
Abstract
Special AT-rich sequence-binding protein 1 (SATB1) is a global chromatin organizer and gene regulator, and high expression of SATB1 is associated with progression and poor prognosis in several malignancies. Here, we examine the expression pattern of SATB1 in glioma. Microarray analysis of 127 clinical samples showed that SATB1 mRNA was expressed at lower levels in highly malignant glioblastoma multiforme (GBM) than in low-grade glioma and normal brain tissue. This result was further confirmed by real-time RT-PCR in the clinical samples, three GBM cell lines, primary SU3 glioma cells and tumor cells harvested by laser-capture microdissection. Consistent with the mRNA levels, SATB1 protein expression was downregulated in high-grade glioma, as shown by western blotting. However, phospho-SATB1 levels showed an opposite pattern, with a significant increase in these tumors. Immunohistochemical analysis of phospho-SATB1 expression in tissue microarrays with tumors from 122 glioma cases showed that phospho-SATB1 expression was significantly associated with high histological grade and poor survival by Kaplan–Meier analysis. In vitro transfection analysis showed that phospho-SATB1 DNA binding has a key role in regulating the proliferation and invasion of glioma cells. The effect of SATB1 in glioma cell is mainly histone deacetylase (HDAC1)-dependent. We conclude that phospho-SATB1, but not SATB1 mRNA expression, is associated with the progression and prognosis of glioma. By interaction with HDAC1, phospho-SATB1 contributes to the invasive and proliferative phenotype of GBM cells.
Similar content being viewed by others

Main
Glioblastoma multiforme (GBM) represents a major health concern because of its increasing incidence and poor prognosis.1 Despite multimodal treatment including surgery, chemotherapy and radiation, the median survival time of glioblastoma patients is ∼14 months.2 The mechanism by which GBM acquires a highly invasive and proliferative phenotype remains unclear, although numerous factors and signaling pathways have been researched.3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13 The malignant progression of GBM is associated with the deregulation of the expression of many genes. However, few studies have examined the role of chromatin organization in GBM.
Special AT-rich sequence-binding protein 1 (SATB1) is essential for chromatin organization, and is involved in the regulation of hundreds of genes.14 SATB1 can be specifically downregulated by miR-155 binding to its 3′ untranslated region15, 16 and it is phosphorylated by protein kinase-C (PKC).17 Furthermore, phosphorylated SATB1 associates with histone deacetylase (HDAC1), whereas dephosphorylated SATB1 interacts with histone acetyltransferase PCAF.17 Recent studies indicated an important role for both HDAC1 and PCAF in the regulation of glioma growth.18, 19 Previous studies reported that high SATB1 expression was correlated with metastasis and unfavorable clinical outcome in several cancer forms including breast carcinoma, gastric carcinoma, colorectal carcinoma and bladder carcinoma.20, 21, 22, 23 Although SATB1 phosphorylation significantly affects its ability to act as a global regulator of gene expression,17, 24 data about the relationship between phospho-SATB1 and prognosis of malignant tumors are not available.
In the present study, we show that phospho-SATB1 but not SATB1 mRNA expression is associated with the progression and prognosis of glioma, and that by interaction with HDAC1, phospho-SATB1 contributes to the invasive and proliferative phenotype of GBM cells.
Results
SATB1 mRNA is expressed at a relatively low level in GBM
The mRNA expression of SATB1 in gliomas of different histological grades was examined by microarray analysis using 127 clinical samples (71P, 8AA, 43A and 5N). The results showed that SATB1 mRNA expression levels were lower in GBM than in low-grade glioma and normal brain tissue, as shown in Figure 1a (P<0.01). SATB1 mRNA expression was not correlated with the prognosis of patients (R=0.056, P=0.497). However, Pearson correlation analysis showed that SATB1 expression was negatively correlated with the expression levels of miR-155 (R=−0.536, P<0.001) and PKC (R=−0.364, P<0.01),(Figures 1b, d and e) which were increased in high-grade gliomas. Our result of SATB1 expression in glioma tissues was validated on two independent data sets that are publicly available25, 26 (Figure 1c).

mRNA expression data obtained from microarray analysis of 127 clinical samples were used for cluster analysis and Pearson correlation analysis. (a) SATB1 mRNA expression was analyzed by cluster analysis and visualized using Treeview software. Compared with low-grade glioma and normal brain tissue, SATB1 mRNA expression was reduced in GBM. (b, d and e) The expression of SATB1 was negatively correlated with the expression of miR-155 and PKC. (c) The expression of SATB1 was validated on the expression data of glioma samples from Henry Ford Hospital (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE4290)
SATB1 levels were further evaluated in three GBM cell lines (U87, U251 and T98G), glioma clinical samples and primary SU3 glioma cells by real-time RT-PCR, which showed a relatively low SATB1 mRNA expression level in high-grade gliomas compared with low-grade gliomas (Figure 2a), confirming the results of microarray analysis. In addition, laser-capture microdissection (LCM) was performed to obtain tumor cells from randomly selected 10 astrocytoma samples and 10 GBM samples and the expression of SATB1 in these cells were examined using real-time quantitative PCR. As shown in Figure 2b, the expression pattern was similar to the PCR results obtained from the same samples without LCM.

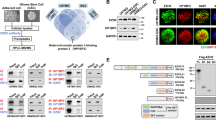
Expression of SATB1 and phospho-SATB1. (a) Real-time RT-PCR showed that SATB1 mRNA was expressed at low levels in GBM clinical samples, the SU3, U87 and T98G cells (AA: anaplastic astrocytoma, (a) astrocytoma). (b) Using real-time quantitative PCR, the SATB1 expression was examined in the homogeneous tumor cells harvested by LCM from randomly selected 10 astrocytoma samples and 10 glioblastoma samples. SATB1 was low-expressed in high-grade glioma compared with low-grade glioma. The expression tendency of tumor cells harvested by LCM (left) was similar to that of the same samples without LCM (right). (c) Western blot analysis showed a low level of total SATB1 protein expression in GBM compared with that in low-grade glioma. (d and e) Western blot analysis showed a marked upregulation of phospho-SATB1 and PKC expressions in GBM compared with low-grade glioma. (f) CoIP of nuclear protein from glioma samples with anti-HDAC1 and anti-PCAF followed by anti-SATB1 immunoblot
PKC and phospho-SATB1 are upregulated in GBM
Consistent with the SATB1 mRNA levels, total SATB1 protein was also downregulated in GBM as shown by western blotting (Figure 2c). However, PKC, which phosphorylates SATB1, was significantly upregulated in GBM (Figure 2e). We therefore examined the expression of phospho-SATB1 in GBM tissues by western blotting, which showed that phospho-SATB1 was markedly upregulated in GBM compared with low-grade glioma (Figure 2d). Moreover, coimmunoprecipitation demonstrated that HDAC1 antibody immunoprecipitated SATB1 efficiently from high-grade glioma compared with low-grade glioma, whereas anti-PCAF showed opposite tendency. Given that phosphorylated SATB1 interacts with HDAC1 and dephosphorylated SATB1 associates with PCAF,17 the results further confirmed that phospho-SATB1 level was higher in high-grade glioma than in low-grade glioma (Figure 2f). These results indicated that despite the reduction in SATB1 mRNA and protein, phospho-SATB1 expression was upregulated in GBM.
Correlation between phospho-SATB1 levels and the malignancy of gliomas
To further determine whether phospho-SATB1 levels were associated with the histological characteristics of gliomas, a glioma tissue microarray was constructed and examined by immunohistochemical (IHC) staining with a specific antibody against phospho-SATB1. Phospho-SATB1 was detected mainly as nuclear staining in all high-grade gliomas (grades III-IV) and at a high level (score >4) in 57% (45/79) of these samples. Semiquantitative analysis indicated that phospho-SATB1 expression was significantly higher in high-grade gliomas than in low-grade gliomas (grade II) (P<0.01), supporting an association between phospho-SATB1 and glioma progression (Figure 3i).

Tissue microarray and IHC images of phospho-SATB1 staining in 122 clinical glioma samples. (a) Negative control; (b) negative; (c–e) weak to moderate staining intensity; (f) strong staining intensity. (g) The deep brown phospho-SATB1 signal was mainly located in the nuclei of GBM cells, and only a few astrocytoma cells were positively stained (h). (i) Semiquantitative analysis of phospho-SATB1 expression showed that phospho-SATB1 levels were significantly higher in high-grade gliomas than in low-grade gliomas. (j) Kaplan–Meier analysis showing the OS of high-grade glioma patients in the low phospho-SATB1 group (green line) and the high phospho-SATB1 group (blue line). A statistically significant difference in OS was observed between the two groups
To determine the prognostic value of phospho-SATB1 in high-grade glioma, we used Cox proportional hazard models and Kaplan–Meier analysis. Patients in the high phospho-SATB1 group had lower overall survival (OS) than those in the low phospho-SATB1 group on univariate analysis (hazard ratio, HR 2.118; 95% CI, 1.328–3.379), and this relation persisted after adjusting for other potential risk factors (multivariate HR 2.896; 95% CI, 1.719–4.878, Table 1). No statistically significant differences were identified between phospho-SATB1 levels in relation to sex, age, tumor size, preoperative Karnofsky performance status (KPS), degree of resection or O(6)-methylguanine-DNA-methyltransferase (MGMT) promoter methylation. Kaplan–Meier analysis was used to assess the discriminative value of phospho-SATB1 levels (low or high) for the survival of patients. We found that phospho-SATB1 levels could effectively discriminate the survival of the two high-grade glioma subgroups, with a mean OS of 12.7 months (median OS, 13.6 months; 95% CI, 12.9–14.3) for the low phospho-SATB1 group versus a mean OS of 9.3 months (median OS, 9.4 months; 95% CI, 7.7–11.0) for the high phospho-SATB1 group (P=0.009) (Figure 3j). Our results indicated that in addition to phospho-SATB1, age and MGMT promoter methylation may have a role in predicting OS in high-grade gliomas (Table 1).
SATB1 knockdown reduces U251 cell proliferation and invasion
On the basis of prior studies that used cell lines to examine the role of SATB1 in malignant tumors, we performed similar in vitro studies to determine the potential role of SATB1 in the proliferative and invasive phenotype of GBM. SATB1 expression was stably knocked down in relatively high SATB1-expressing U251 cells by transfection with a SATB1-specific shRNA as shown in Figure 2a. The loss of SATB1 mRNA expression was confirmed by RT-PCR (Figure 4a). Unlike our in vivo data, SATB1 knockdown resulted in a decrease in phospho-SATB1 levels (Figure 4b). MTT and Matrigel invasion assays showed that the downregulation of SATB1 and phospho-SATB1 in U251-SATB1-shRNA cells resulted in a reproducible significant reduction in cell proliferation and invasion, compared with cells transfected with non-silencing control shRNA plasmid and untransfected U251 cells (Figures 4c–f).

SATB1 knockdown reduces U251 cell proliferation and invasion. (a and b) The loss of SATB1 and phospho-SATB1 expression was confirmed by RT-PCR and western blotting in U251-SATB1-shRNA cells. (c) MTT assay showed a significant reduction in the proliferation of U251-SATB1-shRNA cells. Invasion assay demonstrated that compared with cells transfected with non-silencing control shRNA plasmid (d) and untransfected U251 cells, the invasive ability of U251-SATB1-shRNA cells was markedly decreased (e and f) (*P<0.05)
A SATB1 decoy and HDAC1-silencing inhibit the increase in glioma cell proliferation and invasion caused by SATB1 overexpression
SATB1 expression was stably upregulated in U87 and SU3 cells, which have low endogenous SATB1 mRNA expression, by transfection with the pEGFP-N1-SATB1 plasmid (Figure 2a). Consistent with the results of our knockdown experiment, increased SATB1 mRNA levels were accompanied by an increase in phospho-SATB1 expression in vitro (Figures 5a and b). MTT and invasion assays showed that SATB1 as well as phospho-SATB1 overexpression in U87- and SU3-pEGFP-N1-SATB1 cells led to enhanced cell proliferation and invasion, compared with cells transfected with control vector and untransfected cells (Figures 5c–f). SATB1 upregulation also led to elevated expression of matrix metalloproteinase (MMP)-2 and MMP-9 (Figures 6c and d) but not MMP-3 or MMP-7 in SU3 cells.

SATB1 overexpression increases SU3 and U87 cell proliferation and invasion. (a and b) The upregulation of SATB1 and phospho-SATB1 expression was confirmed by RT-PCR and western blotting in U87- and SU3-pEGFP-N1-SATB1 cells. (c) MTT assay showed that proliferation was significantly increased in U87- and SU3-pEGFP-N1-SATB1 cells. Invasion assay showed that compared with cells transfected with control vector (d and e, left) and untransfected cells, the invasive ability of U87- and SU3-pEGFP-N1-SATB1 cells was markedly increased (d and e (right), f) (*P<0.05)

Effect of HDAC1 or PCAF-silencing on the proliferation and invasion of SU3-pEGFP-N1-SATB1 cells. HDAC1 siRNA, PCAF siRNA or control siRNA were transfected into SU3-pEGFP-N1-SATB1 cells overexpressing SATB1 and phospho-SATB1, and after 24 h cells were subjected to MTT and in vitro invasion assays. (a) MTT assay showed that HDAC1-silencing rather than PCAF-silencing significantly reduced SU3-pEGFP-N1-SATB1 cell proliferation. (b) In vitro invasion assay showed that HDAC1-silencing instead of PCAF-silencing markedly inhibited the invasive ability of SU3-pEGFP-N1-SATB1 cells. (c and d) SATB1 upregulation increases the expression of MMP-2 and MMP-9 in a HDAC1-dependent manner (*P<0.05)
However, a SATB1 decoy designed to interfere with phospho-SATB1 binding to DNA effectively inhibited the proliferative and invasive ability of U87- and SU3-pEGFP-N1-SATB1 cells overexpressing SATB1 and phospho-SATB1 (Figures 7a and b). Moreover, HDAC1-silencing rather than PCAF-silencing greatly reduced the proliferation and invasion of SU3-pEGFP-N1-SATB1 cells (Figures 6a and b). In addition, elevation of MMP-2 and MMP-9 by SATB1 is mainly HDAC1 dependent (Figures 6c and d).

Effect of SATB1-decoy DNA on the proliferation and invasion of U87- and SU3-pEGFP-N1-SATB1 cells. SATB1-decoy DNA or control-decoy DNA was transfected into U87- and SU3-pEGFP-N1-SATB1 cells overexpressing SATB1 and phospho-SATB1, and cells were subjected to MTT and in vitro invasion assays. (a) MTT assay showed that SATB1-decoy DNA significantly reduced U87- and SU3-pEGFP-N1-SATB1 cell proliferation. (b) In vitro invasion assay showed that SATB1-decoy DNA markedly inhibited the invasive ability of U87- and SU3-pEGFP-N1-SATB1 cells. (c) Effect of SATB1-decoy DNA on SATB1 mRNA expression. At 24 h after transfection, 6.1-, 5.2- and 6.4-fold increases in SATB1 mRNA expression were detected in SU3, U87 and T98G cells, respectively (*P<0.05)
SATB1-decoy DNA increases SATB1 expression in glioma cells
To further investigate the effect of phospho-SATB1 on SATB1 mRNA expression, SATB1-decoy DNA was transfected into SU3, U87 and T98G cells. After 24 h, SATB1 mRNA expression was detected by real-time RT-PCR. As shown in Figure 7c, SATB1-decoy DNA, which specifically inhibits the effect of phospho-SATB1,27 significantly increased the expression of SATB1 mRNA by 6.1-, 5.2- and 6.4-fold in SU3, U87 and T98G cells, respectively. These findings indicate that SATB1 DNA binding negatively regulates its own expression, which may explain the opposite tendency of SATB1 expression and its phosphorylation status.
Discussion
The existence of normal cells in glioma clinical samples may affect the analysis of tumor-specific gene expression. To minimize such influence, in this study, only samples containing more than 80% tumor cells were selected for analysis. Moreover, we used the LCM method to obtain RNA that was derived exclusively from a tumor. Using these samples we confirmed that normal cells exhibit negligible influence on the expression tendency of SATB1 in our clinical samples. The combination of microarray analysis, real-time RT-PCR and LCM-based real-time RT-PCR demonstrated that our result of SATB1 expression is reproducible. In addition, we validated our result of SATB1 expression on two independent data sets that are publicly available. All of these results clearly show that SATB1 is low-expressed in high-grade glioma compared with low-grade glioma. In the present study, we show that phospho-SATB1 has a key role in the induction of an aggressive phenotype in glioma, and SATB1 mRNA and phospho-SATB1 showed opposite expression patterns. In high-grade glioma, SATB1 mRNA was expressed at low levels, whereas phospho-SATB1 was significantly upregulated. Similar phenomena also present in regulatory factors, including phosphatase and tensin homolog (PTEN),28 phospholemman29 and TBC1 domain family member 1 (TBC1D1),30 which showed opposite expression patterns. In our GBM samples, decreased SATB1 expression was associated with increased miR-155 expression, which can specifically downregulate SATB1. This may partly explain the downregulation of SATB1 in GBM, and confirmed our results. A similar phenomenon was reported in breast cancer.16 As an important post-translational modification, phosphorylation significantly affects the ability of SATB1 to bind with DNA and act as a global regulator of gene expression.17, 24 The fact that PKC expression was significantly increased in high-grade gliomas may explain the high levels of phospho-SATB1 detected in these samples, despite the reduction in SATB1 mRNA levels.
The role of SATB1 in the proliferation and invasion of GBM cells has been investigated in cell lines;31 therefore, in the present study, we performed similar analyses by specifically knocking down or upregulating SATB1 expression in GBM cells. Our results showed that SATB1 knockdown reduced U251 cell proliferation and invasion, whereas SATB1 overexpression promoted U87 cell proliferation and invasion. To exclude the bias of long-term cultured cell line and confirm our result, primary SU3 glioma cells derived from fresh clinical sample was used as model cells. Upregulation of SATB1 expression also promotes proliferation and invasion in SU3 cells. However, this result must be interpreted with caution as it may lead to two different conclusions. First, similar to previous studies, the upregulation of SATB1 mRNA expression could promote GBM cell proliferation and invasion. However, this was not supported by our in vivo data showing decreased SATB1 mRNA and protein levels in association with increased phospho-SATB1 levels in highly proliferative and invasive GBM specimens. The lack of correlation between SATB1 mRNA and phospho-SATB1 levels in vivo indicated the second conclusion that phospho-SATB1 but not SATB1 expression may have a role in proliferation and invasion. This hypothesis was further confirmed by in vitro transfection of SATB1-decoy DNA, which specifically disturbs phospho-SATB1 binding to DNA.17, 27, 32 Transfection of cells with decoy SATB1 resulted in the inhibition of proliferation and invasion in SATB1-overexpressing glioma cells. Consistent with our results, Asako et al.27 found that decoy-SATB1 DNA inhibits the growth and invasive ability of human breast cancer. Barboro et al.33 reported that the expression of SATB1 is higher in less aggressive prostate cancer cell lines, and binding of SATB1 to matrix attachment regions (MARs) of DNA is increased in highly aggressive cell lines.
Transfection of SU3, U87 and T98 cells with SATB1-decoy DNA significantly upregulated the expression of SATB1 mRNA, indicating that SATB1 DNA binding negatively regulates its own expression. High SATB1 phosphorylation status, which leads to high SATB1-DNA binding, may thus suppress SATB1 expression and result in the low expression of this gene in high-grade glioma. On the contrary, low SATB1 phosphorylation status may induce high expression of SATB1 mRNA in low-grade gliomas. However, the exact regulating mechanism needs further investigation, and other factors such as miR-155 may also be involved in this regulating process.
In our in vitro study, the SATB1 gene was exogenously overexpressed, thus abolishing the modulation of its expression by phospho-SATB1. Therefore, the induction of SATB1 overexpression may lead to an increase in SATB1 phosphorylation. In the analysis of the underlying mechanisms, data obtained by in vitro experiments need to be interpreted with caution because the exogenous overexpression or silencing of these genes is not an exact representation of the in vivo phenomena and regulatory mechanisms. The use of data derived from in vitro experiments to explain in vivo phenomena should therefore be done with caution.
Contrary to the findings reported by Chu et al.,31 in the present study, analysis of a large sample size showed that SATB1 mRNA and protein levels were relatively low and phospho-SATB1 levels were high in GBM in comparison with low-grade glioma and normal brain tissues. Moreover, the assessment of phospho-SATB1 levels is necessary to accurately determine the effect of SATB1 on the progression and prognosis of glioma, and in the absence of such data, the results may lead to misinterpretation. Prior studies on the expression level of SATB1 and its role in tumor development have reported contradictory results. Han et al.20 proposed that SATB1 expression levels are correlated with poor prognosis and promote tumor growth and metastasis in breast cancer. On the other hand, Iorns et al.34 reported that there was no association between SATB1 and breast cancer pathogenesis, and showed the loss of SATB1 expression in squamous preinvasive lesions and in non-small cell lung cancers. In another study, high SATB1 expression was not associated with poor clinical outcome.35 Nevertheless, research about the function of phospho-SATB1 in human cancers has been limited. Our present findings suggest that further investigation is necessary to clarify the role of SATB1 in malignancies and that future studies should focus on the role of SATB1 phosphorylation status.
We further investigated how SATB1 regulates the aggressive phenotype of glioma cells, and demonstrate that SATB1 mainly interacts with HDAC1 in high-grade glioma and majorly associated with PCAF in low-grade glioma. Meanwhile, SATB1 upregulation induced proliferation and invasion, and expression of MMP-2 and MMP-9 is HDAC1-dependent but PCAF-independent. As SATB1 interacts with either HDAC1 or PCAF depending on its phosphorylation status,17 our results suggest that interaction between phospho-SATB1 and HDAC1 has a key role in the regulation of glioma malignant phenotype.
In the present study, phospho-SATB1 level rather than SATB1 mRNA expression was shown to be a prognostic factor for the survival of patients with glioma. Furthermore, phospho-SATB1 upregulation was associated with the progression of glioma. These results are important for several reasons. First, they show that phospho-SATB1 rather than SATB1 mRNA and total SATB1 protein could be a valuable prognostic marker for glioma. Second, they indicate that the molecular targets for glioma therapy should be chosen with caution. Targeting SATB1 mRNA or total SATB1 protein may alter the phosphorylation status of SATB1 depending on the local microenvironment, which may lead to the failure of therapy. Our results indicated that phospho-SATB1 may be a better candidate as a therapeutic target, although further investigation is necessary to clarify the role of phospho-SATB1 in the development of malignant diseases.
In summary, our study showed that phospho-SATB1 but not SATB1 mRNA expression is associated with the progression and prognosis of glioma and its interaction with HDAC1 may contribute to the invasive and proliferative phenotype of GBM cells.
Materials and Methods
Patients and samples
A total of 127 clinical samples was obtained from the Chinese Glioma Genome Atlas (CGGA) specimen bank, including 71 primary GBMs (P, grade IV), 8 anaplastic astrocytomas (AA, grade III), 43 astrocytomas (A, grade II) and 5 normal brain tissue samples (N). After obtaining informed consent, three normal brain samples were collected from patients with severe brain trauma who needed surgery, and two normal samples were collected from patients who underwent surgery for primary epilepsy. All the patients underwent surgical resection from January 2006 to December 2009, and all high-grade glioma (grades III-IV) patients subsequently received radiation therapy and chemotherapy according to the Stupp protocol.36 Surgical resection was performed by neurosurgeons using similar surgical techniques and principles. After resection, samples were immediately snap-frozen in liquid nitrogen. The histological diagnosis was established and verified by two neuropathologists according to the 2007 World Health Organization (WHO) classification guidelines. Before the study, the percentage of tumor cells was assessed in each sample using hematoxylin and eosin-stained frozen sections. Only samples containing more than 80% tumor cells were selected for analysis. This study was approved by the institutional review boards of the First Hospital of China Medical University.
Microarray analysis
The mirVana miRNA Isolation kit (Ambion, Austin, TX, USA) was used for total RNA extraction according to the manufacturer’s protocol. RNA concentration and quality were measured with the NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Waltham, MA, USA).
Microarray analysis was performed in 127 samples (71P, 8AA, 43A and 5N) using the Agilent Whole Human Genome Array according to the manufacturer’s instructions. The integrity of total RNA was surveyed using an Agilent 2100 Bioanalyzer. Complementary DNA (cDNA) and biotinylated cRNA were synthesized and hybridized to the array. The Agilent G2565BA Microarray Scanner System and Agilent Feature Extraction Software (version 9.1) were used for data acquisition. Probe intensities were normalized using GeneSpring GX 11.0 (Agilent, Palo Alto, CA, USA).
Cell culture
The human GBM cell lines U87 and U251 were obtained from the Chinese academy of sciences cell bank (Shanghai, China), and T98G was purchased from American type culture collection (ATCC). Cells were maintained in Dulbecco’s modified Eagle’s medium (Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS, Invitrogen, Carlsbad, CA, USA). Primary SU3 glioma cells 37 were kindly provided by Professor Dong Jun, The Second Hospital Affiliated to Soochow University. SU3 cells were cultured in DMEM supplemented with 10% FBS as attached cells. Cells were incubated at 37 °C with 5% CO2.
LCM and RNA extraction
LCM was performed in randomly selected 10 astrocytoma samples and 10 GBM samples to obtain a homogeneous population of tumor cells. Two 8-μm-thick frozen sections were cut from each cryopreserved tissue: one section for hematoxylin and eosin staining to confirm the orientation of the tissue, and the other section for microdissection. Sections for microdissection were stained with Cresyl Violet using a LCM Staining Kit (Ambion) according to the manufacturer’s protocol. LCM was performed with a PixCell II Microscope (Arcturus Engineering, Mountain View, CA, USA) using a 7.5 m laser beam at 50–100 mW. The selected tumor areas were confirmed to be cleared from non-tumor cells (e.g., vascular endothelial cells, necrotic tissues and normal astrocytes). Approximately 10000 cells were captured from each specimen by LCM and further processed for RNA isolation using the RNAqueous-Micro Kit (Ambion) according to the manufacture’s instruction. Isolates were processed into cDNA and used for real-time PCR analysis to assess the SATB1 expression.
Real-time RT-PCR
Total RNA was isolated from SU3, U87, U251 and T98G cells and glioma clinical samples using the TRIzol reagent (Invitrogen) according to the manufacturer’s protocol. The extracted total RNA was then reverse transcribed into cDNA and used for PCR amplification. Real-time RT-PCR was performed on a rotor Gene 6000 Real-Time PCR System using a SYBR PrimeScript RT-PCR Two-Step Kit according to the manufacturer’s instructions. The PCR cycling conditions were as follows: 95 °C for 10 s, 95 °C for 5 s and 60 °C for 20 s for 40 cycles. Gene-specific primers for real-time RT-PCR were designed and synthesized by TaKaRa Inc. Primer sequences were: SATB1 sense 5′-TGCAAAGGTTGCAGCAACCAAAAGC-3′ and SATB1 antisense 5′-AACATGGATAATGTGGGGCGGCCT-3′; MMP-2 sense 5′-GCTCAGATCCGTGGTGAGAT-3′ and antisense 5′-GGTGCTGGCTGAGTAGATCC-3′; MMP-3 sense 5′-GAAAGTCTGGGAAGAGGTGACTCCAC-3′ and antisense 5′-CAGTGTTGGCTGAGTGAAAGAGACCC-3′; MMP-7 sense 5′-AAACTCCCGCGTCATAGAAAT-3′ and antisense 5′-TCCCTAGACTGCTACCATCCG-3′; MMP-9 sense 5′-CAAACCCTGCGTATTTCC-3′ and antisense 5′-AGAGTACTGCTTGCCCAGGA-3′. A standard melting-curve cycle was used to verify the quality of amplification, such as the absence of primer dimer formation. Samples were normalized by comparison with GAPDH gene expression, and quantitative analysis was performed using the Comparative Delta-delta Ct method.
Western blot analysis
Total cell protein was extracted with a total cell protein extraction kit (Millipore, Billerica, MA, USA). Protein concentrations were determined using the Coomassie protein assay (Bradford). An equivalent amount of protein from each sample was electrophoresed by 10% SDS-PAGE and transferred to nitrocellulose membranes. Membranes were blocked for 30 min in 5% nonfat dried milk in TBST/0.1% Tween-20, and incubated overnight at 4 °C with rabbit polyclonal anti-SATB1 (1 : 1000, Santa Cruz Biotechnology, Santa Cruz, CA, USA), rabbit polyclonal anti-phospho-SATB1 (Bioss Inc., Woburn, MA, USA), rabbit polyclonal anti-PKC (LifeSpan BioSciences, Seattle, WA, USA) or rabbit anti-β-actin (Santa Cruz Biotechnology) antibodies. Membranes were then washed three times with TBST/0.1% Tween-20 (5 min each) and incubated with secondary anybody. Bands were detected using a chemiluminescence ECL kit (Santa Cruz Biotechnology).
Coimmunoprecipitation
The procedure has been described previously.17 Briefly, protein (100 μg) from nuclear extracts was diluted by adding two volumes of IP-150 buffer and precleared with rabbit-IgG (Sigma, St. Louis, MO, USA) and protein A/G plus beads (Pierce, Rockford, IL, USA). Precleared extracts were then incubated with either anti-PCAF (Abcam, Cambridge, UK) or anti-HDAC1 (Abcam, ab46985). Protein–antibody complexes were analyzed by western blot analysis with anti-SATB1.
Tissue microarrays and IHC staining
Tissue microarrays were constructed using 122 glioma clinical samples (71P, 8AA and 43A) and analyzed by IHC staining as previously described.38 Briefly, paraffin-embedded sections were deparaffinized with xylene and rehydrated. After antigen retrieval, the sections were treated with 3% hydrogen peroxide, followed by incubation with 1% FBS. Anti-phospho-SATB1 (Bioss Inc., 1 : 100 dilution) was added and incubated overnight at 4 °C. For negative controls, the primary antibody was replaced by normal rabbit serum. Samples were then incubated with the horseradish peroxidase (HRP) labeled secondary antibody in the IHC kit (KIT-5930, MaxVision, Fu Zhou, China) for 30 min at room temperature. DAB was used for color development and hematoxylin as counterstain.
IHC staining was assessed by two independent investigators who were blinded to clinical and prognostic data. The proportion of positively stained tumor cells was graded as follows: 0 (no positive tumor cells), 1 (<10% positive tumor cells), 2 (10–50% positive tumor cells) and 3 (>50% positive tumor cells). The intensity of staining was recorded on a scale of 0 (no staining), 1 (weak staining, light yellow), 2 (moderate staining, yellowish brown) and 3 (strong staining, brown).31, 38 A combined staining index was generated by multiplying the proportion of positive staining and staining intensity. A staining index >4 was defined as tumors with high phospho-SATB1 level, and a staining index ≤4 as tumors with low level.
MGMT promoter methylation analysis by MSP
Methylation-specific PCR (MSP) was performed as described previously 39 to detect MGMT promoter methylation. Briefly, tissue samples were lysed with 490 μl lysis buffer containing 20 mM Tris-Cl (pH 8.0), 5 mM EDTA (pH 8.0), 400 mM NaCl and 1% (w/v) SDS, and digested with 10 μl proteinase K at 10 mg/ml at 37 °C for 12 h. Genomic DNA was purified from the lysate by phenol/chloroform extraction. One microgram of DNA was denatured by NaOH and modified by sodium bisulfite. MSP was performed using primer sequences for MGMT as follows: 5′-TTTGTGTTTTGATGTTTGTAGGTTTTTGT-3′ (forward) and 5′-AACTCCACACTCTTCCAAAAACAAAACA-3′ (reverse) for the unmethylated reaction; and 5′-TTTCGACGTTCGTAGGTTTTCGC-3′ (forward) and 5′-GCACTCTTCCGAAAACGAAACG-3′ (reverse) for the methylated reaction. Each PCR reaction (10 μl) was loaded onto nondenaturing 6% polyacrylamide gels, stained with ethidium bromide and visualized under UV illumination. The PCR reaction was repeated at least three times.
SATB1 gene expression knockdown
Specific SATB1-shRNA sequences were synthesized as previously described.20 The SATB1-shRNA sequence was 5′-GTCCACCTTGTCTTCTCTC-3′ and the control-shRNA sequence was 5′-GAAGCAGCACGACTTCTTC-3′, which was cloned into pSUPER-puro and transfected into U251 cells. After 24 h, transfected cells were selected for 10 days with 2 μg/ml puromycin. Cells stably expressing SATB1-shRNA were selected for further research. The effectiveness of SATB1 silencing was assessed using RT-PCR and western blotting.
Construction of SATB1-overexpressing cells
The human SATB1 cDNA was cloned into the pEGFP-N1 plasmid (Clontech, Moutain View, CA, USA), and the resulting pEGFP-N1-SATB1 plasmid or control pEGFP-N1 was transfected into SU3 and U87 cells using Lipofectamine. Stable cell lines were selected by G418 screening. The efficiency of SATB1 overexpression was evaluated using RT-PCR and western blotting.
Decoy SATB1 transfection
SATB1-decoy and control-decoy DNA were obtained from Genedetect (Bradenton, FL, USA). The sequence for the SATB1-decoy DNA was 5′-CGGCTATTAGTAATAA-3′ (sense) and 3′-GCCGATAATCATTATT-5′ (antisense). The sequence for the control-decoy DNA was 5′-TATTAGCGGCTAATAA-3′ (sense) and 3′-ATAATCGCCGATTATT-5′ (antisense).27 An equimolar mixture of each pair of decoy DNAs was dissolved in DNase- and RNase-free TE buffer at a concentration of 20 μM and denatured at 90 °C for 5 min, then slowly cooled to room temperature. SugarFect (Medgel, Kyoto, Japan) was used for transfecting decoy DNAs into U87-pEGFP-N1-SATB1, SU3- pEGFP-N1-SATB1, SU3, U87 and T98G cells with a fixed SugarFect/oligonucleotide charge ratio of 2. Decoy-treated cells were used for further studies.
Silencing HDAC1 and PCAF
SU3-pEGFP-N1-SATB1 cells were transfected with siRNAs against HDAC1 or PCAF using Lipofectamine 2000 (Life Technologies, Carlsbad, CA, USA). Total protein was isolated after transfection, and western blot analysis was performed as described above. When we checked the expression levels of HDAC1 and PCAF, we found that the expression was decreased from 24 h and peaked at 48 to 72 h after transfection (data not shown).
The control siRNA sequence was 5′-CUGAUGACCUGAGUGAAUGdTdT-3′; the HDAC1 siRNA sequence was 5′-AACCCGGAGGAAAGTCTGTTA-3′; and the PCAF siRNA sequence was 5′-UCGCCGUGAAGAAAGCGCAtt-3′.
MTT assay
Cell proliferation was assessed using the MTT (3-(4, 5-dimethylthiazolyl-2)-2, 5-diphenyltetrazolium bromide) assay. Briefly, cells were seeded in 96-well plates at a density of 2 × 103/well and incubated for 24, 48, 72, 96 or 120 h. At each time point, 20 μl of 5 mg/ml MTT solution was added to each well. After incubation for 4 h, the medium was removed from the wells by aspiration, and the formazan crystals were dissolved in 150 μl dimethyl sulfoxide (DMSO). Color intensity was measured at 490 nm with an enzyme-linked immunosorbent assay plate reader (Tecan Sunrise Remote, Maennedorf, Austria). The test was repeated three times and the results were presented as the mean±S.E.
In vitro invasion assay
The invasion assay was performed in 24-well Transwell chambers (Corning, Corning, NY, USA) with an 8 μm pore membrane coated with Matrigel (BD Bioscience, San Jose, CA, USA). Cells were plated at a density of 2 × 103 per upper well in 200 μl DMEM with 1% FBS; the lower chamber was filled with 500 μl DMEM with 20% FBS. The cells were allowed to invade for 24 h, and the noninvading cells were then removed from the upper surface of the membrane by scrubbing with a cotton-tipped swab. Cells on the lower surface of the filter were fixed for 30 min in a methanol and glacial acetic acid mixture (3 : 1), air-dried briefly and stained with Giemsa Stain (Sigma). The number of invaded cells was counted from five randomly selected microscopic fields at × 200 magnification. Each experiment was repeated three times.
Statistical analysis
Cox proportional hazards models were used to calculate HRs of death according to phospho-SATB1 level in high-grade gliomas, unadjusted and adjusted for age, sex, preoperative KPS, tumor size, degree of resection and MGMT promoter methylation. To adjust for potential confounders, age, preoperative KPS and tumor size were used as continuous variables and all of the other covariates were used as categorical variables. We dichotomized MGMT promoter methylation status (methylation versus unmethylation), and phospho-SATB1 level (high versus low). Tumor resection was defined as follows: (0) gross total resection; (1) partial removal with residual tumor <30%; and (2) residual tumor >30% or biopsy. Tumor size was calculated based on preoperative MRI scans as follows: longest diameter × widest diameter × thickness (section thickness × the number of layers) × 1/2. Clinical data were retrospectively collected from medical records (Table 2). At the end of follow-up, all patients with high-grade glioma died of the disease. We used Kaplan–Meier analysis to describe the distribution of OS time, and the log-rank test was performed.
The mRNA expression data sets (http://www.cgga.org.cn) of SATB1, PKC and miR-155 obtained from the microarray analysis were used for cluster analysis (Cluster 2.20) and Pearson correlation analysis (SPSS 13.0). Two publicly available data sets were also used to verify the expression of SATB1 in gliomas, which can be accessed at http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE4290 and http://www.ebi.ac.uk/arrayexpress/experiments/E-MEXP-567/. Cluster analysis was performed using the hierarchical clustering method with average linkage. The results of cluster analysis were visualized by Tree View software (Stanford University, Palo Alto, CA, USA). The χ2 test and ANOVA were used to determine statistical significance. Each experiment was performed in triplicate. All data are presented as the mean±S.E. of three independent experiments. A two-tailed P-value of <0.05 was regarded as significant.
Abbreviations
- SATB1:
-
special AT-rich sequence-binding protein 1
- GBM:
-
glioblastoma multiforme
- PKC:
-
protein kinase-C
- HDAC:
-
histone deacetylase
- PCAF:
-
p300/CBP-associated factor
- LCM:
-
laser-capture microdissection
- OS:
-
overall survival
- KPS:
-
Karnofsky performance status
- MGMT:
-
O(6)-methylguanine-DNA-methyltransferase
- HR:
-
hazard ratio
- MMP:
-
matrix metalloproteinase
- PTEN:
-
phosphatase and tensin homolog
- TBC1D1:
-
TBC1 domain family member 1
- MARs:
-
matrix attachment regions
References
Yan W, Zhang W, You G, Zhang J, Han L, Bao Z et al. Molecular classification of gliomas based on whole genome gene expression: a systematic report of 225 samples from the Chinese Glioma Cooperative Group. Neuro Oncol 2012; 14: 1432–1440.
Van Meir EG, Hadjipanayis CG, Norden AD, Shu HK, Wen PY, Olson JJ . Exciting new advances in neuro-oncology: the avenue to a cure for malignant glioma. CA Cancer J Clin 2010; 60: 166–193.
Zhang JX, Zhang J, Yan W, Wang YY, Han L, Yue X et al. Unique genome-wide map of TCF4 and STAT3 targets using ChIP-seq reveals their association with new molecular subtypes of glioblastoma. Neuro Oncol 2013; 15: 279–289.
Rath BH, Fair JM, Jamal M, Camphausen K, Tofilon PJ . Astrocytes enhance the invasion potential of glioblastoma stem-like cells. PLoS One 2013; 8: e54752.
Jia Z, Wang K, Wang G, Zhang A, Pu P . MiR-30a-5p antisense oligonucleotide suppresses glioma cell growth by targeting SEPT7. PLoS One 2013; 8: e55008.
Romao LF, Mendes FA, Feitosa NM, Faria JC, Coelho-Aguiar JM, de Souza JM et al. Connective Tissue growth factor (CTGF/CCN2) Is negatively regulated during neuron-glioblastoma interaction. PLoS One 2013; 8: e55605.
Lee HK, Bier A, Cazacu S, Finniss S, Xiang C, Twito H et al. MicroRNA-145 Is downregulated in glial tumors and regulates glioma cell migration by targeting connective tissue growth factor. PLoS One 2013; 8: e54652.
Huveldt D, Lewis-Tuffin LJ, Carlson BL, Schroeder MA, Rodriguez F, Giannini C et al. Targeting SRC family kinases inhibits bevacizumab-induced glioma cell invasion. PLoS One 2013; 8: e56505.
Qi Q, He K, Liu X, Pham C, Meyerkord C, Fu H et al. Disrupting the PIKE-A/Akt interaction inhibits glioblastoma cell survival, migration, invasion and colony formation. Oncogene 2013; 32: 1030–1040.
Wang Y, Moncayo G, Morin P Jr, Xue G, Grzmil M, Lino MM et al. Mer receptor tyrosine kinase promotes invasion and survival in glioblastoma multiforme. Oncogene 2013; 32: 872–882.
Galeano F, Rossetti C, Tomaselli S, Cifaldi L, Lezzerini M, Pezzullo M et al. ADAR2-editing activity inhibits glioblastoma growth through the modulation of the CDC14B/Skp2/p21/p27 axis. Oncogene 2013; 32: 998–1009.
Cartron PF, Loussouarn D, Campone M, Martin SA, Vallette FM . Prognostic impact of the expression/phosphorylation of the BH3-only proteins of the BCL-2 family in glioblastoma multiforme. Cell Death Dis 2012; 3: e421.
Rampazzo E, Persano L, Pistollato F, Moro E, Frasson C, Porazzi P et al. Wnt activation promotes neuronal differentiation of glioblastoma. Cell Death Dis 2013; 4: e500.
Galande S, Purbey PK, Notani D, Kumar PP . The third dimension of gene regulation: organization of dynamic chromatin loopscape by SATB1. Curr Opin Genet Dev 2007; 17: 408–414.
Beyer M, Thabet Y, Muller RU, Sadlon T, Classen S, Lahl K et al. Repression of the genome organizer SATB1 in regulatory T cells is required for suppressive function and inhibition of effector differentiation. Nat Immunol 2011; 12: 898–907.
McInnes N, Sadlon TJ, Brown CY, Pederson S, Beyer M, Schultze JL et al. FOXP3 and FOXP3-regulated microRNAs suppress SATB1 in breast cancer cells. Oncogene 2012; 31: 1045–1054.
Pavan KP, Purbey PK, Sinha CK, Notani D, Limaye A, Jayani RS et al. Phosphorylation of SATB1, a global gene regulator, acts as a molecular switch regulating its transcriptional activity in vivo. Mol Cell 2006; 22: 231–243.
Jin H, Liang L, Liu L, Deng W, Liu J . HDAC inhibitor DWP0016 activates p53 transcription and acetylation to inhibit cell growth in U251 glioblastoma cells. J Cell Biochem 2013; 114: 1498–1509.
Malatesta M, Steinhauer C, Mohammad F, Pandey DP, Squartrito M, Helin K . Histone acetyltransferase PCAF is required for Hedgehog-Gli-dependent transcription and cancer cell proliferation. Cancer Res 2013; 73: 6323–6333.
Han HJ, Russo J, Kohwi Y, Kohwi-Shigematsu T . SATB1 reprogrammes gene expression to promote breast tumour growth and metastasis. Nature 2008; 452: 187–193.
Cheng C, Lu X, Wang G, Zheng L, Shu X, Zhu S et al. Expression of SATB1 and heparanase in gastric cancer and its relationship to clinicopathologic features. APMIS 2010; 118: 855–863.
Zhang J, Zhang B, Zhang X, Sun Y, Wei X, McNutt MA et al. SATB1 expression is associated with biologic behavior in colorectal carcinoma in vitro and in vivo. PLoS One 2013; 8: e47902.
Liu CX, Wen Y, Xu K, Zheng SB, Xu YW, Chen BS . Expression of special AT-rich sequence-binding protein in bladder urothelial carcinoma and its clinical significance. Nan Fang Yi Ke Da Xue Xue Bao 2010; 30: 1389–1391 1394.
Tan JA, Song J, Chen Y, Durrin LK . Phosphorylation-dependent interaction of SATB1 and PIAS1 directs SUMO-regulated caspase cleavage of SATB1. Mol Cell Biol 2010; 30: 2823–2836.
Sun L, Hui AM, Su Q, Vortmeyer A, Kotliarov Y, Pastorino S et al. Neuronal and glioma-derived stem cell factor induces angiogenesis within the brain. Cancer Cell 2006; 9: 287–300.
Margareto J, Leis O, Larrarte E, Idoate MA, Carrasco A, Lafuente JV . Gene expression profiling of human gliomas reveals differences between GBM and LGA related to energy metabolism and notch signaling pathways. J Mol Neurosci 2007; 32: 53–63.
Yamayoshi A, Yasuhara M, Galande S, Kobori A, Murakami A . Decoy-DNA against special AT-rich sequence binding protein 1 inhibits the growth and invasive ability of human breast cancer. Oligonucleotides 2011; 21: 115–121.
Yang Z, Yuan XG, Chen J, Luo SW, Luo ZJ, Lu NH . Reduced expression of PTEN and increased PTEN phosphorylation at residue Ser380 in gastric cancer tissues: a novel mechanism of PTEN inactivation. Clin Res Hepatol Gastroenterol 2013; 37: 72–79.
Bossuyt J, Ai X, Moorman JR, Pogwizd SM, Bers DM . Expression and phosphorylation of the na-pump regulatory subunit phospholemman in heart failure. Circ Res 2005; 97: 558–565.
Sainz N, Rodriguez A, Catalan V, Becerril S, Ramirez B, Lancha A et al. Leptin reduces the expression and increases the phosphorylation of the negative regulators of GLUT4 traffic TBC1D1 and TBC1D4 in muscle of ob/ob mice. PLoS One 2012; 7: e29389.
Chu SH, Ma YB, Feng DF, Zhang H, Zhu ZA, Li ZQ et al. Upregulation of SATB1 is associated with the development and progression of glioma. J Transl Med 2012; 10: 149.
Notani D, Limaye AS, Kumar PP, Galande S . Phosphorylation-dependent regulation of SATB1, the higher-order chromatin organizer and global gene regulator. Methods Mol Biol 2010; 647: 317–335.
Barboro P, Repaci E, D'Arrigo C, Balbi C . The role of nuclear matrix proteins binding to matrix attachment regions (Mars) in prostate cancer cell differentiation. PLoS One 2012; 7: e40617.
Iorns E, Hnatyszyn HJ, Seo P, Clarke J, Ward T, Lippman M . The role of SATB1 in breast cancer pathogenesis. J Natl Cancer Inst 2010; 102: 1284–1296.
Patani N, Jiang W, Mansel R, Newbold R, Mokbel K . The mRNA expression of SATB1 and SATB2 in human breast cancer. Cancer Cell Int 2009; 9: 18.
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005; 352: 987–996.
Wan Y, Fei XF, Wang ZM, Jiang DY, Chen HC, Yang J et al. Expression of miR-125b in the new, highly invasive glioma stem cell and progenitor cell line SU3. Chin J Cancer 2012; 31: 207–214.
Wang C, Cao S, Yan Y, Ying Q, Jiang T, Xu K et al. TLR9 expression in glioma tissues correlated to glioma progression and the prognosis of GBM patients. BMC Cancer 2010; 10: 415.
Esteller M, Hamilton SR, Burger PC, Baylin SB, Herman JG . Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res 1999; 59: 793–797.
Acknowledgements
We thank Jingpu Shi at the Department of Clinical Epidemiology, The First Affiliated Hospital of China Medical University and Yujie Zhao at China Medical University for superb technical assistance with statistical and epidemiological analyses. We also thank Dong Jun at The Second Hospital Affiliated to Soochow University for the supply of SU3 cells. This work was supported by grants from the National High Technology Research and Development Program of China (863) (No.2012AA02A508), National Natural Science Foundation of China (No.81172409) and Science and Technology Department of Liaoning Province (No.2011225034).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by A Verkhratsky
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Han, S., Xia, J., Qin, X. et al. Phosphorylated SATB1 is associated with the progression and prognosis of glioma. Cell Death Dis 4, e901 (2013). https://doi.org/10.1038/cddis.2013.433
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2013.433
Keywords
This article is cited by
-
SATB1, genomic instability and Gleason grading constitute a novel risk score for prostate cancer
Scientific Reports (2021)
-
Computational identification and characterization of glioma candidate biomarkers through multi-omics integrative profiling
Biology Direct (2020)
-
Analysis of cellular and molecular antitumor effects upon inhibition of SATB1 in glioblastoma cells
BMC Cancer (2017)
-
Lithium enhances the antitumour effect of temozolomide against TP53 wild-type glioblastoma cells via NFAT1/FasL signalling
British Journal of Cancer (2017)
-
Pre-treatment neutrophil-to-lymphocyte ratio is associated with neutrophil and T-cell infiltration and predicts clinical outcome in patients with glioblastoma
BMC Cancer (2015)
