
Abstract
The E3 ubiquitin ligase MULE (Mcl-1 Ubiquitin Ligases E3) targets myeloid cell leukemia factor 1 (Mcl-1) and tumor suppressor p53 for proteasomal degradation. Although Mcl-1 and p53 have been implicated in trophoblast cell death in preeclampsia (PE) and intrauterine growth restriction (IUGR), the mechanisms regulating their expression in the human placenta remains elusive. Herein, we investigated MULE’s involvement in regulating Mcl-1 and p53 degradation during normal and abnormal (PE, IUGR) placental development. MULE expression peaked at 5–7 weeks of gestation, when oxygen tension is low and inversely correlated with that of Mcl-1 and p53. MULE efficiently bound to Mcl-1 and p53 and regulated their ubiquitination during placental development. Exposure of first trimester villous explants to 3% O2 resulted in elevated MULE expression compared with 20% O2. Low-oxygen-induced MULE expression in JEG3 choriocarcinoma cells was abolished by hypoxia-inducible factor (HIF)-1α siRNA. MULE was overexpressed in both PE and IUGR placentae. In PE, MULE preferentially targeted p53 for degradation, allowing accumulation of pro-apoptotic Mcl-1 isoforms. In IUGR, however, MULE targeted pro-survival Mcl-1, allowing p53 to accumulate and exert its apoptotic function. These data demonstrate that oxygen regulates Mcl-1 and p53 stability during placentation via HIF-1-controlled MULE expression. The different preferential targets of MULE in PE and IUGR placentae classify early-onset PE and IUGR as distinct molecular pathologies.
Similar content being viewed by others


Main
Preeclampsia (PE) and intrauterine growth restriction (IUGR) are human pregnancy-related disorders that affect 3–7% and 5–10% of all pregnancies, respectively, and are one of the leading causes of fetal and maternal mortality and morbidity.1, 2 PE manifests itself with a wide variety of maternal symptoms including hypertension and proteinuria,3 whereas IUGR is defined as failure of the fetus to achieve its genetically-determined growth potential.2 The placenta plays a key role in the genesis of both diseases. In PE-defective remodeling of the maternal spiral arteries by trophoblast cells is believed to be the cause for insufficient uteroplacental circulation,4 leading to placental hypoxia5 and/or oxidative stress,6, 7 and in the most severe cases to fetal growth restriction. IUGR may be caused by a variety of conditions such as infections, maternal diseases and chromosomal disorders, but more often, as in PE, its genesis is related to a defect in proper placental development originating early in pregnancy. Typical features of both severe IUGR and PE are altered trophoblast cell proliferation and reduced chorionic vessel development, leading to impaired angiogenesis.8 In addition, increased rates of trophoblast cell death and turnover are characteristic for both pathologies.9, 10, 11, 12 The underlying mechanisms for these changes in trophoblast cell fate are unknown.
Bcl-2 family proteins form a complex network of cell death inducers and suppressors that via their interactions determine cell fate.13 Bcl-2 pro-apoptotic proteins act mainly by localizing permanently or transiently to the outer mitochondrial membrane where they regulate or form pores. Opening of these pores results in transitional changes in mitochondrial membrane potential accompanied by release of death-inducing factors from the mitochondria to the cytoplasm. Pro-survival Bcl-2 family members act by keeping death-inducing Bcl-2 proteins in an inactive state, thereby blocking the decrease in mitochondrial membrane potential and inhibiting the activation of the apoptotic cascade.14 The balance between pro-survival and pro-apoptotic molecules is tightly regulated and involves the ubiquitin–proteasome system that targets specific proteins for degradation. Ubiquitination requires the concerted action of three different enzymes: E1 ubiquitin activators, E2 ubiquitin-conjugating enzymes and E3 ubiquitin ligases that determine the specificity of the process by recognizing the different substrates.15 MULE (Mcl-1 Ubiquitin Ligases E3) is an E3 ligase that specifically targets pro-survival Mcl-1L for ubiquitination, thereby promoting its degradation via the proteasome.16, 17 Besides the classical HECT and ubiquitin-associated domains typical of E3 ligases, MULE also contains a Bcl-2 homology domain 3 (BH3) that functions as a specific binding site for Mcl-1.16, 17 In placentae from pregnancies complicated by early-onset preeclampsia (E-PE) Mcl-1L levels are decreased, while those of its pro-apoptotic spliced variant Mcl-1S and its cleaved product Mcl-1c are increased.18 Whether MULE plays a role in reducing Mcl-1L levels in PE placentae remains to be elucidated. MULE also targets c-Myc, CDC-6, E3Histone and, in particular, the tumor suppressor protein p53 for ubiquitination.19 P53 plays a key role in the rapid cellular response to stress20 and exerts its anti-proliferative activity via a variety of mechanisms, including cell cycle arrest, apoptosis and cellular senescence/aging. Beside MULE, p53 protein stability is also regulated by two other well-characterized ubiquitin ligases, such as murine double minute-2 (Mdm2) and murine double minute-X (MdmX).21, 22 Expression of p53 is elevated in placentae from pregnancies complicated by IUGR.9, 12 As placental pathologies, including PE and IUGR, are characterized by altered expression of key regulators of trophoblast cell fate such as Mcl-1 and p53, we hypothesized that changes in MULE expression and activity may contribute to the altered balance in pro-survival and death-inducing molecules typical of these pathologies.
Results
MULE expression inversely correlates with that of Mcl-1 and p53
We first investigated the expression of MULE in human placental tissue throughout early pregnancy. Real-time PCR (Figure 1a) and western blot analysis (Figure 1b, upper left panel) showed that MULE expression peaked at 5–7 weeks and declined with advancing gestation. In agreement with our previous observations,26 Mcl-1 expression was maximal at 11–13 weeks of gestation (Figure 1b, bottom left panel) and inversely correlated with that of MULE (Pearson’s coefficient r=−0.712). The specificity of the MULE antibody was confirmed by western blot analysis using MULE blocking-peptides. A 500 kDa band corresponding to the main variant of MULE and its two main proteolytic (approximately 240 and 270 kDa) products present in 293T cell lysates23 were efficiently outcompeted (Figure 1b, right panel). As MULE targets Mcl-1 for degradation, we examined Mcl-1/MULE association and Mcl-1 ubiquitination. MULE association to Mcl-1 was maximal at 5–6 weeks (Figure 1c, right panel) and Mcl-1 ubiquitination peaked at 7–9 weeks (Figure 1c, left panel): at the time when Mcl-1 levels are low (Figure 1b, bottom left panel). As MULE has been shown to target p53 for degradation, we also investigated p53 expression, p53/MULE association and p53 ubiquitination during human placental development. Real-time PCR showed no significant changes in p53 mRNA expression throughout early placental development (not shown, P>0.10). In contrast, placental p53 protein expression inversely correlated to that of MULE (Pearson’s coefficient r=−0.739), that is, low p53 protein levels at 5–7 weeks, which increased with advancing gestation (Figure 1d, bottom left panel). The association between p53 and MULE was maximal at 5 and 6 weeks, whereas p53 ubiquitination levels peaked around 7–9 weeks, as well as 12 and 13 weeks of gestation (Figure 1d).

Expression and association of MULE, Mcl-1 and p53 during human placental development (5–7 weeks, n=19; 8–10 weeks, n=14; 11–15 weeks, n=24). (a) MULE mRNA expression during placental development assessed by real-time PCR (5–7 weeks, n=7; 8–10 weeks, n=6; 11–15 weeks, n=12). Values are mean±S.E.M., *P<0.05. (b) Representative immunoblots for MULE (upper left panel) and Mcl-1 (lower left panel) protein expression during placenta development and MULE antibody validation (right panel) assessed by MULE immunoblot using 293T cells in the presence or absence of competing peptide (CP). (c) Representative immunoblots for Mcl-1 ubiquitination (left panel) and MULE/Mcl-1 association (right panel) during placental development. (d) Representative immunoblots for p53 protein expression (left lower panel), ubiquitination (left upper panel) and MULE/p53 association (right panel) during placental development. (b and d) Actin immunoblots demonstrates equal protein loading (5–7 weeks, n=12; 8–10 weeks, n=8; 11–15 weeks, n=12)
MULE, Mcl-1 and p53 placental localization
We then examined the spatial localization of MULE and its targets Mcl-1 and p53 in first trimester placental tissue. At 5 weeks of gestation, a strong positive nuclear and cytoplasmic signal for MULE was detected in cytotrophoblastic cells and to a lesser extent in syncytiotrophoblast (ST) cells (Figure 2a). At 12 weeks of gestation, immunoreactivity for MULE was markedly decreased in both trophoblast layers (Figure 2d). This decrease in trophoblastic MULE signal at 12 weeks was associated with major increases in immunoreactivity for Mcl-1 (cytoplasm of ST cells) and p53 (nuclei of CT cells) (Figures 2e and f). No positive immunoreactivity for MULE was detected in section stained with control IgG (Figure 2g).

MULE, Mcl-1 and p53 spatial localization in first trimester human placental sections. MULE (a and d), Mcl-1 (b and e) and p53 (c and f) spatial localization at 5 and 12 weeks of gestation assessed by immunofluorescent staining (5–7 weeks, n=7; 8–10 weeks, n=6; 11–15 weeks, n=12). Cell nuclei are showed in blue by DAPI signal. ST, syncytiotrophoblast cells. Control IgG (g). Original magnifications, × 20; insets, × 100
MULE oxygen-dependent expression is mediated by HIF-1 α
As MULE expression is high during the gestational time of physiological low oxygenation, we examined its regulation by oxygen using first trimester placental explants exposed to 3% O2, standard 20% O2 or to hypoxia/reoxygenation (HR). MULE protein levels were significantly increased in low oxygen (3% versus 20% O2, P=0.038, 8.6-fold increase) and HR (HR versus 3% O2, P=0.002, 3.5-fold increase; HR versus 20% O2, P=0.001, 30.7-fold increase) (Figure 3a, upper left and right panels). Although exposure of villous explants to 3% O2 resulted in increased Mcl-1L protein levels, HR conditions showed a reduction of pro-survival Mcl-1L (HR versus 20% O2, P=0.04, 1.4-fold decrease) (Figure 3a, middle left panel), in line with our previous findings.18 Protein levels of p53 were unchanged among the different conditions (Figure 3a, bottom left panel). As hypoxia-inducible factor (HIF)-1α is a major player in the physiological response to hypoxia, we next examined HIF-1α protein expression during placental development and found that it paralleled that of MULE (Figure 1b, left panel), showing a peak of expression at 5–7 weeks that decreases with advancing gestation (Figure 3b, right upper panel). Next, we investigated whether MULE’s upregulation in low oxygen was mediated via HIF-1α using HIF-1α siRNA. In preliminary experiments, we observed that fluorescent-labeled siRNA complexes efficiently (>90%) transfected JEG3 cells and that the relative lactate dehydrogenase (LDH) amount released into the media (indication of toxicity) was comparable among siRNA-treated cells (D1 and D2), control (scrambled sequence (SS)) siRNA-treated cells and control cells treated with Lipofectamine 2000 vehicle alone (data not shown). Real-time PCR showed that the HIF-1α gene was efficiently silenced by both D1 and D2 siRNA duplexes relative to control SS siRNA (D1 versus SS: 4.3-fold decrease, P=0.0095; D2 versus SS: 5.2-fold decrease, P=0.0095) (Figure 3b, left upper panel). Also, MULE mRNA expression was significantly downregulated in HIF-1α siRNA-treated cells relative to control SS siRNA-treated cells (D1 versus SS: 1.7-fold decrease, P=0.03; D2 versus SS: 1.8-fold decrease, P=0.03) (Figure 3b, left bottom panel). These results were confirmed at the protein level, that is, decrease of both HIF-1α and MULE protein levels in HIF-1α siRNA (D1)-treated cells relative to control (untreated) cells and cells treated with control SS siRNA (Figure 3b, bottom right panel). Thus, conditions of low oxygen/oxidative stress promote MULE accumulation via HIF-1α. We and others reported that low oxygen promotes TGF-β3 expression via HIF-1 and that TGF-β3 is a key factor for proper placentation.24, 25, 26 Hence, we next investigated whether MULE expression was regulated by TGF-β3. We found that TGF-β3, but not TGF-β1, markedly increased MULE protein levels in placental villous explants (Figure 3c).

HIF-1α-mediated oxygen regulation of MULE expression. (a) Representative immunoblots for MULE, Mcl-1 and p53 protein expression in first trimester villous explants (n=18) exposed at 3% pO2, 20% pO2 and HR conditions (left panel). β-Actin immunoblot demonstrates equal protein loading. Densitometric analysis of MULE protein levels at 20 and 3% pO2 and in condition of HR as assessed by western blot assay (right panel). Values are mean±S.E.M., *P<0.05. (b) HIF-1α and MULE (left panels) mRNA expression levels in HIF-1α siRNA JEG-3 cells assessed by real-time PCR (values are mean±S.E.M., *P<0.05). Representative immunoblots for HIF-1α protein expression during early placental development (right upper panel). Representative immunoblots for HIF-1α and MULE protein expression in siRNA JEG-3 cells (right lower panels). Actin immunoblot demonstrates equal protein loading. (c) Representative MULE immunoblots in JEG-3 cells following exposure to TGF-β1/TGF-β3
MULE, Mcl-1 and p53 expression in E-PE and IUGR placentae
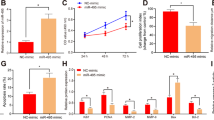
PE and IUGR are pathologies characterized by placental hypoxia and increased trophoblast cell death and turnover due to accumulation of pro-apoptotic molecules. Hence, we examined MULE, Mcl-1 and p53 expression in both disorders. MULE protein levels were increased in both E-PE and IUGR placentae relative to age-matched controls (AMCs) (Figure 4a, upper left and right panels). In line with our previous observations,18 we found a significant decrease in pro-survival Mcl-1L in E-PE placentae relative to AMC controls (2.17-fold decrease, P=0.0003; Figure 4a, left panel). Total p53 protein levels were also significantly decreased in E-PE placentae relative to AMC controls (1.42-fold decrease, P=0.0179) (Figure 4a, left panel), whereas no significant changes in p53 mRNA levels were found by real-time PCR analysis (not shown, P=0.19). In contrast, both p53 mRNA and protein levels were markedly increased in IUGR placentae relative to AMC controls (p53 mRNA: 6.1-fold increase, P=0.007; p53 protein: 1.5-fold increase; Figure 4a, right panel). Notably, while in IUGR Mcl-1L mRNA levels were significantly increased (2.35-fold increase, P=0.010), Mcl-1L protein levels were decreased (2.3-fold decrease, P=0.005) (Figure 4a, right panel). We next investigated whether the differences in Mcl-1 and p53 protein expression between E-PE and IUGR placentae were due to differences in MULE binding and ubiquitination of these proteins. No apparent differences were found in Mcl-1/MULE association, but Mcl-1 ubiquitination was slightly elevated in E-PE versus AMC placentae (Figure 4b, left panel). In contrast, p53/MULE association and p53 ubiquitination were increased in E-PE placentae relative to AMC placentae (Figure 4c, left panel). In IUGR placentae, Mcl-1/MULE association and Mcl-1 ubiquitination were increased relative to AMC placentae (Figure 4b, right panel), whereas p53/MULE association was reduced and ubiquitination of p53 was unchanged (Figure 4c, right panel). Terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick-end labeling (TUNEL) analysis confirmed the augmented levels of trophoblast cell death in E-PE and IUGR placentae relative to AMC (Figures 5a, b versus c). As anticipated, MULE’s nuclear and cytoplasmic immunoreactivity in trophoblastic cells was significantly increased in E-PE and IUGR placentae relative to normotensive AMC controls (Figures 5d, e versus f). Although cytoplasmic Mcl-1 immunoreactivity was slightly reduced in the ST cells of E-PE placentae compared with AMC placentae (Figure 5g versus i), hardly any signal was detectable in IUGR placentae (Figure 5h versus i). The nuclear p53 signal was markedly decreased in E-PE placentae relative to controls (Figure 5j versus l), whereas it was increased in the syncytium of IUGR placentae (Figure 5k versus l).

MULE, Mcl-1 and p53 protein expression, MULE/Mcl-1-p53 association and Mcl-1/p53 ubiquitination levels in E-PE, IUGR and AMC placentae. (a) Representative immunoblots for MULE, Mcl-1 and p53 in E-PE (n=25, left panel) and IUGR (n=18, right panel) versus AMC (n=19) placentae. Actin immunoblot demonstrates equal protein loading. (b) Representative immunoblots for MULE/Mcl-1 association and Mcl-1 ubiquitination (left panels) in E-PE (n=25) versus AMC (n=19) placentae. Representative immunoblots for MULE/Mcl-1 association and Mcl-1 ubiquitination in IUGR (n=18) versus AMC (n=19) placentae (right panel). (c) (left panels). Representative immunoblot for MULE/p53 association and p53 ubiquitination levels (left panels) in E-PE (n=25) versus AMC (n=19) placentae. Representative immunoblot for MULE/p53 association and p53 ubiquitination levels in IUGR (n=18) versus AMC (n=19) placentae (right panels)

TUNEL assay and spatial localization of MULE, Mcl-1 and p53 in E-PE, IUGR and AMC human placental sections. TUNEL assay in placental sections from E-PE (a), IUGR (b) and AMC (c). MULE (d–f; green), Mcl-1 (g–i; red) and p53 (j–l; red) spatial localization in E-PE, IUGR and AMC placental sections. ST, syncytium/syncytiotrophoblast cells. Original magnifications, × 20; insets, × 100
Discussion
PE and IUGR are serious disorders of pregnancy characterized by altered expression and function of pro- and antiapoptotic molecules, leading to increased rates of trophoblast cell death and turnover. The precise mechanism(s) underpinning impaired cell fate in these placental pathologies remain elusive. In this study, we demonstrate for the first time that MULE, a novel E3 ubiquitin ligase,16, 17, 19 is temporally and spatially expressed in the human placenta, where it targets both pro-survival Mcl-1 and tumor suppressor p53 for degradation. In addition, we show that MULE expression is regulated by oxygen via HIF-1α and TGF-β3. Of clinical relevance, we report aberrant MULE expression in severe preeclamptic and IUGR placentae. Notably, our data demonstrate that MULE differentially targets Mcl-1 and p53 for degradation in IUGR and preeclamptic placentae. Hence, distinct mechanism(s) regulating Mcl-1 and p53 protein stability, in part, contribute to the increased trophoblast cell death and turnover typical of both pregnancy-related disorders.
Bcl-2 proteins comprise a family of death-inducer and pro-survival molecules, which by balancing their relative concentrations determine cell fate during development and normal physiological conditions.13 Loss of this balance is central to a variety of diseases including cancer- and pregnancy-related disorders.11, 18 Among Bcl-2 family members, pro-survival Mcl-1 was first described as an ‘early induction’ gene27 and later characterized as a potent antiapoptotic molecule.28 Disruption of Mcl-1 gene in mouse leads to pre-implantation lethality implicating a role for Mcl-1 in early development.29 In contrast, constitutive expression of Mcl-1 in transgenic mice results in the development of hematological malignancies.28 Indeed, high levels of Mcl-1 in cancer are indicative of poor prognosis. We previously demonstrated that a balance between pro-survival Mcl-1 and its death-inducing partner Mtd/Bok is essential in shaping proper placental development and that an alteration in the Mcl-1/Mtd rheostat typifies PE.11, 18 Specifically, decreased pro-survival Mcl-1L expression (accompanied by accumulation of its pro-apoptotic variants Mcl-1c and Mcl-1S) and elevated Mtd/Bok expression contributes to the aberrant trophoblast cell death seen in this disorder.18
Mcl-1L is a very short-lived protein (t1/2=2–4 h), belonging to a group of immediate response genes upregulated in immature cells upon exposure to differentiating agents and antiapoptotic cytokines.30 Hence, Mcl-1 levels must be tightly controlled to regulate cell survival. Mcl-1 protein stability and activity are regulated by several mechanisms, including synthesis, suppression via microRNA, phosphorylation, caspases-mediated proteolysis and/or proteasomal degradation.31, 32, 33 In addition, a recent study showed that de-ubiquitinase USP9X inhibits Mcl-1 ubiquitination, thereby stabilizing protein levels and promoting tumor cell survival.34 MULE, also known as ARF-BP1, UREB1, LASU1 and E3histone, has been identified as the first E3 ligase that specifically regulates Mcl-1L protein stability.16, 17 MULE possesses a BH3 domain that specifically recognizes and binds to the BH3 domain located on Mcl-1, conferring MULE specificity for this Bcl-2 protein.16 Because of its BH3 domain, MULE can be considered a BH3-only pro-apoptotic protein; however, its effect on cell death is indirect via regulation of pro-survival Mcl-1 stability.16 Our findings of an inverse correlation between Mcl-1L and MULE expression and the temporal profiles of MULE binding to Mcl-1 and Mcl-1 ubiquitination highlight a role for MULE as a key regulator of Mcl-1 stability during the early critical stages of placental development. Later on in pregnancy, mechanisms other than MULE-mediated proteasomal degradation may regulate Mcl-1 stability. Indeed, we have previously reported that at the end of first trimester, when MULE levels are lower, active caspase-3 cleaves Mcl-1L, thereby generating pro-apoptotic Mcl-1c.18
Besides binding to Mcl-1 and facilitating its degradation, MULE also interacts with the tumor suppressor p53 and promotes its proteasomal degradation.23 Similarly to Mcl-1L, we found that MULE and p53 expression profiles during placental development are inversely correlated and that MULE actively regulates p53 stability. It has been reported that Mdm2 and MdmX are the primary E3 ligases targeting p53 for degradation.21, 22 However, MULE action on p53 is Mdm2-independent19 and a simple inactivation of endogenous MULE has been shown to stabilize and activate p53,19 indicating that MULE could possess a stronger affinity for p53 than Mdm2. In addition, recent in vitro studies conducted using placental villous explants cultured in 1% O2 reported that hypoxic conditions could stabilize p53 protein by inhibiting its binding to Mdm2.35 Hence, it is plausible that Mdm2 may account for p53 ubiquitination at 12 and 13 weeks of gestation when MULE expression is low.
Subcellular localization studies in rat testis revealed that nuclear MULE regulates p53 stability, while cytoplasmatic MULE preferentially targets Mcl-1 for degradation.23 Our observation of a nuclear and cytoplasmic localization of MULE in both trophoblast cell layers at 5 weeks of gestation implicates a role for this E3 ligase in regulating both p53 and Mcl-1 stability during early human placental development. Hence, early in pregnancy, MULE in part contributes to the regulation of Mcl-1 and p53 homeostasis in a spatial and temporal manner, thereby fine-tuning trophoblast cell fate.
Surprisingly, Mule association with both Mcl-1 and p53 was high early on (5–6 weeks) in placentation, but ubiquitination of Mcl-1 and p53 peaked at 7–9 weeks. We speculate that the low ubiquitination levels of Mcl-1/p53 at 5–6 weeks seen in the ubiquitination assay are due to the low amounts of Mcl-1 and p53 protein present in the placental lysates. The low amount of protein is likely due the rapid proteosomal degradation of ubiquitinated Mcl-1/p53 at this particular gestation. The placental lysates at 7–9 weeks contained relatively more ubiquitinated Mcl-1/p53 protein. We hypothesize that the proteosomal degradation machinery functions less efficient or is maxed out at this particular gestational window, which results in the accumulation of Mcl-1/p53 ubiquitinated earlier (weeks 6 and 7). However, we have to clarify that the developmental expression profiles of Mule, Mcl-1 and p53 represent a snap-shot in time of the in vivo scenario as human placental tissues were snap-frozen immediately after delivery. Thus, it is challenging to interpret from these findings the exact dynamics of the processes taking place in vivo.
Oxygen acts as the driving force for proper placenta development by regulating the expression of several genes involved in trophoblast cell proliferation and death. A variety of Bcl-2 family members, including Mcl-1,18 have been shown to be directly regulated by oxygen via HIF-1. Here we show for the first time that MULE expression in JEG-3 cells is regulated by oxygen via HIF-1α. Moreover, MULE’s expression profile in situ, that is, high expression at 5–7 weeks of gestation, when oxygen tension is low, is consistent with oxygen-mediated MULE regulation. Thus, MULE can be added to the fast-expanding list of HIF-1 gene targets. Other E3 ligases have also been shown to be regulated by oxygen. In 293T cells, low oxygen directly induces the expression and accumulation of Seven In Absentia Homologs 2 (Siah2), an E3 ligase that positively regulates HIF-1α stability. In HeLa and 293T cells, hypoxia is able to downregulate indirectly the expression of the von Hippel–Lindau tumor suppressor protein (targets HIF-1α for degradation in normoxic conditions) via a post-transcriptional mechanisms that involves APC/CCdh1 E3 ligases.36 In addition to being a driving force in guiding proper placental development, oxygen also plays a causal role in the etiopathogenesis of PE and IUGR, and elevated HIF-1α expression in both pathologies is central to the aberrant expression of molecules responsible for altered trophoblast cell proliferation and death.5, 37, 38 Our present observation of elevated MULE levels in E-PE and IUGR placentae is consistent with placental hypoxia and upregulated HIF-1α expression.5 Elevated HIF-1α levels during early placental development and in E-PE are associated with high levels of TGF-β3, a known HIF-1α target.24, 25, 26 These finding of MULE regulation by TGF-β3 further underscores the importance of the HIF/TGF-β3 axis in development and disease of the human placenta.
Despite the fact that PE and IUGR are often clinically associated and share several morphological and molecular features indicative of altered cell death rates, it is still unclear whether or not they are distinct pathologies. In this study, we show for the first time that increased MULE expression in placentae from severe preeclamptic pregnancies is associated with low p53 and Mcl-1 levels. Importantly, our study revealed that MULE preferentially binds and degrades p53 in E-PE placentae, whereas in placentae from pure IUGR pregnancies (with no clinical signs of hypertension) MULE preferentially binds and targets Mcl-1L for degradation. Our IUGR results agree with studies of others9, 12 who reported that augmented trophoblast apoptosis in IUGR pregnancies was due to p53 accumulation and not to increased Bcl-2 pro-apoptotic molecules.9, 12 We recently reported that the oxidative stress environment in severe PE placentae promotes formation and accumulation of pro-apoptotic Mcl-1 variants (Mcl-1c), which are generated via a caspase-3-mediated cleavage of pro-survival Mcl-1L.18 Hence, reduced pro-survival Mcl-1L levels in E-PE placentae are primarily due to cleavage by caspase-3. In contrast, reduced pro-survival Mcl-1L in IUGR are likely due to increased MULE activity. This differential regulation of two key molecules involved in cell fate by MULE in placentae from IUGR and PE pregnancies suggest that PE and IUGR are distinct molecular pathologies.
In conclusion, we demonstrate a role for MULE as regulator of Mcl-1L and p53 stability during early placental development. Furthermore, we provide evidence for a ‘double life’ of MULE in placental pathologies. In PE, MULE selectively targets p53 for degradation, thereby allowing accumulation of pro-apoptotic Mcl-1 isoforms. Conversely in IUGR, increased levels of MULE selectively promote degradation of pro-survival Mcl-1L, whereas p53 levels increase.
Materials and Methods
Patient population and tissue collection
First and second trimester human placenta tissues (5–7 weeks, n=44; 8–10 weeks, n=23, 11–15 weeks, n=39) were obtained from elective termination of pregnancies. Pathological (PE and IUGR) and normotensive AMC placental samples were obtained after informed consent in accordance with the ethics guidelines of University of Toronto and Mount Sinai Hospital (Toronto, Canada) and O.I.R.M-Sant’Anna Hospital, University of Turin (Turin, Italy). Patients clinical data are summarized in Table 1. The study groups included singleton pregnancies complicated by severe E-PE (n=58) and pregnancies complicated by severe IUGR (n=29) with abnormal Doppler flow velocity waveforms in the umbilical artery (AEDF or REDF) and without any evidence of maternal hypertensive disorder, renal disease or diabetes. The diagnosis of PE was made according to the following criteria: presence of pregnancy-induced hypertension (systolic ≥140 mm Hg, diastolic ≥90 mm Hg) and proteinuria (≥300 mg/24 h) after the 20th week of gestation in previously normotensive women.4 IUGR was defined as birth weight below the 3rd percentile for gestational age and fetal sex, according to the local normograms. In all, 58 AMC placentae were obtained from normal pregnancies that did not show any signs of PE, IUGR or other placental disease. Maternal age, gestational age and parity were comparable between E-PE, IUGR and AMC groups; 19.6% of the babies from E-PE pregnancies were growth restricted. Pregnancies with congenital malformations, chromosomal anomalies (of number and/or structure) or notable intrauterine infection were excluded from the study. Patients with diabetes, infections and kidney disease were also excluded. Samples were collected randomly from central and peripheral placental areas and snap frozen immediately after delivery. Calcified, necrotic and visually ischemic areas were excluded from collection.
RNA isolation and real-time PCR
Total RNA, extracted from placental tissues and explant cultures,26 was treated with DNAse I to remove genomic DNA contamination. Total RNA of 1 μg was reverse transcribed using random hexamers (Applied Biosystems (ABI), Foster City, CA, USA). The resulting templates (30 ng of cDNA for Mcl-1L, Mcl-1S, MULE, p53 and HIF-1α and 1.5 ng for 18S) were quantified by real-time PCR (DNA Engine Opticon2 R system, MJ Research, Waltham, MA, USA). TaqMan primers and probes for ribosomal 18S, MULE, p53 and HIF-1α were purchased from ABI as Assays-on-Demand for human genes. Quantitative PCR for Mcl-1 was performed using SYBR Green (Bio-Rad Laboratories, Inc., Hercules, CA, USA) as described previously.18 For each probe, a dilution series determined the efficiency of amplification of each primer/probe set and the relative quantification method was employed.39 For the relative quantitation, PCR signals were compared among groups after normalization using 18S as internal reference. Relative expression and fold change was calculated according to Livak and Schmittgen.39
Western blot analysis
Western blot analyses were performed as described previously.40 Primary antibodies were rabbit polyclonal anti-human MULE (1:1500 dilution; Bethyl Laboratories, Montgomery, TX, USA), rabbit polyclonal anti-human Mcl-1 (S-19; 1 : 500 dilution; Santa Cruz Biotechnology, Santa Cruz, CA, USA), goat polyclonal anti-human p53 (1 : 1000 dilution; Cell Signaling Technology, Beverly, MA, USA) and rabbit polyclonal anti-human HIF-1α (1 : 1000; Cell Signaling Technologies). Horseradish peroxidase-conjugated secondary antibodies were donkey anti-rabbit for MULE (1 : 10 000), Mcl-1 (1 : 10 000), p53 (1 : 10 000) and HIF-1α (1 : 2000). Specificity of MULE antibody was determined using MULE blocking-peptides (Bethyl Laboratories), while a 293T cell lysate was used as positive control (Bethyl Laboratories).
Mcl-1/MULE and p53/MULE associations
The associations between Mcl-1, p53 and MULE were examined as described previously.40 Following immunoprecipitation of Mcl-1 or p53 using anti-Mcl-1 and anti-p53 antibodies, respectively, imunoprecipitates were subjected to SDS-PAGE and immunoblotted with anti-MULE antibody.
Mcl-1 and p53 ubiquitination
In all, 300 μg of total protein was diluted in RIPA buffer to a concentration of 1 μg/μl. Two micrograms of specific rabbit-polyclonal Mcl-1 (Santa Cruz, S-19) or p53 (Cell Signaling) antibodies were added to each sample and the mixtures were incubated at 4 °C on a rotator overnight. A measure of 30 μl of Protein-Aagarose beads (specific for IgG) were added to each lysate and the mixture was incubated at 4 °C on a rotator for 2 h. Agarose beads were collected by centrifugation and washed once with RIPA buffer and twice with PBS. The beads were then resuspended in 2 × SDS loading buffer, boiled for 5 min and centrifuged. The supernatants were collected, subjected to SDS-PAGE and analyzed by western blotting using mouse monoclonal anti-human ubiquitin antibody (1 : 500 dilution; Covance Research Products, Berkeley, CA, USA) in TBST and 5% non-fat milk. A 1 : 10 000 dilution of anti-mouse–Ig–horseradish peroxidase was used as secondary antibody. Immunoblots were visualized using enhanced chemiluminescence.
Immunofluorescence staining
Placental tissues from first trimester (5–12 weeks gestation, n=25), PE (n=15), IUGR (n=10) and AMC (n=15) were fixed in 4% paraformaldehyde and embedded in paraffin. Before immunofluorescence (IF) analysis, every 10th section was stained with hematoxylin and eosin to assess the quality of the tissue and the most representative sections were selected. Sodium citrate antigen retrieval was performed, followed by Sudan Black treatment (0.1% Sudan Black in 70% EtOH) to quench endogenous fluorescence. Sections were pre-incubated in 5% horse serum in PBS (contained 0.04% sodium azide and 0.008% gelatin) to block nonspecific binding and incubated with primary antibodies overnight at 4 °C. Rabbit polyclonal antibodies anti-human MULE (1 : 400 dilution; Bethyl Laboratories), anti-human Mcl-1 (1 : 200 dilution; Santa Cruz Biotechnology) and anti-human p53 (1 : 200 dilution; Cell Signaling Technologies) were used. Control IgG were used as negative controls. Slides were treated with 0.4% DAPI (4′,6-diamidino-2-phenylindole) for nuclear detection. Fluorescence images were viewed and captured using a DeltaVision Deconvolution microscope (Applied Precision, LLC, Issaquah, WA, USA).
Human first trimester villous explant culture
First trimester (5–8 weeks of gestation, n=18) villous explant cultures were established as described previously.25 Villous explants were maintained in standard tissue culture conditions of 5% CO2 in 95% air (20% O2 environment) or in an atmosphere of 3% O2/92% N2/5% CO2 (3% O2 environment) for 48 h at 37 °C. These oxygen concentrations were chosen because they represent the standard culturing condition and the physiological placental low oxygen environment before 10 weeks of gestation, respectively. In parallel experiments, explants were also exposed to HR as described previously.11 In some experiments, villous explants were incubated with 5 ng/ml TGF-β1 or TGF-β3 for 48 h. For each treatment, tissue samples from the same placenta were used and in each experiment, explant cultures were set up in triplicate.
HIF-1 α siRNA experiments
Human JEG-3 cells, cultured at 20% O2, were transfected with 40 nM validated HIF-1α siRNA (Invitrogen, Carlsbad, CA, USA) using Lipofectamine 2000. Scrambled siRNA sequences were used as negative control and fluorescent-tagged siRNA duplexes were used to test transfection efficiency. After transfection, cells were maintained at 3% O2 for 24 h before mRNA and protein collection. To assess siRNA cytotoxicity, LDH release was measured using the LDH-Cytotoxicity Assay Kit (Biovision Inc., Mountain View, CA, USA) following the manufacturer’s instructions.
TUNEL assay
Programmed cell death in control, E-PE and IUGR placentae was assessed by TUNEL as described previously.18 Briefly, tissue sections were de-waxed, rehydrated and treated for 10 min with 10 μg/ml proteinase K. Sections were then treated with 0.1% Sudan Black, pre-incubated with 0.1% Triton X-100 in OPA (One-Phor-All buffer, Pharmacia, Stockholm, Sweden), incubated for 90 min in TdT reaction mixture (TdT plus biotin-dUTP) and processed for IF using streptavidin fluorescein. Images were captured using a DeltaVision Deconvolution microscope (Applied Precision, LLC).
Statistical analysis
All data are presented as mean±S.E.M. For comparison of data between multiple groups, we used one-way analysis of variance (ANOVA) with post-hoc Dunnett’s test and Kruskal–Wallis one-way ANOVA as appropriate. For comparison between two groups, we used paired and unpaired Student’s t-test as appropriate. Correlations between groups was calculated using Pearson’s coefficient. Statistical tests were carried out using Prism statistical software and significance was accepted at P<0.05.
Abbreviations
- Mcl-1:
-
myeloid cell leukemia factor 1
- PE:
-
preeclampsia
- IUGR:
-
intrauterine growth restriction
- MULE:
-
Mcl-1 Ubiquitin Ligases E3
- BH3:
-
Bcl-2 homology domain 3
- Mdm2:
-
murine double minute-2
- Mdmx:
-
murine double minute-X
- HR:
-
hypoxia/reoxygenation
- HIF:
-
hypoxia-inducible factor
- LDH:
-
lactate dehydrogenase
- AMC:
-
age-matched control
- A/REDF:
-
absent/reverse end diastolic flow
- IF:
-
immunofluorescence
- TUNEL:
-
terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick-end labeling
References
Cunningham FG, Lindheimer MD . Hypertension in pregnancy. N Engl J Med 1992; 326: 927–932.
Cetin I, Foidart JM, Miozzo M, Raun T, Jansson T, Tsatsaris V et al. Fetal growth restriction: a workshop report. Placenta 2004; 25: 753–757.
ACOG practice bulletin.Diagnosis and management of preeclampsia and eclampsia. Obstet Gynecol 2002; 99: 159–167.
Pijnenborg R, Vercruysse L, Verbist L, Van Assche FA . Interaction of interstitial trophoblast with placental bed capillaries and venules of normotensive and pre-eclamptic pregnancies. Placenta 1998; 19: 569–575.
Soleymanlou N, Jurisica I, Nevo O, Ietta F, Zhang X, Zamudio S et al. Molecular evidence of placental hypoxia in preeclampsia. J Clin Endocrinol Metab 2005; 90: 4299–4308.
Burton GJ, Yung HW, Cindrova-Davies T, Charnock-Jones DS . Placental endoplasmic reticulum stress and oxidative stress in the pathophysiology of unexplained intrauterine growth restriction and early onset preeclampsia. Placenta 2009; 30(Suppl A): S43–S48.
Roberts JM, Hubel CA . Is oxidative stress the link in the two-stage model of pre-eclampsia? Lancet 1999; 354: 788–789.
Jackson MR, Walsh AJ, Morrow RJ, Mullen JB, Lye SJ, Ritchie JW . Reduced placental villous tree elaboration in small-for-gestational-age pregnancies: relationship with umbilical artery Doppler waveforms. Am J Obstet Gynecol 1995; 172: 518–525.
Levy R, Smith SD, Yusuf K, Huettner PC, Kraus FT, Sadovsky Y et al. Trophoblast apoptosis from pregnancies complicated by fetal growth restriction is associated with enhanced p53 expression. Am J Obstet Gynecol 2002; 186: 1056–1061.
Leung DN, Smith SC, To KF, Sahota DS, Baker PN . Increased placental apoptosis in pregnancies complicated by preeclampsia. Am J Obstet Gynecol 2001; 184: 1249–1250.
Soleymanlou N, Wu Y, Wang JX, Todros T, Ietta F, Jurisicova A et al. A novel Mtd splice isoform is responsible for trophoblast cell death in pre-eclampsia. Cell Death Differ 2005; 12: 441–452.
Heazell AE, Sharp AN, Baker PN, Crocker IP . Intra-uterine growth restriction is associated with increased apoptosis and altered expression of proteins in the p53 pathway in villous trophoblast. Apoptosis 16: 135–144.
Hsu SY, Hsueh AJ . Tissue-specific Bcl-2 protein partners in apoptosis: an ovarian paradigm. Physiol Rev 2000; 80: 593–614.
Gross A, McDonnell JM, Korsmeyer SJ . BCL-2 family members and the mitochondria in apoptosis. Genes Dev 1999; 13: 1899–1911.
Hershko A, Ciechanover A, Varshavsky A . Basic Medical Research Award. The ubiquitin system. Nat Med 2000; 6: 1073–1081.
Zhong Q, Gao W, Du F, Wang X . Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell 2005; 121: 1085–1095.
Warr MR, Acoca S, Liu Z, Germain M, Watson M, Blanchette M et al. BH3-ligand regulates access of MCL-1 to its E3 ligase. FEBS Lett 2005; 579: 5603–5608.
Soleymanlou N, Jurisicova A, Wu Y, Chijiiwa M, Ray JE, Detmar J et al. Hypoxic switch in mitochondrial myeloid cell leukemia factor-1/Mtd apoptotic rheostat contributes to human trophoblast cell death in preeclampsia. Am J Pathol 2007; 171: 496–506.
Chen D, Kon N, Li M, Zhang W, Qin J, Gu W . ARF-BP1/Mule is a critical mediator of the ARF tumor suppressor. Cell 2005; 121: 1071–1083.
Levine AJ . P53, the cellular gatekeeper for growth and division. Cell 1997; 88: 323–331.
Iwakuma T, Lozano G . MDM2, an introduction. Mol Cancer Res 2003; 1: 993–1000.
Haupt Y, Maya R, Kazaz A, Oren M . Mdm2 promotes the rapid degradation of p53. Nature 1997; 387: 296–299.
Liu Z, Miao D, Xia Q, Hermo L, Wing SS . Regulated expression of the ubiquitin protein ligase, E3(Histone)/LASU1/Mule/ARF-BP1/HUWE1, during spermatogenesis. Dev Dyn 2007; 236: 2889–2898.
Caniggia I, Mostachfi H, Winter J, Gassmann M, Lye SJ, Kuliszewski M et al. Hypoxia-inducible factor-1 mediates the biological effects of oxygen on human trophoblast differentiation through TGFbeta(3). J Clin Invest 2000; 105: 577–587.
Caniggia I, Grisaru-Gravnosky S, Kuliszewsky M, Post M, Lye SJ . Inhibition of TGF-beta 3 restores the invasive capability of extravillous trophoblasts in preeclamptic pregnancies. J Clin Invest 1999; 103: 1641–1650.
Nishi H, Nakada T, Hokamura M, Osakabe Y, Itokazu O, Huang LE et al. Hypoxia-inducible factor-1 transactivates transforming growth factor-beta3 in trophoblast. Endocrinology 2004; 145: 4113–4118.
Kozopas KM, Yang T, Buchan HL, Zhou P, Craig RW . MCL1 a gene expressed in programmed myeloid cell differentiation, has sequence similarity to BCL2. Proc Natl Acad Sci USA 1993; 90: 3516–3520.
Zhou P, Qian L, Bieszczad CK, Noelle R, Binder M, Levy NB et al. Mcl-1 in transgenic mice promotes survival in a spectrum of hematopoietic cell types and immortalization in the myeloid lineage. Blood 1998; 92: 3226–3239.
Rinkenberger JL, Horning S, Klocke B, Roth K, Korsmeyer SJ . Mcl-1 deficiency results in peri-implantation embryonic lethality. Genes Dev 2000; 14: 23–27.
Craig RW . MCL1 provides a window on the role of the BCL2 family in cell proliferation, differentiation and tumorigenesis. Leukemia 2002; 16: 444–454.
Herrant M, Jacquel A, Marchetti S, Belhacene N, Colosetti P, Luciano F et al. Cleavage of Mcl-1 by caspases impaired its ability to counteract Bim-induced apoptosis. Oncogene 2004; 23: 7863–7873.
Mott JL, Kobayashi S, Bronk SF, Gores GJ . Mir-29 regulates Mcl-1 protein expression and apoptosis. Oncogene 2007; 26: 6133–6140.
Inoshita S, Takeda K, Hatai T, Terada Y, Sano M, Hata J et al. Phosphorylation and inactivation of myeloid cell leukemia 1 by JNK in response to oxidative stress. J Biol Chem 2002; 277: 43730–43734.
Schwickart M, Huang X, Lill JR, Liu J, Ferrando R, French DM et al. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature 463: 103–107.
Heazell AE, Lacey HA, Jones CJ, Huppertz B, Baker PN, Crocker IP . Effects of oxygen on cell turnover and expression of regulators of apoptosis in human placental trophoblast. Placenta 2008; 29: 175–186.
Liu W, Xin H, Eckert DT, Brown JA, Gnarra JR . Hypoxia and cell cycle regulation of the von Hippel–Lindau tumor suppressor. Oncogene 30: 21–31.
Nevo O, Many A, Xu J, Kingdom J, Piccoli E, Zamudio S et al. Placental expression of soluble fms-like tyrosine kinase 1 is increased in singletons and twin pregnancies with intrauterine growth restriction. J Clin Endocrinol Metab 2008; 93: 285–292.
Nevo O, Soleymanlou N, Wu Y, Xu J, Kingdom J, Many A et al. Increased expression of sFlt-1 in in vivo and in vitro models of human placental hypoxia is mediated by HIF-1. Am J Physiol Regul Integr Comp Physiol 2006; 291: R1085–R1093.
Livak KJ, Schmittgen TD . Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001; 25: 402–408.
Ietta F, Wu Y, Winter J, Xu J, Wang J, Post M et al. Dynamic HIF1A regulation during human placental development. Biol Reprod 2006; 75: 112–121.
Acknowledgements
We thank Dr. Dragica Curovic for placental collection and the BioBank Program of the CIHR Group in Development and Fetal Health (CIHR no. MGC-13299), the Samuel Lunenfeld Research Institute and the MSH Department of Obstetrics and Gynecology for the human specimens used in this study. We also acknowledge Megan Melland-Smith and Antonella Racano for the technical contribution with the immunofluorescence staining using control IgG and western blotting of p53 in placental development. This work was supported by the Canadian Institutes of Health Research (CIHR) Grant (MOP-89813) to Isabella Caniggia. Isabella Caniggia is the recipient of a mid-career CIHR award administered through the Ontario Women’s Health Council. Martin Post is the holder of a Canadian Research Chair (tier 1) in Fetal, Neonatal and Maternal Health.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by A Stephanou
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Rolfo, A., Garcia, J., Todros, T. et al. The double life of MULE in preeclamptic and IUGR placentae. Cell Death Dis 3, e305 (2012). https://doi.org/10.1038/cddis.2012.44
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2012.44
Keywords
This article is cited by
-
Ubiquitin ligases: guardians of mammalian development
Nature Reviews Molecular Cell Biology (2022)
-
Downregulation of p53 drives autophagy during human trophoblast differentiation
Cellular and Molecular Life Sciences (2018)
-
Dichotomous role of pancreatic HUWE1/MULE/ARF-BP1 in modulating beta cell apoptosis in mice under physiological and genotoxic conditions
Diabetologia (2014)
