
Abstract
The proapoptotic protein, prostate apoptosis response-4 (Par-4), acts as a tumor suppressor in prostate cancer cells. The serine/threonine kinase casein kinase 2 (CK2) has a well-reported role in prostate cancer resistance to apoptotic agents or anticancer drugs. However, the mechanistic understanding on how CK2 supports survival is far from complete. In this work, we demonstrate both in rat and humans that (i) Par-4 is a new substrate of the survival kinase CK2 and (ii) phosphorylation by CK2 impairs Par-4 proapoptotic functions. We also unravel different levels of CK2-dependent regulation of Par-4 between species. In rats, the phosphorylation by CK2 at the major site, S124, prevents caspase-mediated Par-4 cleavage (D123) and consequently impairs the proapoptotic function of Par-4. In humans, CK2 strongly impairs the apoptotic properties of Par-4, independently of the caspase-mediated cleavage of Par-4 (D131), by triggering the phosphorylation at residue S231. Furthermore, we show that human Par-4 residue S231 is highly phosphorylated in prostate cancer cells as compared with their normal counterparts. Finally, the sensitivity of prostate cancer cells to apoptosis by CK2 knockdown is significantly reversed by parallel knockdown of Par-4. Thus, Par-4 seems a critical target of CK2 that could be exploited for the development of new anticancer drugs.
Similar content being viewed by others


Main
Prostate apoptotis response-4 (Par-4) is a ubiquitously expressed protein characterized by its ability to induce apoptosis in cancer cells only but not in normal or immortalized cells.1, 2, 3 The proapoptotic role of Par-4 was first discovered in rat prostate cancer cells undergoing apoptosis in response to calcium inducers. The essential role of Par-4 in apoptosis was later confirmed in diverse cell types in response to cellular insults, such as ultraviolet, cytokines, serum withdrawal, hormone deprivation and exposure to cytotoxic drugs.4, 5, 6 Consistent with an important role of Par-4 in apoptosis, ectopic expression of Par-4 in various cultured cells, including NIH3T3 cells, neurons, melanoma, prostate and colon cancer cells, has been found to sensitize these cells to apoptotic stimuli or chemotherapeutic drugs.7, 8, 9, 10, 11 In addition, the in vivo delivery of Par-4 plasmid by nanoliposomes or Par-4 adenovirus injection into tumors growing in nude mice (HT29 and PC-3 cells, respectively) induces tumor regression1 and/or tumor sensitization to therapeutic agents.12
The par-4 gene maps to human chromosome 12q21, in a region often deleted in pancreatic and gastric cancers, as well as in tumors of male germ cell.13, 14 In addition, Par-4 is downregulated in many cancers, including breast cancer,15 lymphoma,16 renal and pancreatic carcinoma.17 Most important, Par-4 was recently described as a major determinant underlying breast cancer recurrence, and its reduced expression was associated with poor prognosis.15, 18 Oncogenes like Ras, Raf or Src are involved in the reduction of Par-4 abundance, and restoration of Par-4 levels in those models leads to tumor cell apoptosis.3, 19 Notably, Par-4 null mice develop spontaneous tumors in the prostate, endometrium, liver or lungs or carcinogen-induced tumors in the bladder and endometrium, providing strong evidence of Par-4’s tumor-suppressor activity.20
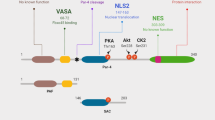
Functionally, the critical domains of Par-4 include the C-terminus leucine zipper domain, which mediates interaction with its partners (e.g., Wilm’s tumor suppressor, atypical protein kinase C (PKCζ), ZIP kinase) and the core domain designated SAC (selective for apoptosis in cancer cells, spanning amino acids 137–195), which is essential for Par-4 apoptotic function.21 Human Par-4 can be cleaved by caspases at the D131 site and the generated C-terminus cleaved form (132–340 aa) includes the fully active SAC domain.22, 23, 24 The proapoptotic properties of Par-4 depend on both caspase activation and inhibition of survival factors, including nuclear factor kappa B (NF-κB),8, 11 PKCζ7 and/or Akt kinase.25 Par-4 is also spontaneously secreted from mammalian cells via the classical endoplasmic reticulum (ER)-Golgi pathway.26 Following secretion, Par-4 is able to induce apoptosis both in vitro and in vivo through the activation of the 78-kDa glucose-regulated protein receptor/caspases pathway.26, 27 Par-4 functions are regulated by different kinases, including Akt and protein kinase A (PKA). Akt induces Par-4 phosphorylation and this triggers its binding and sequestration in the cytoplasm by the chaperone proteins 14–3–3, thus preventing apoptosis in prostate cancer cells.28 In contrast, PKA phosphorylates Par-4 (on residue T155 in rat Par-4 and T163 in human Par-4) to activate the proapoptotic functions of Par-4.29
Casein kinase 2 (CK2) is a constitutive, highly conserved serine/threonine kinase that recognizes the acidic consensus motif ‘S/T-X-X-Asp/Glu/pSer’ (X denotes any non-basic amino acid).30, 31 Generally, CK2 acts as a tetrameric holoenzyme, which comprises two catalytic (CK2α) and two regulatory (CK2β) subunits. CK2β mediates interactions between the catalytic subunits and many of its substrates, thereby modulating substrate selectivity and catalytic activity.30, 31 The activity and/or expression of CK2 has been found to be significantly enhanced in many human cancers, including prostate, head and neck, colon and lung32, 33, 34, 35 as well as in experimental rodent tumors.36 Although the mechanistic understanding of how CK2 exerts its protective role on cell survival remains far from complete, increasing evidence links CK2 to the dual function of pro-survival and apoptotic pathways. Indeed, CK2 has been reported to support, on the one hand, cell viability via activation of Wnt, Akt and NF-κB signaling pathways and, on the other hand, apoptosis by: (i) protecting some proapoptotic proteins from caspase-mediated degradation and (ii) inhibiting the mitochondrial pathway.37, 38, 39 The anti-apoptotic function of CK2 is further supported by the fact that inhibitors of CK2 enhance the susceptibility of cancer cells to apoptosis induced by death receptors (such as tumor necrosis factor (TNF-α), TNF-related apoptosis-inducing ligand (TRAIL) and Fas Ligand) or drugs (such as etoposide, diethylstilbestrol, resveratrol). Conversely, increased activity and/or expression of CK2, observed in cancer cells, including prostate cancer cells, protects from cell death induced by cytotoxic insults.9, 40, 41, 42, 43, 44
Par-4 and CK2 activities have opposing effects on apoptosis and tumor growth, particularly in prostate cancer.9, 6, 18, 28, 39, 45 Given that Par-4 sequence analysis revealed potential sites for CK2 phosphorylation that are overall evolutionarily conserved (database http://scansite.mit.edu), we hypothesized that Par-4 could be a target of CK2. We demonstrate, both in human and rodents, that CK2 directly associates with and phosphorylates Par-4. We also show that the phosphorylation by CK2 of S231 in human Par-4 and S124/S223 in rodent Par-4 inhibits the proapoptotic functions of Par-4. Furthermore, our results underline a species-dependent regulation of Par-4 cleavage through phosphorylation by CK2 at residue S124 of rodent Par-4.
Importantly, a higher phosphorylation of the human Par-4 residue S231 was observed in prostate cancer cells – which display elevated CK2 activity – compared with their normal counterparts. Finally, we showed that the anti-apoptotic effect of CK2 in human prostate cancer cells depends on Par-4 inhibition, thereby identifying Par-4 as a functionally critical substrate of CK2.
Results
Both human and rodent Par-4 are new substrates of CK2
To study whether Par-4 is a target of CK2, we first assessed the ability of Par-4 to interact with CK2 subunits both in human and rodent cells. Given the extremely high homology between human and rodent CK2, we used human CK2 for all our experiments. Rat green fluorescent protein (GFP)-Par-4 and the hemagglutinin (HA)-tag CK2 subunits (CK2α and/or CK2β) were co-transfected in COS cells and immunoprecipitated either with anti-GFP (Par-4) or anti-HA (CK2 subunit) antibodies. As commonly noted with other CK2 substrates,30 Par-4 was co-immunoprecipitated with the α subunit of CK2 but not with the GFP-tag (Figure 1a, left panels). We further evaluated the ability of Par-4 to co-immunoprecipitate with the CK2β subunit (Figure 1a, right panels). As shown in Figure 1a (upper right panel), we did not detect any interaction between Par-4 and the CK2β subunit, whereas Par-4 co-immunoprecipitated with the CK2 holoenzyme. We next examined the ability of endogenous human Par-4 to associate with CK2. Similar to rat GFP-Par-4, human Par-4 interacts with the α subunit of the CK2 holoenzyme but not with the CK2β subunit (Figure 1b). To determine whether the interaction was direct,35 S-labeled subunit CK2α and/or CK2β were incubated with rat glutathione-S-transferase (GST)-Par-4 or GST alone, and immunoprecipitation was performed using an anti-GST antibody (Supplementary Figure S1a). The resolution of the immunoprecipitated complexes in sodium dodecyl phosphate polyacrylamide gel electrophoresis (SDS-PAGE) confirmed that Par-4 interacts directly with CK2.

Both rodent and human Par-4 interact and are substrates of CK2. (a) COS cells were co-transfected with rat GFP-tagged Par-4 or empty GFP vector together with the CK2α and/or the CK2βHA-tagged subunits. In the left panel, immunoprecipitation of GFP vector, Par-4 (GFP) or CK2α subunits (HA). In right panel, immunoprecipitation of Par-4 (GFP) or CK2α and/or CK2β subunits (HA). The immunoprecipitations were followed by immunodetection of either CK2 (HA) or Par-4 (GFP). (b) Immunoprecipitation, from a same PC-3 cells extract, of endogenous human Par-4 (hPar-4) or CK2 subunits was followed by immunodetection of endogenous human CK2α, CK2β or Par-4. Inputs: proteins in total cell lysates. IP IgG: immunoprecipitation with a non-relevant antibody (IgG mouse). *HC, LC immunoglobulin heavy chain and light chain respectively. (c) The GFP-tagged CK2 kinase or GFP alone (as a control, Mock) were immunoprecipitated from transfected COS cells and used for an in vitro kinase assay in the presence of [γ−32P]ATP with either recombinant GST-Par-4 or GST alone, as substrates (left panel). Reaction products were resolved by SDS-PAGE on 10% gels, stained with Coomassie blue to verify that equal amounts of GST-Par-4 or GST alone were used in each reaction (lower panel), and autoradiography was performed (upper panel). In parallel, the amount of GFP (Mock) or CK2 subunits immunoprecipitated was analyzed by immunoblotting (right panel). (d) Recombinant GFP-tagged human Par-4 (GFP-hPar-4) was produced in vitro by the TNT rabbit reticulocyte lysate system (RRL), and lysates were subsequently immunoprecipitated using GFP antibody. Used as substrates, the immunoprecipitates were subjected to an in vitro kinase assay using recombinant CK2 (recCK2) in the presence of [γ−32P]ATP for 30 min. The CK2 inhibitor (TBB, 1 μM) was incubated 5 min before addition of CK2 recombinant (middle panel). After migration, phosphorylated Par-4 (32P-hPar-4) and autophosphorylation of recombinant CK2β (32P- CK2β) was detected by autoradiography. The amount of recombinant GFP-Par-4 produced by RRL was checked by western blotting (input, lower panel)
Next, we determined whether Par-4 could be phosphorylated by CK2 both in human and rodent cells. We performed an in vitro kinase assay, using either the immunoprecipitated GFP-tag CK2α and CK2β subunits in the presence of rat GST-Par-4 (Figure 1c) or the recombinant CK2 in presence of human GFP-Par-4 as a substrate (Figure 1d). These experiments indicate that CK2 phosphorylates Par-4 (Figures 1c and d). As for the majority of CK2 substrates, the presence of the regulatory subunit CK2β was necessary to phosphorylate Par-4 as judged by the effect of increasing doses of CK2β on Par-4 phosphorylation (Supplementary Figure S1b). As Par-4 functions are regulated by phosphorylation,28, 29 we searched for a phosphatase able to dephosphorylate Par-4. By means of phosphatase inhibitors and the use of recombinant protein phosphatase 1 (PP1), we observed that phosphatase PP1 interacts with and leads to an efficient dephosphorylation of Par-4 (Supplementary Figure S2 and Supplementary Data). Accordingly, the phosphatase PP1 has been shown to dephosphorylate several substrates of CK2.46
Collectively, the fully active holoenzyme CK2 phosphory-lates human and rodent Par-4, and this action might be counteracted by PP1 phosphatases.
Phosphorylation of rodent Par-4 by CK2 and consequences on its proapoptotic functions
In order to characterize the CK2 target site(s), we performed a tryptic phosphopeptide map of rat Par-4-GST phosphorylated in vitro with recombinant CK2 (in vitro kinase assay done as in Figure 1c). Autoradiography of the thin layer chromatography (TLC)-plate with the tryptic peptides separated in two dimensions indicates that the in vitro 32P-labeled material has three major CK2-specific spots designated phosphopeptides P1, P′1 and P2 (Figure 2a). MS analysis of these major phosphopeptides (Table 1, spectra shown in Supplementary Figure S3a) allowed identification of the following phosphopeptides P1: 117-GDEEEPDpS(124)APEK-128, P’1: 117-GDEEEPDpS(124)APEKGR-128 and P2: 220-STIpS(223)APEEEILNRYPR-235. Matrix-assisted laser desorption/ionization-time-of-flight-mass spectrometry (MALDI-TOF-MS) analysis indicated that the P1′ phosphopeptide was similar to P1 but lacked a cryptic cleavage site at the C-terminus. The identity of the CK2-specific phosphopeptides P1, P′1 and P2 was further checked by manual Edman degradation (Supplementary Figure S3b). Accordingly, S124 and S223 residues of rat Par-4 were the targeted sites of CK2.

Identification of the CK2 phosphorylated sites S124 and S223 in rodent Par-4. (a) Two-dimensional phosphopeptide map analysis of the phosphorylated recombinant rat GST-Par-4. GST-Par-4 was phosphorylated by recombinant CK2 in the presence of [γ−32P]ATP. After migration, band corresponding to the radiolabeled phospho-Par-4 was excised from the membrane and digested by trypsin. Tryptic fragments were purified and resolved by two-dimensional thin layer electrophoresis and ascending chromatography followed by autoradiography to visualize the phosphopeptides. The separation origin is indicated as ‘0’. Three major phosphopeptides were revealed by autoradiography: P1, P1′, and P2. (b) Transfected wild-type Par-4 (rat) or 124A223A Par-4 mutants (GFP-tagged) were immunoprecipitated from COS cells and subjected to an in vitro kinase assay in the presence of recombinant CK2 and [γ−32P]ATP. Radiolabeled Par-4 was visualized upon autoradiography. Immunoblot analysis confirmed that equal amounts of GFP-Par-4 and GFP-124A223A were immunoprecipitated from each cell lysate (inputs, lower panel)
In order to determine the functional relevance of rat Par-4 phosphorylation by CK2, we constructed two GFP-tagged mutants of Par-4 in which the two serine residues were substituted either by alanine (124A223A) or by aspartic acid (124D223D) that prevent or mimic phosphorylation, respectively. An in vitro kinase assay with the 124A223A mutant confirmed that CK2 was no longer able to induce Par-4 phosphorylation (Figure 2b). Moreover, we observed a decrease in the phosphorylation, with the single mutant 124D or 223D (Supplementary Figure S3c).
Par-4 is known to inhibit the cell survival pathways like the NF-κB pathway and to induce apoptosis in a caspase-dependent manner. In contrast, CK2 has been reported to stimulate the transcriptional activity of NF-κB and to prevent caspase activation.39, 40 We therefore investigated the effect of the two phosphomutants on the proapoptotic functions of Par-4, including the ability to inhibit NF-κB pro-survival pathway.1, 3 Using a luciferase reporter assay, we found that the 124A223A mutant Par-4 (mimicking the non-phosphorylated Par-4) as well as wild-type Par-4 retained the ability to inhibit NF-κB transcriptional activity, while the 124D223D (mimicking the phosphorylated Par-4) lost this capacity (Supplementary Figure S4).
A similar loss of function for 124D223D was observed when we studied Par-4 proapoptotic role. Prostate cancer cells PC-3 were treated either with the death ligand, TRAIL (Figures 3a–c) or the chemotherapeutic drug, Paclitaxel (Figure 3d). We found that cells transfected with wild-type Par-4 or the 124A223A mutant were sensitive to apoptosis while the cells expressing the 124D223D mutant were still resistant (as judged by Hoechst staining, caspase-3, poly ADP ribose polymerase (PARP) and caspase-8 activation; Figures 3a–c). Interestingly, we noticed the appearance of a cleaved form of Par-4 in cells expressing 124A223A or wild-type Par-4 but not in cells expressing 124D223D (Figures 3c and d). This smaller Par-4 form, 50 kDa in size including GFP, most likely corresponds to the caspase-induced cleavage product of Par-4. Indeed, a Par-4 cleaved form was previously reported in human cells during Fas- or cisplatin-induced apoptosis.23, 24 Thus, phosphorylation by CK2 on residues S124 and S223 results in loss of rat Par-4 proapoptotic properties and prevents the generation of the caspase-mediated cleaved form of Par-4.

Phosphorylation of rodent Par-4 by CK2 blocks Par-4 proapoptotic functions. (a–c) PC-3 cells were transfected with empty GFP vector control (Mock), rat GFP-tagged Par-4, GFP-tagged 124A223A mutant (mimicking non-phosphorylated Par-4 form) or 124D223D mutant (mimicking phosphorylated Par-4 form) and then treated with recombinant death ligand TRAIL (500 ng/ml, 3 h). (a) Green fluorescent (transfected) cells were gated (represent around 20% of total cell) and further analyzed for apoptosis by Hoechst staining. Bars represent the mean±S.D. of at least four independent experiments. (b) FACS analysis of the caspase-3 activity was assessed by red fluorescent signal FLICA (Fluorogenic inhibitors of caspase-3 activation). Bars represent the mean±S.D. of at least four independent experiments. (c) Immunoblot analysis of caspase-8 and PARP cleavage (included both GFP-positive and -negative cells) at the indicated times. *P<0.05, **P<0.01. (d) PC-3 cells, transfected as above, were treated 8 h with 200 nM of Paclitaxel, and then apoptosis was determined by detection of caspases 8 and PARP cleavage. Hsp90 and 14-3-3 were used as a loading control
Protective role of CK2-induced phosphorylation of S124 by preventing the caspase-mediated cleavage of Par-4
We noticed that the rodent Par-4 site S124 is the most strongly CK2 phosphorylated site (Figure 2a, phosphopeptides P1 and P1′ versus P2), and it is in the vicinity of D123, the ortholog for human D131 that corresponds to the human caspase cleavage site24 (Supplementary Figures S5a and b). As the Par-4 cleavage product observed in cells transfected with Par-4 wild type (Figures 3c and d) disappeared with the Par-4 mutant D124, we wondered whether CK2 phosphory-lation on S124 in rodents could impair caspase-3-mediated Par-4 cleavage. To test this hypothesis, we first verified that rodent Par-4 is also cleaved by caspases after an apoptotic stimulus (Figure 4a). We show that (i) benzyloxycarbonylvalyl-alanyl-aspartic acid (O-methyl)-fluoro-methylketone (zVAD-FMK), a pan inhibitor of caspases, prevents rat Par-4 cleavage, (ii) the Par-4 mutant D123A (potential rat caspase site) was not cleavable (Supplementary Figure S6a, left panel). As previously reported for the Par-4 mutant D131A human (human caspase site24), mutant D123A also impaired rat Par-4 proapoptotic function (Supplementary Figure S6a, right panel, and 6b).

Rodent Par-4 phosphorylation by CK2 prevents caspase-mediated Par-4 cleavage. (a) PC-3 cells transfected with the GFP-tagged constructs wild-type rat Par-4, 124A/D, 223A/D, 124A223A or 124D223D mutants of Par-4 were treated with TRAIL (500 ng/ml, 3 h) in the presence or absence of Z-VAD (15 μM). Par-4 cleavage was determined by western blotting using anti-GFP antibody. Hsp90 was used as a loading control. (b) Wild-type Par-4, 124A/D, 223A/D, 124D223D and 124A223A mutant proteins (GFP tagged) were produced in vitro by a TNT RRL system. Recombinant caspase-8 (upper panel) or caspase-3 and -7 (Casp3, Casp7, lower panel) were incubated with the indicated Par-4 proteins for 3 h at 37 °C, and Par-4 cleavage was assessed by western blotting using GFP antibody. (c) Caspase assay was performed on pre-phosphorylated rat GST-Par-4 (in vitro kinase assay using RecCK2 as described above Figure 2), and cleaved Par-4 was detected by immunoblot. *NS: non-specific bands are probably due to the presence of recombinant Par-4 degraded forms. To note: cleaved GST Par-4 migrates at lower molecular weight than cleaved GFP-Par-4, given GST tag is located in the N-terminus of Par-4 and not on C-terminus as the GFP-tag. (d–f) PC-3 cells transfected with Par-4 wild-type or FLAG-tagged Par-4 (124–332) were treated as above with TRAIL and analyzed for apoptosis by Hoechst staining (d) or FACS analysis of the caspase-3 activity (FLICA) (e) and by immunoblotting analysis of caspase-8 and PARP cleavage (f). Bars represent the mean±S.D. of at least three independent experiments. Flag-Par-4 (124–332) and GFP were revealed in the same membrane using corresponding antibodies (lower panel). Endogenous Par-4 was used as a loading control. *P<0.05, **P<0.01
We then examined whether Par-4 cleavage was regulated by phosphorylation of S124 by CK2. We first performed an in vitro caspase assay using purified wild-type Par-4 or its mutants (S124A, S124D, S223A, S223D, S124A, S223A or S124D/S223D) in the presence of recombinant caspases. We observed that caspase-3 and caspase-8 cleaved wild-type Par-4 and the mutants S124A and S124A/S223A, whereas they were unable to cleave Par-4 mutant S124D and S124D/S223D (Figure 4b and data not shown). Notably, mutation of rat Par-4 S223 did not by itself affect caspase cleavage (Figure 4b). Then, to confirm that loss of Par-4 cleavage was a direct consequence of CK2 phosphorylation and not an artifact introduced by the mutation, we performed an in vitro kinase assay on rat GST-Par-4 using recombinant CK2, followed by an in vitro caspase assay using recombinant caspase 3. As shown in Figure 4c, phosphorylation of Par-4 by CK2 prevented its cleavage by caspase-3, but this effect was specific to rodent Par-4 as phosphorylated human Par-4 was still cleaved (Supplementary Figure S6c). Finally, we show that the proapoptotic function of S124D (mimicking CK2 phosphorylation) like D123A mutants (non-cleavable caspase site) of rat Par-4 was impaired (Supplementary Figure S7). Therefore, in rat, phosphorylation by CK2 at residue S124 inhibits proapoptotic functions of Par-4 by preventing caspase-mediated Par-4 cleavage (D123), whereas in human the proteolytic cleavage of Par-4 (D131) is apparently not dependent on CK2 phosphorylation.
Recently, Chaudhry et al.24 have shown that the caspase-cleaved form of human Par-4 displays proapoptotic activity. Therefore, we tested whether rat Par-4 caspase-cleaved fragment retained the apoptotic properties of wild-type Par-4. We found that expression of rat Par-4 (124–332) cleaved form sensitizes cells to apoptosis induced by TRAIL more efficiently than wild-type Par-4, as measured by its ability to induce chromatin condensation, caspase-3 activation and cleavage of PARP and caspase-8 (Figures 4d–f).
Par-4 is known to be spontaneously secreted by normal and cancer cells in culture. Once secreted, Par-4 can induce apoptosis of surrounding cancer cells through the activation of the caspases’ signaling pathway.26 Therefore, we first studied whether cleaved Par-4 (124–332) could be secreted. COS cells were transfected with GFP alone, wild-type GFP-Par-4 or Par-4 (124–332), and the presence of those proteins in the conditioned media was determined by western blotting analysis. We found both Par-4 and Par-4 (124–332) in the cell-conditioned media, while GFP alone was not detectable (Supplementary Figure S8a). To address the exogenous function of caspase-cleaved Par-4, we added Par-4 (124–332) or wild-type Par-4 (same volume from the concentrated conditioned media) into PC-3 cell culture medium for 24 h, and the rate of apoptosis was evaluated. As shown in Supplementary Figure S8b, extracellular Par-4 (124–332) exerted a stronger apoptotic effect than wild-type Par-4. Collectively, these findings indicate that CK2 regulates the proapoptotic functions of rodent Par-4 by preventing its caspase-mediated cleavage in a phosphorylation-dependent manner.
Phosphorylation of human Par-4 S231 site by CK2 and its protective function
By comparative analysis of human and rodent Par-4 sequences, we determined the human phosphosite orthologs of rodents S124 and S223 (Figure 5a, Supplementary Figure S5). Unlike the phosphosite S124 site (Supplementary Figure S5b), the corresponding S223 phosphosite, S231 in humans, is well conserved between species (Figure 5a) and was also detected by mass spectrometry analysis on leukemia cell lines (www.phos-phosite.com), thereby supporting a functional role for this site.

Human Par-4 is phosphorylated both in vitro and in vivo on S231 (ortholog of rodent S223) by CK2 and this phosphorylation impairs apoptosis. (a) Conservation of the CK2 recognition motif (including the serine 231 of human Par-4) was evaluated in different species. (b) Recombinant Myc-tagged human Par-4 (hPar-4) or 231D mutant proteins were produced with TNT RRL system. An in vitro kinase assay was performed with the immunoprecipitates as substrates, in the presence of recombinant CK2 and [γ−32P]ATP. Phosphorylated Par-4 was detected by autoradiography. Production of recombinant Myc tagged proteins were checked by western blotting (right panel). (c) Phosphorylation of endogenous Par-4 was detected by western blotting (left panel), using the human anti-phosphoserine231-Par-4 antibody (Ph231 hPar-4), in PC-3 cells treated (+) or not (−) with TRAIL for 9 h, and densitometry analysis was done. The blocking peptide was used in order to test the specificity of the phospho-antibody. In parallel, the percentage of apoptosis induced by TRAIL (9 h) was assessed by Hoetsch staining. Bars represent the mean±S.D. of at least three independent experiments (right panel). (d–f) HCT116 cells were transfected with empty Myc vector control (Mock), human Myc-tagged Par-4, 231A or 231D mutant Par-4 (mimicking unphosphorylated and phosphorylated hPar-4, respectively) and then treated with recombinant TRAIL (150 ng/ml, 3 h). Expression of the different constructs was assessed by western blotting using Myc antibody, and apoptosis was monitored by immunoblot analysis of caspase-8 and PARP cleavage (d) and by DAPI staining (e and f). Bar graph shows semi-quantified densitometry from PARP and Caspase-8 western blotting analysis. Bars represent the mean±S.D. of at least four independent experiments. Hsp90 was used as a loading control. Bar=10 μm, magnification × 63; *P<0.05. a.u, arbitrary unit
To determine whether S231 is a target of CK2, we performed an in vitro kinase assay with recombinant CK2 in the presence of immunoprecipitated human wild-type Par-4 or S231D Par-4 mutant produced by in vitro translation. We found that mutation of the S231 site markedly decreased the phosphorylation of human Par-4 by CK2, suggesting that S231 is a target site for CK2 (Figure 5b).
To validate the role of Par-4 phosphorylation in prostate cancer, we generated an antibody directed against the phosphoserine 231 residue of human Par-4. We detected an expected protein band in prostate cancer PC-3 cells corresponding to phosphorylated Par-4 (Figure 5c). This band was not detectable when the blocking peptide was added, showing the specificity of the anti-phospho-S231 antibody (Figure 5c). Interestingly, the phosphorylation of endogenous human Par-4 site S231 increased in the resistant PC-3 cells following TRAIL treatment (Figure 5c). To further study the functionality of S231, we transfected the human cancer cell line HCT116, which allowed high transfection efficiency, with the S231D and S231A Par-4 mutants and determined their effect on apoptosis. The cells transfected with the S231A mutant showed enhanced sensitivity to TRAIL, as judged by caspase-8, PARP cleavage and DAPI staining. Conversely, cells transfected with the constitutively phosphorylated S231D mutant were less sensitive to TRAIL (Figures 5d–f). These results indicate that phosphorylation by CK2 at residue S231 of human Par-4 strongly impairs Par-4 apoptotic properties and seems to be more efficient than the phospho mimicking mutant S223D of rat Par-4 (Supplementary Figure S7).
Par-4 phosphorylation by CK2 in prostate cancer cells
CK2 activity is elevated in prostate cancer cells compared with their normal counterparts.35 Therefore, we assessed by immunoblotting the phosphorylation status of Par-4, in parallel with CK2 expression and activity, in human prostate cancer cells (PC-3, LNCaP) versus normal cells (PNT2C2, PrEC) (Figure 6a). Our results revealed that Par-4 phosphorylation at the CK2 site S231 occurs preferentially in cancer cells, which display a higher CK2 activity compared with normal cells. Accordingly, the CK2 protein level is increased in prostate cancer (especially the CK2β subunit) as compared with their normal counterparts (Figure 6a). Furthermore, CK2 depletion in PC-3 cells by siRNA significantly impaired Par-4 phosphorylation at S231 (Figure 6b). Accordingly, we noted a coordinated regulation of CK2α protein levels and Par-4 phosphorylation, that is, both showed approximately 50% decrease (Figure 6b). Similar results were obtained using the selective inhibitor of CK2, 4,5,6 tetrabromobenzotriazole (TBB; Supplementary Figure S9).47, 48

Par-4 is highly phosphorylated on S231 (ortholog of rodent S223) in human prostate cancer cells compared with normal counterparts. (a) Upper panel, phosphorylation of Par-4 on S231 was studied by western blotting in prostate cancer cells (PC-3, LnCap cells) and in normal prostate cells (PrCE, PNT2C2). In parallel, endogenous expression of Par-4 and CK2 subunits in the different cell lines was determined (middle panel). Hsp90 was used as a loading control. Lower panel, analysis of CK2 activity in the different prostate cells studied using Cyclex CK2 screening kit. Bars represent the mean±S.D. of at least two independent experiments. (b) PC-3 cells were transfected with CK2 siRNA (siCK2T, ThermoFischer) or scrambled siRNA fluorescently labeled with FAM (Scr siRNA) for 48 h and then treated or not with TRAIL (500 ng/ml, 3 h). Endogenous phosphorylated human Par-4 was detected by western blotting using the anti-phospho231 Par-4 antibody (Ph231-hPar-4) (left panel). Par-4 expression (left panel) and downregulation of CK2α protein (right panel) were evaluated by immunoblotting using the corresponding antibodies. Bar graph shows semi-quantified densitometry from western blotting analysis
As previously reported,43, 49, 50, 51 CK2-depleted cells are more sensitive to apoptosis. In line with these results, resistant PC-3 cells were more sensitive to TRAIL treatment when they were transfected with two different CK2 siRNA (siCK2T and siCK2AB, Figures 7a–d and Supplementary Figure S10). Most importantly, when we co-depleted CK2 and Par-4 by siRNA (two different siRNAs tested for each), we restored PC-3 cell resistance to TRAIL-induced apoptosis (Figures 7a–d and Supplementary Figure S10). Collectively, these results suggest that CK2 anti-apoptotic effect involves Par-4 phosphorylation and inhibition of Par-4 proapoptotic functions.

Anti-apoptotic role of CK2 is dependent on Par-4 in human prostate cancer cells. (a–d) PC-3 cells were transfected with different CK2 siRNAs (siCK2T from ThermoFischer (a) or siCK2AB from Ambion (b)) together with scrambled siRNA fluorescently labeled with FAM (Scr siRNA) or different Par-4 siRNA (siPar-4T from ThermoFisher or siPar-4sc from Santa Cruz) for 48 h. Then, cells were treated or not with TRAIL (500 ng/ml, 3 h). The downregulation of Par-4 and CK2α proteins was confirmed by immunoblot using the corresponding antibodies. Apoptosis was monitored (a and b) by immunoblot analysis of caspase-8 and PARP (long and short cleaved PARP forms) cleavage and (c and d) by DAPI staining. Graphs represent semi-quantified densitometry analysis from PARP and caspase-8 western blotting analysis of panel (a). See also Supplementary Figure S10A for the densitometric analysis of panel (b). Bars represent the mean±S.D. of at least four independent experiments. Hsp90 was used as a loading control. Bar=10 μm, magnification × 63; *P<0.05. a.u, arbitrary unit
Discussion
Par-4 induces tumor regression in prostate tumors, and this action is associated with both Par-4 intracellular and extracellular proapoptotic functions. Indeed, Par-4 has been shown to be secreted and to induce apoptosis in the surrounding cells.26 In this study, we noted both in rat and human that: (i) Par-4 is a new substrate of the survival kinase CK2, and (ii) phosphorylation by CK2 impairs Par-4 proapoptotic functions (hypothetical model, Supplementary Figure S11).
We also unraveled another level of CK2-dependent regulation of Par-4 apoptotic functions. Cleavage of Par-4 by caspases is a crucial step that leads to the production of a Par-4-cleaved form ((132–340) in humans and (124–332) in rats) that displays full apoptotic activity. Phosphorylation of rat Par-4 at S124 by CK2 inhibits caspase-mediated cleavage at residue D123, thus dramatically reducing its apoptotic function. Notably, this inhibitory effect of CK2 on Par-4 cleavage occurs in rat Par-4 but not in human Par-4, underlying species-specific effects of CK2 phosphorylation. Similarly to rat Par-4, the proteolytic cleavage of caspase-9 is specifically blocked in rodent cells by CK2 phosphorylation.52 In both species, caspase-dependent cleavage of other substrates such as Bid, Max or apoptosis repressor with caspase recruitment domain, has been also shown to be blocked by CK2 phosphorylation.40, 53 In the case of Bid, phosphorylation by CK2 prevents the generation of a Bid caspase-cleaved form that, as for Par-4, is active and can induce apoptosis. Therefore, CK2 is able to inhibit the activity of its substrates across species, yet by acting through different mechanisms in a species-dependent manner.
The caspase-cleavage product of Par-4 is also secreted and induces a ‘bystander’ effect that results in the killing of the surrounding cells (Supplementary Figure S8), which might create an amplification loop in cell apoptotic process. The proapoptotic effect of the rat Par-4 cleaved form (124–332), as for human Par-4, likely relies on the presence of the SAC domain that is an essential domain for extracellular and intracellular Par-4 proapoptotic activity.21, 26 In contrast, the N-terminal rat Par-4 truncated form (1–163) is devoid of proapoptotic function.10, 21
In this work, we observed that S231D mutant of human Par-4 is weakly phosphorylated by CK2, thus suggesting that although this is the major phosphosite for CK2 other minor phosphorylation sites are present. Among the potential sites, residues S233 and S110 deserve further considerations. However, we have demonstrated the essential regulatory function of residue S231, via its ability to impair the proapoptotic function of human Par-4 following phosphorylation by CK2. Consistently, CK2 knockdown is associated with diminished S231 phosphorylation, and this results in an increased sensitivity to apoptosis. In contrast to phosphorylation of S124 in rat Par-4 protein, phosphorylation of S231 did not affect human Par-4 cleavage by caspases. The mechanism by which phosphorylated S231 prevents Par-4-induced apoptosis deserves further investigations. Because S231 is located in the C-terminal domain of Par-4 – which mediates interaction of Par-4 with its partners (i.e., NF-κB, PKCζ) – we hypothesize that S231 phosphorylation may prevent these interactions. CK2 is known to be a partner of p6254 and PKCζ,55 and p62 prevents the inhibitory effect of Par-4 on PKCζ activity during apoptosis.56 During apoptosis, the p62-CK2-PKCζ-Par-4 complex may further increase the phosphorylation of Par-4 by CK2, leading to a strong inhibition of Par-4 proapoptotic functions. Consistent with this hypothesis, we observed an increase in human Par-4 phosphorylation during TRAIL-induced apoptosis in the resistant PC-3 cancer cells (Figure 5c). Importantly, S231 is highly phosphorylated in human prostate cancer cells compared with normal cells, thus suggesting that CK2 phosphorylated Par-4 could be a potential biomarker of prostate cancer. Similar to other tumor suppressors, such as phosphatase tensin homolog or promyelocytic leukemia, Par-4 is a key CK2 target (this paper), whose proapoptotic activity must be blocked to allow tumor progression.18, 19, 20 This observation is consistent with our findings indicating that increased sensitivity of PC-3 cells to apoptosis following CK2 depletion is hampered by siRNA-mediated Par-4 depletion. Several other observations support the fact that the pro-tumorigenic function of CK2 may depend on the inhibition of Par-4. First, the overexpression of Par-4, as well as treatment with CK2 inhibitors, showed barely any effect in normal cells, yet resulted in a potent induction of apoptosis in cancer cells.49, 57 Second, CK2 inhibitors were reported to induce ER-mediated apoptosis, while ER stress favors Par-4 secretion and apoptosis.26 Third, CK2 antisense delivery into prostate cancer xenografts or intra-tumor injection of a peptide preventing CK2 phosphorylation, both associated with significant reduction of CK2 activity, demonstrated a potent induction of apoptosis and tumor regression.51 A similar induction of apoptosis and tumor regression of prostate cancer is observed when Par-4 is injected intra-tumorally in nude mice.1
In summary, our findings shed light on a new regulatory role of CK2 on Par-4 functions in prostate cancer cells and delineate Par-4 as a key target that could be exploited for the development of anti-tumoral therapeutics in prostate cancer.
Materials and Methods
Cells and reagents
Human colon cancer HCT116, prostate cancer PC3, LnCap cells and benign prostate PNT2C2 cells were cultured in RPMI 1640 (BioWittaker, Fontenay sous-bois, France) and PrEC cells in medium Bullet kit (Lonza, Walkersville, USA). The authentication of cell lines was conducted by ATCC (Molsheim, France). COS7 cells were grown in Dubelcco’s Modified Eagle Medium (BioWittaker). The selective inhibitor of CK2, TBB (Sigma-Aldrich, St Quentin Fallavier, France) was pre-incubated for 30 min and used at 20 or 40 μM. The recombinant human His-TRAIL was produced as described previously.58 The pan caspase inhibitor zVAD-FMK and the phosphatase inhibitors Calyculin A and Okadaic acid (Calbiochem and Merck KGaA, Darmstadt, Germany) were used at 15 μM, 20 nM and 0.5 or 1 μM, respectively.
Plasmids and transfection
GST-Par-4 was a gift from Dr. Robert (University of Manchester); GFP-CK2α and β constructs and their corresponding mutants were previously described.59 The rat constructs pcDNA3.1 TOPO GFP-Par-4 mutants (124A223A, 124D223D, 124A, 124D, 223A, 223D, 123A), the Flag or GFP deletant rat Par-4 (124–332) and the human constructs pcDNA3.1 Myc-Par-4 (wild-type, 231A and 231D mutants) were performed by directed mutagenesis following the manufacturer’s instructions (Stratagene, Agilent Technology, Massy, France). Cell lines were transfected for 15 h (no significant apoptosis due to Par-4 overexpression is detected at that time29 and data not shown) using Nanojuice (Millipore, Molsheim, France) or Jetpei (Polyplustransfection, Ozyme, St Quentin en Yvelines, France). PC3 cells were transfected with interferin reagent using two different human CK2 siRNA (siCK2T (L-003475-00-0005) ON-TARGETplus SMARTpool from Thermo Fisher Scientific (Waltham, MA, USA) or siCK2AB from Ambion (Life Technology, Saint Aubin, France, S3638)) and/or two different human Par-4 siRNA (siPar-4T (L-004434-00-0005) ON-TARGETplus SMARTpool from Thermo Fisher Scientific (or siPar-4sc (SC-36190) from Santa Cruz (Tebu-Bio, Le Perray en Yveline, France)) together with scrambled siRNA fluorescently labeled with FAM (Scr siRNA, Santa Cruz) for 48 h.
Recombinant protein purification
Escherichia coli BL21 bacteria expressing GST-Par-4 were grown 3 h with 1 mM isopropyl β-D-1-thiogalactopyranoside. After centrifugation at 5000 × g for 10 min, bacteria were lysed in 50 mM Tris-HCl pH 8, 300 mM sodium chloride (NaCl), 1% triton, 0.2 mM EDTA, 1 mM dithiothreitol (DTT), 1 mM phenylmethylsulfonyl fluoride (PMSF) and protease inhibitors for 30 min. Gluthatione sepharose 4B beads (Sigma, Saint-Quentin Fallavier, France) were added to the cleared supernatants and subjected to rotation for 30 min at 4 °C. Beads were washed four times in phosphate-buffered saline, 1 mM DTT and 1 mM PMSF, and the recombinant protein was eluted in 50 mM Tris-HCl, 0.3% Triton, 20 mM NaCl, 50 mM Glutathione, 1 mM DTT for 15 min at 4 °C with agitation. The protein concentration was evaluated using the Bradford method.
Proteins using TNT Quick Coupled Transcription/Transcription System (Promega, Charbonnières-les-Bains, France) were produced as follows: 1 μg of template plasmid DNA was added to the reaction mixture, which was afterwards incubated at 30 °C for 90 min. Twenty microliters of the in vitro translated proteins were used for immunoprecipitation.
Immunoprecipitation and immunoblotting
Cells were lysed in lysis buffer (50 mM Hepes pH 7.4, 140 mM NaCl, 5 mM EDTA, 0.2% NP40, 10 mM sodium fluoride, protease inhibitor cocktail (Roche, Neuilly sur Seine, France)). In all, 800 μg of each lysate was incubated with 3 μg of CK2α, CK2β (Santa Cruz), Par-4 (Sigma-Aldrich), GFP-tag (Millipore) or HA-tag (Biomol, Hamburg, Germany) antibodies with constant agitation at 4 °C. Then, the immuno-complexes were precipitated with protein G (Amersham Bioscience, GE Healthcare, Orsay, France), and the beads were washed in lysis buffer and resuspended in Laemmli buffer.
Proteins were separated on SDS-PAGE 8–10%. The membranes were probed with primary antibodies and then incubated with the appropriate secondary antibody. Proteins were visualized using the ECL western blotting kit (Pierce, ThermoScientific, Rockford, IL, USA). The rabbit anti-Par-4 was purchased from Sigma-Aldrich, the mouse CK2β, rabbit anti-14-3-3τ and -actin antibodies were from Santa Cruz and the mouse anti-CK2α was purchased from Millipore. The monoclonal Myc-tag antibody and the polyclonal rabbit antibodies anti-PP1 and -GST were purchased from Cell Signaling (Danvers, MA, USA). The rabbit anti-human phospho-serine 231 Par-4 antibody was produced by Covalab (Dijon, France). Hsp90, Hsp110 were from Stressgen (Tebu-Bio).
Luciferase assay
Cos cells were transfected with the different Par-4 mutants in the presence or absence of the NF-κB promoter and Beta galactosidase (Gift from Professor L Sistonen, Finland) as an internal control. Cells were lysed 24 h post transfection and, after dosage, assayed using Promega’s Luciferase Assay System kit (Promega, Charbonnières-les-Bains, France). Luciferase activity was measured using a Luminometer (Promega, Mannheim, Germany, LUMAT LB 9507). To normalize for transfection efficiency, the activity of co-transfected β-galactosidase was also assayed.
Caspase cleavage assay
In vitro translated Par-4 protein and the corresponding mutants were produced using TNT Quick Coupled Transcription System (Promega, Charbonnières-les-Bains, France). Fourmicroliters of the in vitro translated proteins were incubated for 3 h with one unit of recombinant caspases 8, 3 and 7 (R&D System, Lille, France) in a buffer containing 10 mg CHAPS, 25 mM Hepes (pH 7.4), 10 mM DTT.
In vitro kinase assay
Immunoprecipitated GFP-CK2 from Cos cells or 60 ng of recombinant CK2 (RecCK2) (New England Biolabs, Evry, France) was added in the kinase reaction buffer (20 mM Hepes pH 7.4, 1 mM DTT, 10 mM magnesium chloride, 10 mM MnCl2, 0.2 mM EGTA) containing 200 μM adenosine triphosphate (ATP), 10 μCi [γ32P] ATP in the presence of 5 μg of purified rat GST-Par-4 or the immunoprecipitated recombinant rat wild-type GFP-tagged Par-4 and its corresponding mutants (124A223A, 124A, 223A) or the immunoprecipitated recombinant human GFP- or Myc-tagged wild-type Par-4 and its corresponding mutant (Myc-231A), all produced by in vitro translation as described above. After incubation (25 min, 30 °C), Laemmli buffer was added to stop the reaction, and the samples were resolved on 10% SDS-PAGE gels. The phosphorylated Par-4 protein was visualized by staining with Coomassie blue, and the dried gel was exposed to autoradiography.
Measure of CK2 activity
The determination of CK2 activity was performed as described by the manufacturer (Cliniscience, Nanterre, France). Briefly, 10 μg of cell lysate from the different prostate cell lines were diluted in the CK2 buffer in the presence of ATP and incubated 30 min at 30 °C in a 96-well plate pre-coated with the substrate corresponding to recombinant p53, which contains a serine residue (S46) that was phosphorylated by CK2. After several washings, a specific horseradish peroxidase (HRP)-conjugated antibody recognizing only the phosphorylated serine 46 of p53 was added in each well for 30 min at room temperature. Then, the amount of phosphorylated substrate was determined by addition of the HRP substrate and measurement by spectrophotometry (dual wavelength 450/595 nm). As a positive control, we used recombinant CK2 (Rec.CK2, 20 000 units), and as a baseline control samples treated with CK2-specific inhibitor tetrabromocinnamic acid (20 μM, Calbiochem).
Measure of apoptosis and caspase-3 activity
Cells transfected with Par-4 mutants were either treated with recombinant His-TRAIL (500 ng/ml) or Paclitaxel 200 nM (Calbiochem). Cells were harvested and fixed with 0.2% paraformaldehyde in presence of 1 mg/ml of DAPI or Hoetsch 33342 (Sigma-Aldrich) to stain the nuclei. Apoptosis was measured by counting cells that are condensed and fragmented. Images were acquired using the Cell Observer station (Zeiss, Göttingen, Germany). Briefly, the system consists of an inverted microscope AxioVert 200M (Zeiss) equipped for fluorescence with a CCD camera. All the system is motorized and controlled by the Axiovision software (Zeiss). Apoptosis was also assessed by the determination of PARP and caspase 8 cleavage by western blotting using anti-mouse caspase-8 from R&D and rabbit PARP (detection of the short cleaved form) or mouse PARP (detection of the large fragment) antibodies (Santa Cruz).
For FACS analysis, cells stained with APO and FITC–annexin V conjugate were analyzed by flow cytometry using a FACS Scan flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA). Caspase-3 activity was measured by FACS using the fluorochrome benzyloxycarbonyl-Asp-Glu-Val-Asp (OMe) fluoromethylketone (PromoKine Caspase-3 kit, PromoCell, Heidelberg, Germany).
Tryptic digestion, two-dimensional separation and Edman degradation
In-gel tryptic digestions of 32P-labeled Par-4 followed by a two-dimensional phosphopeptide mapping were carried out as previously described.60 Briefly, the labeled Par-4 band was excised from the dried gel and in-gel digested with sequencing grade trypsin (Promega, Mannheim, Germany). The gel pieces were incubated with 2 ng/μl trypsin in 50 mM ammonium bicarbonate buffer (pH 8) for 16 h at 37 °C. Supernatants were collected, vacuum-dried and resuspended in 15 μl of pH 1.9 buffer (formic acid 2.3%, acetic acid 2.9% v/v). Peptides were then separated in two dimensions by electrophoresis and TLC. The first dimension – electrophoresis – was performed in a pH 1.9 buffer at 750 V for 1.5 h using the Hunter Thin Layer Peptide Mapping System, model no. HTLE-7000 (C.B.S. Scientific). Fifteenmicroliters of the tryptic digest together with the 3 μl of separation markers mixture (Xylene Cyanol FF (blue), 1 mg/ml and Nɛ-DNP-L lysine hydrochloride (yellow), 5 mg/ml, Sigma) were carefully applied on a cellulose sheet (20 × 20 cm2, Merck KgaA) and electrophoretically separated. The sheet was dried and ascending TLC in the second dimension was performed for 13 h in a chromatography tank saturated with a mobile phase containing 30% water, 37.5% nbutanol, 7.5% acetic acid and 25% pyridine. The sheet was dried and the 32P-phosphopeptides were visualized by autoradiography. For MALDI MS and Edman sequencing, corresponding 32P-peptides were extracted from a cellulose sheet by scraping off the powder into an Eppendorf tube and then eluted twice with 200 ml of 30% acetonitrile (Rathbrun, UN1648) and 0.1% trifluoroacetic (Sigma-aldrich) solution. The resulting extract was vacuum-evaporated, and MALDI-MS was performed. For Edman degradation, phosphopeptides were immobilized on arylamine membrane discs (Sequelon-AA membrane; Applied Biosystems, Foster City, CA, USA) using water-soluble carbodiimide. The Sequelon-AA membranes consist of a PVDF matrix that has been derivatized with arylamine groups. Individual Par-4 phosphopeptides are immobilized on the Sequelon-AA discs through their C-terminal carboxyl groups. Therefore, the N termini of such peptides are free, and amino acids are clipped off during 10 Edman degradation cycles. The collected fractions were spotted on a Whatman filter paper, which was then visualized by autoradiography on a Fuji phosphorimager plate (VWR International Oy, Helsinki, Finland) to reveal the cycle at which the radiolabel is released, which corresponds to the position of the phosphorylated amino acid, as counted from the N-terminus.
Statistical methods
Results are expressed as means±S.E.M. from at least three independent experiments. Values were analyzed using the Student’s t-test. All P-values were obtained using the two-tailed tests, and error bars in the graphs represent 95% confidence intervals. Quantitative data were analyzed using the GraphPad Prism program (La Jolla, CA, USA). Blots were treated by Photoshop (San Jose, CA, USA) and quantified by Image J (Bethesda, MD, USA). Statistical significance, P<0.05 and P<0.01, are denoted with single and double asterisks (* and **), respectively.
Abbreviations
- CK2:
-
casein kinase 2
- ER:
-
endoplasmic reticulum
- Par-4:
-
prostate apoptosis response-4
- NF-κB:
-
nuclear factor kappa B
- PKA:
-
protein kinase A
- PKCζ:
-
atypical protein kinase C
- SAC:
-
selective for apoptosis in cancer cells
- TNF:
-
tumor necrosis factor
- ARC:
-
apoptosis repressor with caspase recruitment domain
- TRAIL:
-
TNF-related apoptosis-inducing ligand
- PP1:
-
protein phosphatase 1
- PARP:
-
poly ADP ribose polymerase
- 2-D-PPM:
-
two-dimensional phosphopeptide mapping
- HA:
-
hemagglutinin
- GFP:
-
green fluorescent protein
- GST:
-
glutathione-S-transferase
- SDS-PAGE:
-
sodium dodecyl phosphate polyacrylamide gel electrophoresis
- DTT:
-
dithiothreitol
- IPTG:
-
isopropyl β-D-1-thiogalactopyranoside
- TBB:
-
4,5,6 tetrabromobenzotriazole
- zVAD-FMK:
-
benzyloxycarbonylvalyl-alanyl-aspartic acid (OMe)-fluoro-methylketone
- PMSF:
-
phenylmethylsulfonyl fluoride
- ATP:
-
adenosine triphosphate
- NaCl:
-
sodium chloride
- HRP:
-
horseradish peroxidase
- MALDI-TOF-MS:
-
matrix-assisted laser desorption/ionization-time-of-flight-mass spectrometry
References
Chakraborty M, Qiu SG, Vasudevan KM, Rangnekar VM . Par-4 drives trafficking and activation of Fas and Fasl to induce prostate cancer cell apoptosis and tumor regression. Cancer Res 2001; 61: 7255–7263.
El-Guendy N, Rangnekar VM . Apoptosis by Par-4 in cancer and neurodegenerative diseases. Exp Cell Res 2003; 283: 51–66.
Nalca A, Qiu SG, El-Guendy N, Krishnan S, Rangnekar VM . Oncogenic Ras sensitizes cells to apoptosis by Par-4. J Biol Chem 1999; 274: 29976–29983.
Sells Stephen F, Wood DP, Joshi-barve SS, Muthukumar S, Jacob RJ, Crist SA et al. Commonality of apoptosis of the gene programs in androgen-dependent prostate cells. Cell Growth Differs 1994; 5: 457–466.
Mattson MP, Duan W, Chan SL, Camandola S . Par-4: an emerging pivotal player in neuronal apoptosis and neurodegenerative disorders. J Mol Neurosci 1999; 13: 17–30.
Gurumurthy S, Rangnekar VM . Par-4 inducible apoptosis in prostate cancer cells. J Cell Biochem 2004; 91: 504–512.
Díaz-Meco MT, Municio MM, Frutos S, Sanchez P, Lozano J, Sanz L et al. The product of par-4, a gene induced during apoptosis, interacts selectively with the atypical isoforms of protein kinase C. Cell 1996; 86: 777–786.
Diaz-Meco MT, Lallena MJ, Monjas A, Frutos S, Moscat J . Inactivation of the inhibitory kappaB protein kinase/nuclear factor kappaB pathway by Par-4 expression potentiates tumor necrosis factor alpha-induced apoptosis. J Biol Chem 1999; 274: 19606–19612.
Guo C, Yu S, Davis AT, Wang H, Green JE, Ahmed K . A potential role of nuclear matrix-associated protein kinase CK2 in protection against drug-induced apoptosis in cancer cells. J Biol Chem 2001; 276: 5992–5999.
Sells SF, Han SS, Muthukkumar S, Maddiwar N, Johnstone R, Boghaert E et al. Expression and function of the leucine zipper protein Par-4 in apoptosis. Mol Cell Biol 1997; 17: 3823–3832.
Wang B-D, Kline CLB, Pastor DM, Olson TL, Frank B, Luu T et al. Prostate apoptosis response protein 4 sensitizes human colon cancer cells to chemotherapeutic 5-FU through mediation of an NF kappaB and microRNA network. Mol Cancer 2010; 9: 98.
Kline CL, Shanmugavelandy SS, Kester M, Irby RB . Delivery of PAR-4 plasmid in vivo via nanoliposomes sensitizes colon tumor cells subcutaneously implanted into nude mice to 5-FU. Cancer Biol Ther 2009; 8: 1831–1837.
Johnstone RW, Tommerup N, Hansen C, Vissing H, Shi Y . Mapping of the human PAWR (par-4) gene to chromosome 12q21. Genomics 1998; 53: 241–243.
Schneider BG, Rha SY, Chung HC, Bravo JC, Mera R, Torres JC et al. Regions of allelic imbalance in the distal portion of chromosome 12q in gastric cancer. Mol Pathol 2003; 56: 141–149.
Nagai MA, Gerhard R, Salaorni S, Humberto J, Guerreiro T, Nonogaki S et al. Down-regulation of the candidate tumor suppressor gene PAR-4 is associated with poor prognosis in breast cancer. Int J Oncol. 2010; 37: 41–49.
Boehrer S, Chow KU, Puccetti E, Ruthardt M, Godzisard S, Krapohl A et al. Deregulated expression of prostate apoptosis response gene-4 in less differentiated lymphocytes and inverse expressional patterns of par-4 and bcl-2 in acute lymphocytic leukemia. Hematol J 2001; 2: 103–107.
Cook J, Krishnan S, Ananth S, Sells SF, Shi Y, Walther MM et al. Decreased expression of the pro-apoptotic protein Par-4 in renal cell carcinoma. Oncogene 1999; 18: 1205–1208.
Alvarez JV, Pan T-C, Ruth J, Feng Y, Zhou A, Pant D et al. Par-4 downregulation promotes breast cancer recurrence by preventing multinucleation following targeted therapy. Cancer Cell 2013; 24: 30–44.
Barradas M, Monjas A, Diaz-Meco MT, Serrano M, Moscat J . The downregulation of the pro-apoptotic protein Par-4 is critical for Ras-induced survival and tumor progression. EMBO J 1999; 18: 6362–6369.
Garcia-Cao I, Duran A, Collado M, Carrascosa MJ, Martin-Caballero J, Flores JM et al. Tumour-suppression activity of the proapoptotic regulator Par-4. EMBO Rep 2005; 6: 577–583.
El-Guendy N, Zhao Y, Gurumurthy S, Burikhanov R, Rangnekar VM . Identification of a unique core domain of par-4 sufficient for selective apoptosis induction in cancer cells. Mol Cell Biol 2003; 23: 5516–5525.
de Thonel A, Bettaieb A, Jean C, Laurent G, Quillet-Mary A . Role of protein kinase C zeta isoform in Fas resistance of immature myeloid KG1a leukemic cells. Blood 2001; 98: 3770–3777.
Leroy I, de Thonel A, Laurent G, Quillet-Mary A . Protein kinase C zeta associates with death inducing signaling complex and regulates Fas ligand-induced apoptosis. Cell Signal 2005; 17: 1149–1157.
Chaudhry P, Singh M, Parent S, Asselin E . Prostate apoptosis response 4 (Par-4), a novel substrate of caspase-3 during apoptosis activation. Mol Cell Biol 2012; 32: 826–839.
Lee TJ, Jang JH, Noh HJ, Park EJ, Choi KS, Kwon TK . Overexpression of Par-4 sensitizes TRAIL-induced apoptosis via inactivation of NF-kappaB and Akt signaling pathways in renal cancer cells. J Cell Biochem 2010; 109: 885–895.
Burikhanov R, Zhao Y, Goswami A, Qiu S, Schwarze SR, Rangnekar VM . The tumor suppressor Par-4 activates an extrinsic pathway for apoptosis. Cell 2009; 138: 377–388.
Zhao Y, Burikhanov R, Brandon J, Qiu S, Shelton BJ, Spear B et al. Systemic Par-4 inhibits non-autochthonous tumor growth. Cancer Biol Ther 2011; 12: 152–157.
Goswami A, Burikhanov R, de Thonel A, Fujita N, Goswami M, Zhao Y et al. Binding and phosphorylation of par-4 by akt is essential for cancer cell survival. Mol Cell 2005; 20: 33–44.
Gurumurthy S, Goswami A, Vasudevan KM, Rangnekar VM . Phosphorylation of Par-4 by protein kinase A is critical for apoptosis. Mol Cell Biol 2005; 25: 1146–1161.
Allende JE, Allende CC . Protein kinases. 4. Protein kinase CK2: an enzyme with multiple substrates and a puzzling regulation. FASEB J 1995; 9: 313–323.
Meggio F, Pinna L . One-thousand-and-one substrates of protein kinase CK2? FASEB J 2003; 17: 349–368.
Daya-Makin M, Sanghera JS, Mogentale TL, Lipp M, Parchomchuk J, Hogg JC et al. Activation of a tumor-associated protein kinase (p40TAK) and casein kinase 2 in human squamous cell carcinomas and adenocarcinomas of the lung. Cancer Res 1994; 54: 2262–2268.
Gapany M, Faust RA, Tawfic S, Davis A, Adams GL, Ahmed K . Association of elevated protein kinase CK2 activity with aggressive behavior of squamous cell carcinoma of the head and neck. Mol Med 1995; 1: 659–666.
Munstermann U, Fritz G, Seitz G, Lu YP, Schneider HR, Issinger OG . Casein kinase II is elevated in solid human tumours and rapidly proliferating non-neoplastic tissue. Eur J Biochem 1990; 189: 251–257.
Yenice S, Davis AT, Goueli SA, Akdas A, Limas C, Ahmed K . Nuclear casein kinase 2 (CK-2) activity in human normal, benign hyperplastic, and cancerous prostate. Prostate 1994; 24: 11–16.
Landesman-Bollag E, Song DH, Romieu-Mourez R, Sussman DJ, Cardiff RD, Sonenshein GE et al. Protein kinase CK2: signaling and tumorigenesis in the mammary gland. Mol Cell Biochem 2001; 227: 153–165.
Ruzzene M, Pinna LA . Addiction to protein kinase CK2: a common denominator of diverse cancer cells? Biochim Biophys Acta 2010; 1804: 499–504.
Ahmed K, Gerber DA, Cochet C . Joining the cell survival squad: an emerging role for protein kinase CK2. Trends Cell Biol 2002; 12: 226–230.
Wang G, Ahmad KA, Ahmed K . Role of protein kinase CK2 in the regulation of tumor necrosis factor-related apoptosis inducing ligand-induced apoptosis in prostate cancer cells. Cancer Res 2006a; 66: 2242–2249.
Desagher S, Osen-Sand A, Montessuit S, Magnenat E, Vilbois F, Hochmann A et al. Phosphorylation of bid by casein kinases I and II regulates its cleavage by caspase 8. Mol Cell 2001; 8: 601–611.
Izeradjene K, Douglas L, Delaney A, Houghton J . Casein kinase II (CK2) enhances death-inducing signaling complex (DISC) activity in TRAIL-induced apoptosis in human colon carcinoma cell lines. Oncogene 2005; 24: 2050–2058.
Ravi R, Bedi A . Sensitization of tumor cells to Apo2 ligand/TRAIL-induced apoptosis by inhibition of casein kinase II. Cancer Res 2002; 62: 4180–4185.
Wang G, Unger G, Ahmad KA, Slaton JW, Ahmed K . Downregulation of CK2 induces apoptosis in cancer cells—a potential approach to cancer therapy. Mol Cell Biochem 2005; 274: 77–84.
Wang G, Ahmad KA, Unger G, Slaton JW, Ahmed K . CK2 signaling in androgen-dependent and -independent prostate cancer. J Cell Biochem 2006b; 99: 382–391.
Wang G, Ahmad KA, Harris NH, Ahmed K . Impact of protein kinase CK2 on inhibitor of apoptosis proteins in prostate cancer cells. Mol Cell Biochem 2008; 316: 91–97.
Popescu M, Gurel Z, Ronni T, Song C, Hung KY, Payne KJ et al. Ikaros stability and pericentromeric localization are regulated by protein phosphatase 1. J Biol Chem 2009; 284: 13869–13880.
Ruzzene M, Penzo D, Pinna LA . Protein kinase CK2 inhibitor 4,5,6,7-tetrabromobenzotriazole (TBB) induces apoptosis and caspase-dependent degradation of haematopoietic lineage cell-specific protein 1 (HS1) in Jurkat cells. Biochem J 2002; 364: 41–47.
Sarno S, Reddy H, Meggio F, Ruzzene M, Davies SP, Donella-Deana A et al. Selectivity of 4,5,6,7-tetrabromobenzotriazole, an ATP site-directed inhibitor of protein kinase CK2 (casein kinase-2). FEBS Lett 2001; 496: 44–48.
Di Maira G, Brustolon F, Bertacchini J, Tosoni K, Marmiroli S, Pinna LA et al. Pharmacological inhibition of protein kinase CK2 reverts the multidrug resistance phenotype of a CEM cell line characterized by high CK2 level. Oncogene 2007; 26: 6915–6926.
Izeradjene K, Douglas L, Delaney A, Houghton J . Influence of casein kinase II in tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in human rhabdomyosarcoma cells. Clin Cancer Res 2004; 10: 6650–6660.
Slaton JW, Unger GM, Sloper DT, Davis AT, Ahmed K . Induction of apoptosis by antisense CK2 in human prostate cancer xenograft model. Mol Cancer Res 2004; 2: 712–721.
McDonnell MA, Abedin MJ, Melendez M, Platikanova TN, Ecklund JR, Ahmed K et al. Phosphorylation of murine caspase-9 by the protein kinase casein kinase 2 regulates its cleavage by caspase-8. J Biol Chem 2008; 283: 20149–20158.
Krippner-Heidenreich A, Talanian RV, Sekul R, Kraft R, Thole H, Ottleben H et al. Targeting of the transcription factor Max during apoptosis: phosphorylation-regulated cleavage by caspase-5 at an unusual glutamic acid residue in position P1. Biochem J 2001; 358: 705–715.
Matsumoto G, Wada K, Okuno M, Kurosawa M, Nukina N . Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol Cell 2011; 44: 279–289.
Bren GD, Pennington KN, Paya CV . PKC-zeta-associated CK2 participates in the turnover of free IkappaBalpha. J Mol Biol 2000; 297: 1245–1258.
Chang S, Kim JH, Shin J . p62 forms a ternary complex with PKCzeta and PAR-4 and antagonizes PAR-4-induced PKCzeta inhibition. FEBS Lett 2002; 510: 57–61.
Ahmad KA, Wang G, Slaton J, Unger G, Ahmed K . Targeting CK2 for cancer therapy. Anticancer Drugs 2005; 16: 1037–1043.
Schneider P . Production of recombinant TRAIL and TRAIL receptor: Fc chimeric proteins. Methods Enzymol 2000; 322: 325–345.
Filhol O, Nueda A, Martel V, Gerber-Scokaert D, Benitez MJ, Souchier C et al. Live-cell fluorescence imaging reveals the dynamics of protein kinase CK2 individual subunits. Mol Cell Biol 2003; 23: 975–987.
Kochin V, Imanishi S Y, Eriksson JE . Fast track to a phosphoprotein sketch—MALDI-TOF characterization of TLC-based tryptic phosphopeptide maps at femtomolar detection sensitivity. Proteomics 2006; 6: 5676–5682.
Acknowledgements
We thank Professor Sistonen (Turku Centre of Biotechnology, Finland), Dr. Downward (London Research institute, England), Dr. Robert (University of Manchester, England) for plasmid gifts, and Professor Schneider (University of Lausanne, Switzerland) for recombinant TRAIL. This work was supported by grants from the ‘Ligue Nationale Contre le Cancer’ and its committees in ‘Nièvre‘and ‘Sâone et Loire’ and in part by funding from the NCI/NIH (grant CA60872 to VMR) AH is recipient of a post-doctoral fellowship from the ‘Conseil regional de Bourgogne’, AdT has a post-doctoral fellowship from the ‘Association pour la Recherche contre le Cancer’. CG’s team has the ‘Label’ from the ‘Ligue Nationale contre le Cancer’.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Author contributions
Conception and design: AdT, AH; development of methodology: VK, OF, GJ; acquisition of data: AH, KI, HM; analysis and interpretation of data: AdT; technical and material support: PR; writing, review and/or revison of the manuscript: VM, OM; study supervision: CG, VR, EE.
Supplementary Information accompanies this paper on Cell Death and Disease website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
de Thonel, A., Hazoumé, A., Kochin, V. et al. Regulation of the proapoptotic functions of prostate apoptosis response-4 (Par-4) by casein kinase 2 in prostate cancer cells. Cell Death Dis 5, e1016 (2014). https://doi.org/10.1038/cddis.2013.532
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2013.532
Keywords
This article is cited by
-
Prostate apoptosis response-4 and tumor suppression: it’s not just about apoptosis anymore
Cell Death & Disease (2021)
-
Coordinated targeting of CK2 and KIT in gastrointestinal stromal tumours
British Journal of Cancer (2020)
-
Inhibition of AKT promotes FOXO3a-dependent apoptosis in prostate cancer
Cell Death & Disease (2016)
-
A journey beyond apoptosis: new enigma of controlling metastasis by pro-apoptotic Par-4
Clinical & Experimental Metastasis (2016)
