PCO. In this model, gain-of-function study was conducted using miR-26a and -26b agomirs, which were applied to stably induce endogenous expression of miRNAs in vivo. As expected, after miR-26a and -26b agomirs were injected into the anterior chamber of the mice, they could markedly induce the expression of miR-26a and 26b in the lens, and maintained for at least 7 days (Supplementary Figure S3). Strikingly, overexpression of miR-26a and -26b dramatically decreased EMT markers α-SMA, FN, Col I and vimentin expression induced by injury of lens anterior capsular for 1, 3 and 7 days (Figure 3a). Lens anterior capsule whole-mount staining also showed lenses developed remarkable multilayered LECs opacities around the capsular break and beneath the lens anterior capsule when the lenses were injured for 7 days, and strong staining of α-SMA, Col I and vimentin could be seen in the multilayered LECs opacities. However, in the miR-26a and -26b overexpression lens, the volumes of the subcapsular plaques and the expression of α-SMA, Col I and vimentin were obviously decreased compared with the NC agomir group (Figures 3b and c). Together, these findings further prove that miR-26a and -26b are negative regulators in LEC–EMT and ASC development in vivo.

MiR-26a and -26b prevent injury-induced ASC in vivo. The anterior capsules of mouse lens were punctured with a needle and 1 μl of 1 nM of miRNA negative control (NC) agomir, miR-26a agomir or miR-26b agomir were injected into the anterior chamber of the eye immediately after injury with a microsyringe. (a) One, 3 and 7 days later, lenses were harvested for real-time PCR analysis of α-SMA, FN, Col I and vimentin mRNA levels. *P<0.05; **P<0.01. (b) Representative confocal microscopy 3D images of lens capsule whole mounts show regions of subcapsular plaques, and EMT markers vimentin, Col I, and α-SMA staining after 7 days. Scale bar, 20 μm. (c) Quantification of the subcapsular plaques volumes, and EMT markers vimentin, Col I and α-SMA distributions (n=6 lenses per group). *P<0.05
MiR-26a and -26b inhibit LEC–EMT via directly targeting Jagged-1 and suppressing Jagged-1/Notch signaling
To clarify the underlying mechanisms of the inhibitory effects of miR-26a and -26b on LEC–EMT, we used two miRNA target identification tools PicTar (http://pictar.mdc-berlin.de/) and TargetScan 4.2 (http://www.targetscan.org/vert_42/) to search for potential target genes of miR-26a and -26b. We found that Jagged-1 is a potential target of miR-26a and -26b (Figure 4a). In light of the critical role of the Jagged/Notch pathway in EMT during embryonic development, fibrotic diseases and cancer metastasis,33, 34 we then examined whether miR-26a and -26b inhibit LEC–EMT via regulating Jagged-1/Notch pathway. To confirm this, luciferase reporter carrying two miR-26a and -26b potential-binding sites or mutant-binding sites of Jagged-1 3′-UTR were constructed and co-transfected with miR-26a, -26b or NC mimic into the 293 T cells. Compared with the NC mimic group, transfection with miR-26a and -26b mimics reduced the luciferase activities significantly in the wild-type and site 1 mutant construct (Figure 4b). However, NC, miR-26a and -26b mimics did not affect the luciferase activities in the site 2 mutant and the site 1 plus site 2 mutant constructs (Figure 4b). These findings indicate that miR-26a and -26b directly interacts with the 3′-UTR of Jagged-1 via binding to site 2, but not site 1.

MiR-26a and -26b inhibit LEC–EMT via directly targeting Jagged-1/Notch signaling, and Jagged-1 siRNA and Notch pathway specific inhibitor DAPT reverse LEC–EMT in vitro. (a) Predicted miR-26a and -26b target two sites of potential-binding sequences in Jagged-1-3′UTR and mutants containing four mutated nucleotides (green) in two sites of Jagged-1-3′UTR, respectively. (b) Normalized luciferase activities of reporters containing wild-type or mutant 3′-UTR of Jagged-1 in 293 T cells co-transfected with miRNA negative control mimic (NC-m), miR-26a mimic (26a-m) or miR-26b mimic (26b-m). *P<0.05. (c) Western blot analysis of Jagged-1, Notch-1 and Notch-3 protein levels in LECs transfected with miRNA negative control mimic, miR-26a mimic or miR-26b mimic, and treated with TGFβ2 (5 ng/ml) for 48 h. (d) Immunofluorescent staining analysis of EMT markers FN, N-cadherin, Col IV and vimentin in LECs transfected and treated as indicated in c. Scale bar, 40 μm. (e) Western blot analysis of Jagged-1, Col IV, FN, Snail, Slug and ZEB1 protein levels in LECs transfected with control siRNA, or Jagged-1 siRNA, and treated with TGFβ2 (5 ng/ml) for 48 h. (f) Western blot analysis of Col IV, FN, α-SMA, N-cadherin, Snail, Slug and ZEB1 protein levels in LECs exposure to TGFβ2 with or without DAPT (2.5 μM) for 48 h
Although Jagged-1/Notch pathway has been extensively studied in cancer and fibrotic diseases, the role of Notch signaling in LEC–EMT is largely unknown. Therefore, we next investigated whether Notch pathway is involved in TGFβ2-stimulated EMT in LECs. As shown in Figure 5, TGFβ2 could significantly induce the expression of Notch pathway ligand Jagged-1, receptors Notch-1, Notch-2 and Notch-3, and the downstream target genes Hes-1 and Hey-1. Jagged-1 was the most significantly upregulated gene in response to TGFβ2 treatment. However, TGFβ2-induced activation of Notch signaling was dramatically inhibited by SB431542, a specific inhibitor for TGFβ/Smad2/3 signaling. These results suggest that TGFβ2 activates Jagged-1/Notch pathway via the canonical TGFβ/Smad2/3 signaling. Next, we confirmed whether miR-26a and -26b inhibit LEC–EMT via directly regulating Jagged-1/Notch signaling. The results from real-time RCR and western blot showed that the mRNA and protein levels of Jagged-1 were markedly decreased in miR-26a and -26b mimics-transfected LECs, but slightly increased in cells transfected with miR-26a and -26b inhibitors (Supplementary Figure S4 and Figure 4c). This implies that miR-26a and -26b regulate the expression of Jagged-1 at the transcriptional and the posttranscriptional levels. Besides, Notch-1 and Notch-3 were also reduced in miR-26a and -26b mimic-transfected cells (Figure 4c), while increased in inhibitor-transfected cells (Supplementary Figure S4d). Furthermore, real-time PCR results revealed that overexpression of miR-26a and -26b in injury-induced ASC model also suppressed Jag-1, Notch-1, Notch-2 and Notch-3 expression in vivo (Supplementary Figure S5). Taken together, these results reveal that the activation of Jagged-1/Notch pathway is involved in LEC–EMT, and the mechanism of the inhibitory effects of miR-26a and -26b on LEC–EMT is via directly targeting Jagged-1 and suppressing Jagged-1/Notch signaling.

TGFβ2 activates Jagged-1/Notch pathway in LECs EMT through canonical Smad2/3 signaling. (a) Real-time PCR analysis of Jagged-1, Notch-1, Notch-2, Notch-3, Hes-1 and Hey-1 mRNA levels in LECs treated with different concentrations of TGFβ2 (2.5, 5, 10 and 30 ng/ml) for 48 h. *P<0.05; **P<0.01. (b) Western blot analysis of Jagged-1, Notch-1, Notch-2 and Notch-3 protein levels in LECs treated as indicated in (a). (c) Real-time PCR analysis of Jagged-1, Notch-1, Notch-2, Notch-3, Hes-1 and Hey-1 mRNA levels in LECs exposure to TGFβ2 with DAPT (2.5 μM) or SB431542 (10 μM) for 48 h. *P<0.05; **P<0.01
Jagged-1 siRNA and blockade of Notch signaling reverse TGFβ2-induced EMT in LECs
To further explore the role of Jagged-1/Notch signaling in LEC–EMT, we study the impacts of blockade of Notch pathway via knockdown of Jagged-1 using siRNA and DAPT (a γ-secretase inhibitor) in TGFβ2-induced LEC–EMT. As illustrated in Figure 4 and Supplementary Figure S6, inhibition of Notch signaling by Jagged-1 siRNA and DAPT both abrogated the upregulation of EMT markers FN, Col IV, N-cadherin, α-SMA and EMT key transcription factors Snail, Slug and ZEB1 expression. Furthermore, wound-healing and EdU-staining assays demonstrated that DAPT dramatically inhibited LECs migration, whereas had no effect on cell proliferation (Supplementary Figure S7). These data clearly demonstrate that downregulation of Jagged-1 and blockade of Notch pathway can reverse TGFβ2-induced EMT phenotype in LECs.
Blockade of Notch pathway abrogates TGFβ2-induced ASC in vitro and injury-induced ASC in vivo
Previous studies have identified that TGFβ can induce the whole lens cultured in vitro to form opacities that contain morphologic and pathological markers for ASC.35 To further investigate whether blockade of Notch signaling by DAPT reverses TGFβ2-induced EMT in the lens in a more complicated system, we utilized the whole lens culture semi-in vivo model. When the lenses from 20- to 22-day-old rats were cultured with 5 ng/ml of TGFβ2 for 7 days, the lenses developed obvious clumpy opacities beneath the lens capsule in each lens, whereas DAPT abrogated TGFβ2-induced ASC and the lenses remained transparent as cultured without TGFβ2 (Figure 6a). As shown in Figures 6b and c, the morphology of the frozen sections showed that the aberrant cells were co-localized with the subcapsular clumpy opacities. The clumps contained accumulations of FN and Col IV, and increases of α-SMA and vimentin expression. In contrast, the lenses cultured with DAPT retained normal lens morphology, and did not have obvious accumulations of FN, Col IV, α-SMA and vimentin. In addition, we also demonstrated that DAPT dramatically reduced the upregulation of α-SMA, Col I, FN, Snail and Slug at mRNA and the protein levels in rat lenses induced by TGFβ2 for 7 days (Figures 6d and e).

Blockade of Notch pathway with DAPT prevents TGFβ2-induced ASC in vitro. Rat lenses were cultured in the absence or presence of TGFβ2 with DAPT (5.0 μM) or DMSO for 7 days. (a) Representative dissecting microscope images of the morphological changes of the lenses cultured for 7 days (n=12 lenses per group). (b) Representative confocal microscopy images of frozen sections staining for α-SMA, Col IV, FN and vimentin (n=6 lenses per group). Scale bar, 20 μm. (c) Quantification of the area of α-SMA, Col IV, FN and vimentin per field (n=24 randomized fields per group). **P<0.01. (d) Seven days later, the lenses were harvested for real-time PCR analysis of α-SMA, Col I, FN, Snail and Slug mRNA levels (n=6 lenses per group). *P<0.05; **P<0.01. (e) Western blot analysis of α-SMA, FN and Col I protein levels in lenses
Finally, we used injury-induced mouse ASC model to further verify the inhibitory effects of DAPT on ASC in vivo. First, we found Notch pathway ligands Jagged-1 and Jagged-2, receptors Notch-1, Notch-2 and Notch-3, and the downstream target genes Hes-1 and Hey-1 were obviously upregulated from day 1 to day 3 after injury (Supplementary Figure S8), and returned to the base lines at day 5 (data not shown). These results show that Notch pathway is actually required for injury-induced EMT of lens epithelium. Next, the results from real-time PCR displayed DAPT treatment notably decreased EMT markers α-SMA, FN, Col I and vimentin expression when the lenses were injured for 1, 3 and 7 days (Figure 7a). Immunofluorescent staining of the lens anterior capsule whole-mounts also showed the volumes of the subcapsular plaques and the expression of α-SMA, Col I and vimentin in the subcapsular plaques were obviously decreased in DAPT treated lenses (Figure 7b). Taken together, these findings indicate that Notch signaling has a vital role in the development of ASC, inhibition of Notch signaling can suppress ASC formation in vitro and in vivo.

Blockade of Notch pathway with DAPT inhibits injury-induce ASC in vivo. The anterior capsules of mouse lens were punctured with a needle and 1 μl of 80 μM of DAPT were injected into the anterior chamber of the eye immediately after injury. (a) One, 3 and 7 days later, the lenses were harvested for real-time PCR analysis of α-SMA, FN, Col I and vimentin mRNA levels. *P<0.05; **P<0.01. (b) Representative fluorescence microscope (left column) and confocal microscopy 3D (right column) images of lens capsule whole mounts show regions of subcapsular plaques, and EMT markers α-SMA, vimentin and Col I staining at 7 day after injury. Scale bar, 40 μm in left column; 20 μm in right column. (c) Quantification of the subcapsular plaques volumes, and EMT markers α-SMA, vimentin and Col I distributions (n=6 lenses per group). *P<0.05; **P<0.01
Discussion
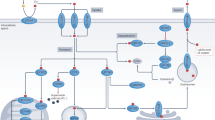
Fibrosis occurs in almost all tissues and organs, which under pathological conditions often impairs organ functions and in some cases significant morbidity and mortality. MiR-26 family has been verified that they have antifibrotic effects in idiopathic pulmonary fibrosis and cardiac fibrosis,28, 29, 30 however, its biological effects on LEC–EMT progression and lens fibrosis remain unclear. Our results from cultured cells in vitro and injury-induced ASC model in vivo revealed that Jagged-1/Notch pathway have a crucial role in LEC–EMT and lens fibrosis. MiR-26a and -26b are EMT and fibrosis suppressors. They markedly inhibit LECs proliferation, migration and EMT, and the development of injury-induced ASC in mice. Notably, we revealed a previously unidentified mechanism that miR-26a and -26b inhibit EMT via directly targeting Jagged-1 and negatively regulating Jagged-1/Notch signaling. Furthermore, blockade of Notch pathway can also reverse LEC–EMT and lens fibrosis (Figure 8).

Schematic illustration of the functions of miR-26a/b-Jagged/Notch axis in modulation of EMT and fibrosis. During the EMT process, TGFβ2 can activate Jagged-1/Notch pathway via the canonical Smad2/3 signaling. Upon ligand binding, the Notch receptors are cleaved by γ-secretase, releasing NICD, which subsequently translocates into the nucleus and regulates downstream target genes expression, leading to EMT and fibrosis. Nevertheless, miR-26a and -26b can directly target Jagged-1 and suppresses Jagged-1/Notch signaling, thus inhibiting EMT and fibrosis. Meanwhile, knockdown of Jagged-1 and using Notch pathway specific inhibitor DAPT can also reverse EMT and fibrosis
MiR-26 family has been reported to be fibrosis suppressors in idiopathic pulmonary fibrosis and cardiac fibrosis,28, 29, 30 however, its role in lens fibrosis is completely unknown. In this study, we found that the expression of miR-26a and -26b were decreased in TGFβ2-induced LEC–EMT. Analysis of the promoter sequence showed there is a potential-binding site for Smad3 in the region upstream of the miR-26a promoter. Previous study has already verified the effect of TGFβ on miR-26a is in fact mediated by Smad3.28 Activation of Smad3 can significantly repress miR-26a expression, meanwhile, knockdown of Smad3 increases miR-26a expression and reverses the downregulation of miR-26a induced by TGFβ. These results indicated that Smad3 negatively regulates transcription of miR-26a.28 Moreover, the complex of Smad2/Smad3/Smad4 also possibly bind to the promotor of miR-26b. Smad4 has been reported to be the target gene of miR-26b,32 so miR-26b downregulation results in upregulation of Smad4, which increases nuclear translocation of p-Smad3 and may further suppresses miR-26b. Therefore, we think miR-26a and -26b downregulations are probably the direct effects of TGFβ. Futhermore, miR-26a and miR-26b were also decreased in injury-induced ASC in mice, which suggesting they may be associated to lens fibrosis.
To gain in-depth understanding of biological functions of miR-26a and -26b in lens fibrosis, we examined their effects on LECs proliferation, migration and EMT using gain- and loss-of-function experiments. Our EdU-staining and wound-healing assays both demonstrated that miR-26a and -26b markedly suppress LECs proliferation and migration. In addition, they exhibit significant inhibition of TGFβ2-induced LEC–EMT in vitro. These are consistent to the previous study.32 With regard to miR-26a, we reported, for the first time, that it suppresses LECs proliferation, migration and EMT as well as miR-26b. Importantly, we further investigated the anti-EMT effects of miR-26a and -26b in vivo using miRNA agomirs. In injury-induced ASC model, miR-26a and -26b agomirs were injected into the anterior chambers of the mouse eyes for the first time. Excitingly, they can successfully induce the expression of miR-26a and -26b in the lens, and maintain for 7 days. Using this model, we found overexpression of miR-26a and -26b dramatically suppressed injury-induced LEC–EMT, ECM components deposition and the development of ASC in vivo. Therefore, miR-26a and -26b both can inhibit LECs proliferation, migration and EMT, and further inhibit injury-induced ASC in vitro and in vivo.
Several target genes of miR-26a and -26b have been identified for fibrosis thus far, including collagen I, CTGF, Smad4 and HMGA2.28, 29, 30 In the current study, we uncovered a novel target gene Jagged-1 for miR-26a and -26b. We confirmed that they can directly interact with the 3′-UTR of Jagged-1, thus negatively regulate Jagged-1 expression. Interestingly, the Notch receptors Notch-1 and Notch-3 were also reduced in the miR-26a and -26b mimic-transfected cells. This implies that miR-26a and -26b inhibit LEC–EMT via directly targeting Jagged-1 and suppressing Jagged-1/Notch signaling. Notch signaling has been reported to have a critical role in EMT during embryonic development, cancer metastasis and various fibrotic diseases.33 However, its functions in LEC–EMT remains poorly understood. Our results demonstrated that Notch pathway is activated by TGFβ2 in LECs through the canonical TGFβ/Smad signaling, and Jagged-1 is the most upregulated gene by TGFβ2 (Figure 8). Subsequently, we found inhibition of Notch signaling entirely reversed TGFβ2-induced LEC–EMT in vitro and TGFβ2-induced ASC formation in the whole lens culture semi-in vivo model. We also further confirmed Notch pathway was actually activated in injury-induced EMT of lens epithelium, and DAPT treatment obviously inhibited injury-induced LEC–EMT and the development of ASC in vivo. Taken together, our data indicate that the activation of Notch signaling has a vital role in LEC–EMT and ASC formation, inactivation of Notch signaling can be useful for the abrogation of LEC–EMT and ASC. It is noteworthy that miR-26a and -26b can both dramatically inhibit LECs proliferation, but blockade of Notch pathway with DAPT has no effect on the proliferation of LECs. This implies that miR-26a and -26b may inhibit the proliferation of LECs via targeting other genes. Such a possibility warrants future studies to explore the mechanism of effect of miR-26 family on cell proliferation.
In summary, using different gain- and loss-of-function assays and multiple model systems, our results provide, for the first time, evidence that miR-26 family and Jagged-1/Notch pathway have important roles in lens fibrosis. MiR-26a and -26b inhibit LECs proliferation, migration, EMT and ASC formation in vitro and in vivo via directly targeting Jagged-1 and negatively regulating Jagged-1/Notch signaling. Our data suggest that miR-26 family and blockade of Notch pathway may be promising strategies in the prevention and treatment of organ fibrosis. These insights into the regulatory relationship between miR-26 family and Notch signaling pathway are not only helpful in understanding the pathogenesis of fibrosis diseases, but also beneficial for studying cancer metastasis, drug resistance and recurrence.
Materials and Methods
Cell culture and treatment
The human LEC line SRA01/04 was kindly provided by Professor Fu Shang at the Laboratory for Nutrition and Vision Research, Tufts University (Boston, MA, USA), and cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, Life Technologies, NY, USA) containing 10% fetal bovine serum (FBS, Gibco, Life Technologies). For TGFβ2 treatment, the cells were seeded in six-well plates and incubated with 5 ng/ml of TGFβ2 (Cell Signaling, Danvers, MA, USA) for 48 h. Pharmacological inhibitors DAPT (a specific inhibitor of Notch receptor cleavage, Sigma-Aldrich, Louis, MO, USA) and SB431542 (a specific inhibitor of TGFβ/Smad2/3 signaling, Sigma-Aldrich) were added 60 min before treatment with TGFβ2.
Microarray analysis
The preparation of RNA samples and microarray analysis were performed commercially by RiboBio Co. Ltd (Guangzhou, China). In brief, total RNA of LECs treatment with or without TGFβ2 was isolated using TRIzol (Invitrogen, Carlsbad, CA, USA) and miRNeasy mini kit (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions, which efficiently recovers all RNA species, including miRNAs. RNA quality and quantity were measured by nanodrop spectrophotometer (ND-1000, Nanodrop Technologies, Wilmington, DE, USA), and RNA integrity was determined by gel electrophoresis. The isolated miRNAs were then labeled with Hy3/Hy5 using the miRCURY Array Power Labeling kit (Exiqon, Vedbaek, Denmark) and hybridized on a miRCURY LNA miRNA Array (v.18.0, Exiqon) according to array manual. Following hybridization, the slides were achieved, washed several times using wash buffer kit (Exiqon) and finally dried by centrifugation for 5 min at 400 r.p.m. Then the slides were scanned using the Axon GenePix 4000B microarray scanner (Axon Instruments, Foster City, CA, USA). The scanned images were then imported into GenePix Pro 6.0 software (Axon Instruments) for grid alignment and data extraction. Bioinformatics analysis and visualization of microarray data were performed with MEV software (v4.6, TIGR). The microarray assays were repeated three times each group.
Model of injury-induced ASC in mouse
All animal experiments were approved by the Animal Use and Care Committee of Zhongshan Ophthalmic Center at the Sun Yat-Sen University, Guangzhou, People’s Republic of China. The model of injury-induced ASC in mouse eyes were performed as described previously.36, 37 Before making the injury, the mice were anesthetized generally with intraperitoneal injection of pentobarbital sodium (70 mg/kg) and topically with dicaine eye drop. After pupillary dilation with compound tropicamide eye drop, a small incision was made in the central anterior capsule with the blade part of a 26-gauge hypodermic needle through the cornea in the right eye of the mouse. The depth of injury was approximately 300 μm or one-fourth of the length of the blade part of the needle. To forced expression of miR-26a and -26b in this model, the mice were randomly divided into three groups to receive 1 μl of 1 nM of miR-26a, miR-26b and NC agomirs (RiboBio; n=30 mice per group). The agomirs were injected into the anterior chambers of the injured eyes immediately after injury with a microsyringe (30-gauge, Hamilton). For DAPT treatment, the mice were randomly divided into two groups to receive 1 μl of 80 μM of DAPT and equivalent amount of DMSO as a control. The animals were allowed to heal for 1, 3 and 7 days. At the end of the treatment period, the mice were killed and the lenses were harvested for real-time PCR and whole-mount staining.
Forced expression and knockdown of miR-26a and -26b in vitro
To examine the functions of miR-26a and -26b in vitro, gain- and loss-of-function experiments were performed. MiRNA mimics and inhibitors were applied to enhance or knockdown the endogenous expression of miR-26a and -26b according to the manufacturer’s protocols. For overexpression of miR-26a and -26b, when LECs grew to 60–70% confluency, the mixture of 100 nM of negative control (NC), miR-26a or -26b mimics (RiboBio) with 5 μl of Lipofectamine 2000 (Invitrogen) was added into the cells in Opti-MEM medium. Conversely to knockdown of miR-26a and -26b, the cells were transfected with 200 nM of NC, miR-26a or -26b inhibitors (RiboBio) with 10 μl of Lipofectamine 2000. After 4–6 h incubation, the medium was replaced and LECs exposed to 5 ng/ml of TGFβ2 for a further 48 h.
Wound-healing assay
After miR-26a and -26b mimics or inhibitors transfection, streaks were created with a 100 μl yellow micropipette tip. For DAPT treatment, LECs were further incubated with or without DAPT (2.5 μM) for 48 h. Progression of migration was monitored and photographed with an inverted phase contrast microscope after incubation for 48 h. For quantitative assessment, the remaining area of the wound in each image was determined by Image-Pro Plus software 5.1 (Media Cybernetics, Inc. Silver Spring, MD, USA).
EdU-staining assay
LECs proliferation was determined using Cell-Light EdU Apollo 488 In Vitro Kit (RiboBio) according to the manufacturer’s protocols. In brief, SRA01/04 cells were seeded in six-well plates and transfected with miR-26a and -26b mimics or inhibitors for 48 h. The culture medium was replaced with fresh medium containing 50 μM EdU for 2 h. After that, the cells were fixed with acetone for 10 min and washed with PBS, and then they were sequentially incubated with Apollo reaction buffer containing FITC-fluorescein for 30 min. Finally, the cells were incubated with DAPI, mounted and examined by a confocal microscopy (LSM510, Carl Zeiss, Overkochen, Germany).
Real-time PCR analysis for gene expression
Total RNA from cultured cells and lenses was extracted using the Trizol reagent according to the manufacturer’s instruction. Then genomic DNA was removed using DNase I. For miRNAs, their cDNAs were synthesized using Mir-X miRNA First-Strand Synthesis kit (Clontech Laboratories, Mountain View, CA, USA), and the expression levels of miR-26a and -26b were quantified by real-time PCR using SYBR Premix Ex Taq II kit (Takara, Siga, Japan). Other gene cDNAs were synthesized using PrimeScript RT Master Mix kit (Takara) and their mRNAs expression were quantitatively analyzed by SYBR PrimeScript RT-PCR kit (Takara). Real-time PCR reactions were performed using an ABI Prism 7000 sequence detection system (Applied Biosystems, Foster City, CA, USA). RNU6B and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were used as internal controls. Human, rat and mouse primers used in this study were listed in Supplementary Table S1.
Western blot analysis for protein expression
The cells were washed twice with PBS and then lysed in 100 μl of lysis buffer with protease inhibitor cocktail for total protein extraction. The protein samples mixed with 5 × SDS sample buffer were subjected to SDS-PAGE, and then electroblotted onto PVDF membranes. The membranes were blocked in 5% non-fat milk and incubated with different primary antibodies at 4 °C overnight. After being washed with PBS containing 0.1% Tween20 (PBST), the membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 h at room temperature (RT) and washed three times with PBST. The bands on the membranes were visualized using the chemiluminescence detection reagents.
The primary antibodies against Jagged-1, Notch-1, Notch-2, Notch-3, p-Smad2, p-Smad3, Snail, Slug, ZEB1, horse anti-mouse and goat anti-rabbit HRP-conjugated secondary antibodies were obtained from Cell Signaling Technology. Antibodies against β-actin, fibronetin (FN), collagen type I (Col I), collagen type IV (Col IV), N-cadherin (N-cad), α-SMA and vimentin were obtained from Abcam (Cambridge, UK).
Immunofluorescence staining for cryosections and cultured cells
Lens cryosections or slides of cultured cells were fixed with acetone for 15 min at RT, permeated with 0.5% Triton X-100 for 10 min and blocked with 1% bovine serum albumin (BSA) for 1 h. Afterwards, the sections or cell slides were incubated with different primary antibodies at 4 °C for overnight in a humidity chamber. Next day, secondary antibodies Alexa Fluor 488-conjugated goat anti-rabbit or Alexa Fluor 555-conjugated donkey anti-mouse antibodies (Cell Signaling Technology) were incubated for 1 h at RT. After washing with PBS, the lens cryosections or cell slides were incubated with DAPI for nuclear staining and mounted with anti-fade mounting medium. The slides were observed using a laser scanning confocal microscopy (LSCM; LSM510, Carl Zeiss).
Lens anterior capsule whole-mount staining
Lens anterior capsule whole-mount staining was performed according to our previously described method.37 Briefly, the injured mice were killed and their eyes were enucleated. And then the lens were isolated under a dissecting microscope and fixed in 100% methanol for 1 h at RT. After that, lenses anterior capsules were separated, and blocked and permeated with 1% Triton X-100 in 1% BSA for 1 h at RT. Primary antibodies diluted in PBS were added to immerse the capsules and incubated for 12–24 h at 4 °C. On the following day, after being washed with PBST, the capsules were incubated with appropriate secondary antibodies for 1 h at RT. After being stained with DAPI, the whole anterior capsules were mounted in anti-fade mounting medium on a microscope slide and examined using a fluorescence microscope (Carl Zeiss) and LSCM within a few days. For 3D images analysis, consecutive images of lens anterior capsule whole mount containing the whole anterior subcapsular plaque were acquired from LSCM, and then reconstructed using the Zeiss LSCM Image Browser software.
Quantitative analysis of the subcapsular plaques and EMT marker distribution in injury-induced ASC model
Quantitative analysis of the sizes of the subcapsular plaques and EMT markers distribution was performed using LSCM Image Browser software as our method previously described.37 The shape of the subcapsular plaque in this model is similar to the trustum of a pyramid, so the volume of the subcapsular plaque in every two images can be calculated according to the formula of the volume of pyramid V1=1/3 × H × [Sup+Sdown +√(Sup × Sdown)], and the total volume of the subcapsular plaque in one sample equal to the sum total of every two images (Vtotal=V1+V2+……+Vn).37 A minimum of six capsules were analyzed for each group.
Jagged-1 3′-UTR luciferase reporter assay
The 3′-UTR region of human Jagged-1 was amplified by PCR from genomic DNA and cloned downstream to the firefly luciferase coding region in the pMIR-RB-REPORT. The 293 T cells were seeded in 96-well plates 24 h before transfection. The following day, to access the effects of miR-26a and -26b on reporter activities, 100 ng/ml of reporter plasmid and 50 nM of miR-26a, -26b or NC mimics were co-transfected into 293 T cells using lipofectamine 2000. Luciferase activities were measured 48 h after transfection using the Dual-Glo Luciferase Assay System (Promega, Madison, WI, USA). All experiments were performed in triplicates.
Jagged-1 siRNA knockdown in cultured cells in vitro
Jagged-1 siRNA was transfected into LECs with transfection reagent (Santa Cruz Biotechnology, Santa Cruz, CA, USA) according to the manufacturer’s instructions. In short, 60 nM of control siRNA or Jagged-1 siRNA was mixed with 6 μl of transfection reagent in transfection medium and incubated for 30 min at RT. The mixture then was added into each well of the six-well plates containing cells in transfection medium, and the cells were incubated for 5–7 h. The transfection medium was subsequently replaced and the cells exposed to 5 ng/ml of TGFβ2 for a further 48 h. For siRNA knockdown efficiency, the mRNA and protein levels of Jagged-1 were determined by real-time PCR and western blot, respectively.
Model of TGFβ2-induced ASC in whole lens culture
Lenses of 20–22 days old Wistar rats were cultured as described previously.35, 38 Shortly, whole lenses were carefully isolated and maintained in 4 ml serum-free M199 medium (two lenses per well) containing 0.1% BSA, 0.1 mg/ml l-glutamine, 50 IU/ml penicillin and 50 mg/ml streptomycin. Next day, cloudy lenses caused by technique were removed and TGFβ2 was added to the culture medium at a final concentration of 5 ng/ml, using 0.1% BSA as a control. The culture medium was renewed every second day throughout the culture period. The lenses were cultured for up to 7 days and photographed using a dissecting microscope (Carl Zeiss). After that, the lenses were harvested for real-time PCR, western blot and cryosection staining.
Statistical analysis
The results presented in the figures are representative of three or more different repetitions. All data are presented as mean±standard error of the mean (S.E.M.). The data were analyzed by using two-tailed Student's t-test and one-way analysis of variance (ANOVA) statistical analysis. A value of P<0.05 was considered statistically significant, P<0.01 was highly significant.
References
McAnulty RJ . Fibroblasts and myofibroblasts: their source, function and role in disease. Int J Biochem Cell Biol 2007; 39: 666–671.
Thiery JP, Acloque H, Huang RY, Nieto MA . Epithelial-mesenchymal transitions in development and disease. Cell 2009; 139: 871–890.
Eldred JA, Dawes LJ, Wormstone IM . The lens as a model for fibrotic disease. Philos Trans R Soc Lond B Biol Sci 2011; 366: 1301–1319.
Nathu Z, Dwivedi DJ, Reddan JR, Sheardown H, Margetts PJ, West-Mays JA . Temporal changes in MMP mRNA expression in the lens epithelium during anterior subcapsular cataract formation. Exp Eye Res 2009; 88: 323–330.
Shin EH, Basson MA, Robinson ML, McAvoy JW, Lovicu FJ . Sprouty is a negative regulator of transforming growth factor beta-induced epithelial-to-mesenchymal transition and cataract. Mol Med 2012; 18: 861–873.
Apple DJ, Solomon KD, Tetz MR, Assia EI, Holland EY, Legler UF et al. Posterior capsule opacification. Surv Ophthalmol 1992; 37: 73–116.
Hodge WG . Posterior capsule opacification after cataract surgery. Ophthalmology 1998; 105: 943–944.
de Iongh RU, Wederell E, Lovicu FJ, McAvoy JW . Transforming growth factor-beta-induced epithelial-mesenchymal transition in the lens: a model for cataract formation. Cells Tissues Organs 2005; 179: 43–55.
Allen JB, Davidson MG, Nasisse MP, Fleisher LN, McGahan MC . The lens influences aqueous humor levels of transforming growth factor-beta 2. Graefes Arch Clin Exp Ophthalmol 1998; 236: 305–311.
Koch U, Radtke F . Notch and cancer: a double-edged sword. Cell Mol Life Sci 2007; 64: 2746–2762.
Rizzo P, Osipo C, Foreman K, Golde T, Osborne B, Miele L . Rational targeting of Notch signaling in cancer. Oncogene 2008; 27: 5124–5131.
Xu K, Moghal N, Egan SE . Notch signaling in lung development and disease. Adv Exp Med Biol 2012; 727: 89–98.
Sirin Y, Susztak K . Notch in the kidney: development and disease. J Pathol 2012; 226: 394–403.
Miele L . Notch signaling. Clin Cancer Res 2006; 12: 1074–1079.
Kadesch T . Notch signaling: the demise of elegant simplicity. Curr Opin Genet Dev 2004; 14: 506–512.
Nyhan KC, Faherty N, Murray G, Cooey LB, Godson C, Crean JK et al. Jagged/Notch signalling is required for a subset of TGFbeta1 responses in human kidney epithelial cells. Biochim Biophys Acta 2010; 1803: 1386–1395.
Murata K, Ota S, Niki T, Goto A, Li CP, Ruriko UM et al. p63 - Key molecule in the early phase of epithelial abnormality in idiopathic pulmonary fibrosis. Exp Mol Pathol 2007; 83: 367–376.
Matsuno Y, Coelho AL, Jarai G, Westwick J, Hogaboam CM . Notch signaling mediates TGF-beta1-induced epithelial-mesenchymal transition through the induction of Snai1. Int J Biochem Cell Biol 2012; 44: 776–789.
Filipowicz W, Bhattacharyya SN, Sonenberg N . Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet 2008; 9: 102–114.
Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol 2008; 10: 593–601.
Ru P, Steele R, Newhall P, Phillips NJ, Toth K, Ray RB . miRNA-29b suppresses prostate cancer metastasis by regulating epithelial-mesenchymal transition signaling. Mol Cancer Ther 2012; 11: 1166–1173.
Siemens H, Jackstadt R, Hunten S, Kaller M, Menssen A, Gotz U et al. miR-34 and SNAIL form a double-negative feedback loop to regulate epithelial-mesenchymal transitions. Cell Cycle 2011; 10: 4256–4271.
Zhou Q, Fan J, Ding X, Peng W, Yu X, Chen Y et al. TGF-{beta}-induced MiR-491-5p expression promotes Par-3 degradation in rat proximal tubular epithelial cells. J Biol Chem 2010; 285: 40019–40027.
Kota J, Chivukula RR, O'Donnell KA, Wentzel EA, Montgomery CL, Hwang HW et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell 2009; 137: 1005–1017.
Li J, Kong X, Zhang J, Luo Q, Li X, Fang L . MiRNA-26b inhibits proliferation by targeting PTGS2 in breast cancer. Cancer Cell Int 2013; 13: 7.
Liu XX, Li XJ, Zhang B, Liang YJ, Zhou CX, Cao DX et al. MicroRNA-26b is underexpressed in human breast cancer and induces cell apoptosis by targeting SLC7A11. FEBS Lett 2011; 585: 1363–1367.
Wu N, Zhao X, Liu M, Liu H, Yao W, Zhang Y et al. Role of microRNA-26b in glioma development and its mediated regulation on EphA2. PLoS ONE 2011; 6: e16264.
Liang H, Xu C, Pan Z, Zhang Y, Xu Z, Chen Y et al. The antifibrotic effects and mechanisms of microRNA-26a action in idiopathic pulmonary fibrosis. Mol Ther 2014; 22: 1122–1133.
Wei C, Kim IK, Kumar S, Jayasinghe S, Hong N, Castoldi G et al. NF-kappaB mediated miR-26a regulation in cardiac fibrosis. J Cell Physiol 2013; 228: 1433–1442.
Liang H, Gu Y, Li T, Zhang Y, Huangfu L, Hu M et al. Integrated analyses identify the involvement of microRNA-26a in epithelial-mesenchymal transition during idiopathic pulmonary fibrosis. Cell Death Dis 2014; 5: e1238.
Wang Y, Li W, Zang X, Chen N, Liu T, Tsonis PA et al. MicroRNA-204-5p regulates epithelial-to-mesenchymal transition during human posterior capsule opacification by targeting SMAD4. Invest Ophthalmol Vis Sci 2013; 54: 323–332.
Dong N, Xu B, Benya SR, Tang X . MiRNA-26b inhibits the proliferation, migration, and epithelial-mesenchymal transition of lens epithelial cells. Mol Cell Biochem 2014; 396: 229–238.
Wang Z, Li Y, Kong D, Sarkar FH . The role of Notch signaling pathway in epithelial-mesenchymal transition (EMT) during development and tumor aggressiveness. Curr Drug Targets 2010; 11: 745–751.
Chen X, Xiao W, Liu X, Zeng M, Luo L, Wu M et al. Blockade of Jagged/Notch pathway abrogates transforming growth factor beta2-induced epithelial-mesenchymal transition in human retinal pigment epithelium cells. Curr Mol Med 2014; 14: 523–534.
Hales AM, Chamberlain CG, McAvoy JW . Cataract induction in lenses cultured with transforming growth factor-beta. Invest Ophthalmol Vis Sci 1995; 36: 1709–1713.
Saika S, Kono-Saika S, Ohnishi Y, Sato M, Muragaki Y, Ooshima A et al. Smad3 signaling is required for epithelial-mesenchymal transition of lens epithelium after injury. Am J Pathol 2004; 164: 651–663.
Xiao W, Chen X, Li W, Ye S, Wang W, Luo L et al. Quantitative analysis of injury-induced anterior subcapsular cataract in the mouse: a model of lens epithelial cells proliferation and epithelial-mesenchymal transition. Sci Rep 2015; 5: 8362.
Hales AM, Chamberlain CG, McAvoy JW . Susceptibility to TGFbeta2-induced cataract increases with aging in the rat. Invest Ophthalmol Vis Sci 2000; 41: 3544–3551.
Acknowledgements
This research was funded by the Program of International Cooperation and Exchanges of the National Natural Science Foundation of China Grant to YL (81320108008), the Doctoral Fund of Guangdong Province Natural Science Foundation to XC (2016A030310232), the Fundamental Research Funds of the State Key Laboratory of Ophthalmology to WX (2016QN07) and the National Natural Science Foundation of China Grant to QB (81400386).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by P Salomoni
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Chen, X., Xiao, W., Chen, W. et al. MicroRNA-26a and -26b inhibit lens fibrosis and cataract by negatively regulating Jagged-1/Notch signaling pathway. Cell Death Differ 24, 1431–1442 (2017). https://doi.org/10.1038/cdd.2016.152
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2016.152
This article is cited by
-
A hsa_circ_001726 axis regulated by E2F6 contributes to metastasis of hepatocellular carcinoma
BMC Cancer (2024)
-
Loss of miR-26b-5p promotes gastric cancer progression via miR-26b-5p-PDE4B/CDK8-STAT3 feedback loop
Journal of Translational Medicine (2023)
-
Long non-coding RNA nuclear paraspeckle assembly transcript 1 downregulation protects lens epithelial cells from oxidative stress-induced apoptosis by regulating the microRNA-124-3p/death-associated protein kinase 1 axis in age-related cataract
International Ophthalmology (2023)
-
The interaction between autophagy and the epithelial-mesenchymal transition mediated by NICD/ULK1 is involved in the formation of diabetic cataracts
Molecular Medicine (2022)
-
A functional map of genomic HIF1α-DNA complexes in the eye lens revealed through multiomics analysis
BMC Genomics (2021)
