
Abstract
Initially identified as an inhibitor of transforming growth factor (TGF)-β mainly owing to its ability to bind TGF-β receptor type I and abrogate TGF-β-driven signaling, Smad7 can interact with additional intracellular proteins and regulate TGF-β-independent pathways, thus having a key role in the control of neoplastic processes in various organs. Genome-wide association studies have shown that common alleles of Smad7 influence the risk of colorectal cancer (CRC), even though the contribution of Smad7 in colon carcinogenesis is not fully understood. In this study, we assessed the expression and role of Smad7 in human and mouse models of sporadic CRC. We document a significant increase of Smad7 in human CRC relative to the surrounding nontumor tissues and show that silencing of Smad7 inhibits the growth of CRC cell lines both in vitro and in vivo after transplantation into immunodeficient mice. Knockdown of Smad7 results in enhanced phosphorylation of the cyclin-dependent kinase (CDK)2, accumulation of CRC cells in S phase and enhanced cell death. Smad7-deficient CRC cells have lower levels of CDC25A, a phosphatase that dephosphorylates CDK2, and hyperphosphorylated eukaryotic initiation factor 2 (eIF2)α, a negative regulator of CDC25 protein translation. Consistently, knockdown of Smad7 associates with inactivation of eIF2α, lower CDC25A expression and diminished fraction of proliferating cells in human CRC explants, and reduces the number of intestinal tumors in Apcmin/+ mice. Altogether, these data support a role for Smad7 in sustaining colon tumorigenesis.
Similar content being viewed by others


Main
Colorectal cancer (CRC) is the third most common form of malignancy and the second leading cause of cancer-related death in the western world.1 CRC rates are also increasing in countries adopting western life style and dietary habits.2 CRC develops in a step-wise manner from normal mucosa, to adenomatous polyps, to carcinoma, a complex and multistage process characterized by accumulation of genetic changes, each conferring a selective growth advantage to tumor cells.3 These changes ultimately result in uncontrolled cell growth and clonal tumor development. Genome-wide association studies have shown that common alleles of Smad7 influence the risk of sporadic CRC.4, 5 Patients with deletion of Smad7 have a significantly better prognosis than patients with two copies of this gene, whereas increased copy numbers of Smad7 are associated with a significantly worse prognosis.6 The Smad7 gene encodes an intracellular protein, which interacts with the transforming growth factor (TGF)-β type I receptor, targeting it for degradation in the proteasome, thus inhibiting TGF-β1-induced phosphorylation of Smad2/Smad3.7 Nonetheless, Smad7 can interact with additional intracellular proteins and control cell function via TGF-β1-independent pathways. Overexpression of Smad7 supports the development of pancreatic cancer, skin cancer, scleroderma and lung cancer.8 In contrast, Smad7 inhibits metastasis of human melanoma cells to bone, metastasis of human breast cancer to the lung or liver, and the growth of hepatocellular carcinoma.8 Thus, a better understanding of the contribution of Smad7 in specific cancer cell types could help optimize personalized strategies to combat neoplastic growth and progression. In this study, we examined the role of Smad7 in CRC to determine if Smad7 could be a molecular target for pharmacological intervention.
Results
Smad7 is upregulated in human CRC and positively controls CRC cell growth
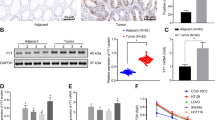
Smad7 expression was analyzed by western blotting in proteins extracted from 14 matched pairs of human CRC and adjacent tissues. Smad7 was significantly increased in CRC samples, compared with nontumor mucosa (Figure 1a). Immunohistochemistry confirmed these results and showed that Smad7 was overexpressed predominantly by cancer cells (Figure 1b). Elevated levels of Smad7 were also seen in four different CRC cell lines (Figure 2a and Supplementary Figures 1a and b). In this context, it is noteworthy that expression of Smad7 in CRC cell lines was not different from that seen in HepG2 cells (Supplementary Figure 1a), which are known to express elevated levels of Smad7. To determine whether Smad7 had a role in CRC proliferation, Smad7 expression was inhibited in HCT-116 and DLD-1 cells with a specific antisense oligonucleotide (Figure 2b and Supplementary Figure 1c). Twenty-four-hour treatment of cells with the Smad7 antisense significantly inhibited CRC growth, regardless of whether the cells were cultured in the presence (not shown) or absence of serum (Figure 2c). Smad7 knockdown caused a progressive accumulation in the number of cells in S phase, and a decrease of cells in G0/G1 (Figure 2d), suggesting that DNA replication is inhibited following Smad7 knockdown. In eukaryotic cells, the initiation of DNA replication and entry into mitosis are orchestrated by cyclin/cyclin-dependent kinase (CDK) complexes, formed at specific stages of the cell cycle, and whose activities are necessary for progression through S phase and mitosis.9 CDK1 is necessary for mitotic onset, and shares an overlapping role with CDK2 in controlling S phase initiation. The enzymatic activity of CDKs is modulated by both inhibitory and activating phosphorylation. In particular, the CDK–cyclin complex is inhibited by phosphorylation of Thr-14 and Tyr-15 residues within the ATP-binding pocket.9 Inhibition of Smad7 in both HCT-116 and DLD-1 cells resulted in a marked phosphorylation of CDK2 on Thr-14 and Tyr-15 residues (Figure 2e).

Smad7 is overexpressed in human sporadic CRC. (a) Total proteins extracted from both tumoral (T) and nontumoral (NT) areas of two patients with sporadic CRC were evaluated for Smad7 expression by western blotting. β-actin was used as loading control. The figure is representative of seven separate experiments in which similar results were obtained. Right inset. Quantitative analysis of Smad7/β-actin protein ratio in total extracts of T and NT tissues taken from fourteen patients with sporadic CRC, as measured by densitometry scanning of western blots. Values are expressed in arbitrary units (a.u.) and indicate the mean±S.E.M. of all experiments (n=14). (b) Smad7 immunostaining in specimens taken from tumoral (T) and non-tumoral (NT) areas of one patient with sporadic CRC. The figure is representative of six separate experiments in which sections of six CRC patients were analyzed. Staining with isotype control IgG is also shown. The scale bars are 100 μm

Smad7 sustains CRC cell proliferation and survival. (a) Total proteins extracted from two CRC cell lines (i.e., DLD-1 and HCT-116) and from normal colonic epithelial cells (CEC) were evaluated for Smad7 expression by western blotting. β-actin was used as loading control. One of two representative experiments in which similar results were obtained is shown (b) Smad7 antisense oligonucleotide (AS) downregulates Smad7 protein expression in HCT-116 cells. HCT-116 cells were transfected with either Smad7 sense oligonucleotide (S) (2 μg/ml) or increasing doses (0.5–2 μg/ml) of Smad7 AS. Smad7 and β-actin were analyzed by western blotting. One of three representative experiments is shown. (c) Smad7 AS inhibits the growth of CRC cell lines. HCT-116 and DLD-1 cells were either left untreated (Untr) or transfected with Smad7 AS or Smad7 S. The percentage of proliferating cells was evaluated by flow cytometry. Data indicate mean±S.D. of four experiments (HCT-116: Smad7 S-transfected cells versus Smad7 AS-transfected cells, *P<0.001; DLD-1: Smad7 S-transfected cells versus Smad7 AS-transfected cells, †P<0.001). Right insets. Representative histograms of HCT-116 cells transfected with SMAD7 S and AS. Numbers above lines indicate the percentages of proliferating cells. (d) Smad7 AS induces HCT-116 cells to arrest in S phase of cell cycle. Cells were either left untreated (Untr) or transfected with Smad7 S or AS. After 24 h cells were washed with PBS and cultured for further 24 h. Cell cycle distribution was assessed by flow cytometry. Values are the percentages of cells in the different phases of cell cycle and indicate mean±S.D. of four experiments. A significant increase in the number of cells that accumulate in S phase (*P<0.001) and a significant decrease in the number of cells in G0/G1 (**P<0.01) was seen in Smad7 AS-transfected cells as compared with Smad7 S-transfected cells. (e) Smad7 AS enhances the expression of p-CDK2. HCT-116 cells were either left untreated (Untr) or transfected with Smad7 AS or S for 24 h. p-CDK2, CDK2 and β-actin expression was analyzed by Western blotting after further 24 h of culture. One of three representative experiments in which similar results were obtained is shown. (f) Inhibition of Smad7 with AS induces HCT-116 cell death at late time points. Cells were either left untreated (Untr) or transfected with Smad7 S or AS (1 μg/ml). After 24 h cells were washed with PBS and cultured for further 24 (left panel) or 48 (middle panel) hours. Data indicate the percentage of cell death as assessed by flow cytometry analysis of Annexin V (AV) and/or propidium iodide (PI)-positive cells and are expressed as mean±S.D. of three experiments. Right panel. Representative dot-plots showing the percentages of AV- and/or PI-positive cells after 48 h
The percentage of viable cells was not changed by Smad7 inhibition at 24 h (Figure 2f, left panel), indicating that Smad7 antisense-mediated cell growth arrest was not secondary to cell death. As the block of cells in S phase is followed by induction of cell death, we next analyzed the fraction of Annexin V/propidium iodide (PI)-positive cells at later time points. As expected, accumulation of Smad7-deficient CRC cells in S phase was accompanied by increased cell death at 48 h (Figure 2f, right panel). The majority of human sporadic CRC carry mutations in genes involved in TGF-β1 signaling such that cell lines (i.e., HCT-116 and DLD-1 cells) are unresponsive to TGF-β1. Indeed, neither TGF-β1 nor a neutralizing TGF- β1 antibody changed the fraction of proliferating HCT-116 and DLD-1 cells (Supplementary Figure 2). Moreover, Smad7 antisense-induced HCT-116 and DLD-1 cell growth inhibition were altered by neither TGF-β1 nor anti-TGF-β1 antibody (Supplementary Figure 2).
Inhibition of Smad7 reduces the in vivo growth of HCT-116-derived xenografts
To confirm the anti-neoplastic effect of Smad7 antisense oligonucleotide, we tested the ability of the Smad7-antisense to inhibit tumor growth in vivo. HCT-116 cells were inoculated subcutaneously into Rag1−/− mice. After one week, the animals received a single intraperitoneal injection of either phosphate-buffered saline (PBS; CTR) or fluorescein isothiocyanate (FITC)-conjugated Smad7 antisense. Twenty-four hours later, mice were killed, tumors excised and distribution of labeled Smad7 antisense assessed by immunofluorescence. Smad7 antisense oligonucleotide was taken up by tumor cells (Figure 3a). Next, xenografts were induced as above and animals were treated intraperitoneally with either Smad7 sense or antisense (both at 100 μg/mouse daily) starting 7 days after the HCT-116 injection until killing (day 21). No weight loss was observed in either Smad7 sense- or antisense-treated groups, and all animals survived until the end of study. HCT-116-derived tumors were evident in all mice, but mice that were given Smad7 antisense oligonucleotide had a significant decrease in tumor volume (Figure 3b). Treatment of mice with Smad7 antisense downregulated Smad7 protein expression and reduced the fraction of proliferating cell nuclear antigen (PCNA)-positive cells in the tumors (Figure 3c).

Effect of Smad7 AS on the in vivo formation of HCT-116-derived xenografts. (a) Representative images showing the uptake of fluorescent-conjugated Smad7 antisense oligonucleotide (AS) in HCT-116-derived xenografts induced in Rag1−/− mice. Smad7 AS-positive cells were evaluated by immunofluorescence. The scale bars are 20 μm. One of three separate experiments is shown. (b) Representative histograms showing the volume of HCT-116-derived tumors taken from mice treated with either Smad7 AS or Smad sense oligonucleotide (S). Data indicate the mean±S.D. of two independent experiments in which at least six mice per group were analyzed. Lower panel: representative photographic images of xenografts developed in mice treated with either Smad7 AS or S. (c) Representative pictures of SMAD7- and PCNA-stained sections of xenografts taken from mice treated as indicated in b. Isotype control stainings are indicated. The scale bars are 20 μm. The scale bar in the inset is 10 μm. One of six separate experiments is shown
Silencing of Smad7 in CRC cells increases eIF2α phosphorylation thus leading to CDC25A protein downregulation
Activation of CDK2 is strictly dependent on the activity of a specialized phosphatase, CDC25, which dephosphorylates and activates CDK–cyclin complexes.10 There are three members of the human CDC25 family. CDC25A controls progression through S phase, whereas CDC25B and CDC25C are involved in the control of the transition from G2 to mitosis.10 Knockdown of Smad7 was accompanied by a marked reduction of CDC25A protein expression while CDC25B and CDC25C levels remain unchanged (Figure 4a).

Smad7 knockdown in CRC cells enhances eIF2α phosphorylation and downregulates CDC25A expression. (a) Transfection of HCT-116 cells with Smad7 antisense oligonucleotide (AS) reduces CDC25A protein expression. HCT-116 cells were either left untreated (Untr) or transfected with Smad7 AS or Smad7 sense oligonucleotide (S). After 24 h cells were washed with PBS and cultured for further 24 h. CDC25A, CDC25B and CDC25C expression was assessed by Western blotting. One of three representative experiments in which similar results were obtained is shown. (b) Smad7 AS treatment does not reduce CDC25A RNA expression. HCT-116 cells were either left untreated (Untr) or transfected with Smad7 AS or S. CDC25A transcripts were evaluated by real-time polymerase chain reaction. Levels are normalized to β-actin. Values are mean±S.D. of three experiments. Smad7 AS-transfected cells versus Smad7 S-transfected cells, *P<0.001. (c) CDC25A protein downregulation in cells treated with Smad7 AS is not reverted by proteasome inhibitors (MG115 and MG132). Representative Western blots for CDC25A and β-actin in extracts of HCT-116 cells transfected with either Smad7 S or AS and then cultured in the presence or absence of MG115 and MG132 for 24 h. One of three representative experiments in which similar results were obtained is shown. (d) Treatment of CRC cells with Smad7 AS enhances eIF2α phosphorylation and this phenomenon precedes the downregulation of CDC25A expression. Representative Western blots for p-eIF2α Ser51, eIF2α, CDC25A and β-actin in extracts of HCT-116 cells transfected with either S or AS and then cultured for the indicated time points. One of 3 three representative experiments in which similar results were obtained is shown. (e) Treatment of CRC cells with Smad7 AS reduces de novo protein synthesis. Representative western blots for puromycin and β-actin in extracts of HCT-116 cells transfected with either Smad7 S or AS and then cultured for 24 h. Where indicated, cells were pulsed with puromycin. One of three representative experiments in which similar results were obtained is shown
The intracellular levels of CDC25A can be regulated in multiple ways.11 Initially, we examined whether CDC25 protein downregulation by the Smad7 antisense resulted from changes in CDC25A transcripts. However, RNA transcripts were upregulated and not inhibited in cells treated with Smad7 antisense (Figure 4b). These data show uncoupling between CDC25A protein and RNA levels in Smad7-deficient CRC cells, suggesting that regulation of CDC25A protein expression by Smad7 occurs through a translational or posttranslational (e.g., proteasome-dependent) mechanism. So, we next determined whether proteasomal degradation was involved in the downregulation of CDC25A in Smad7-deficient cells. Treatment of CRC cells with proteasome inhibitors did not prevent Smad7 antisense oligonucleotide-induced CDC25A protein inhibition (Figure 4c). CDC25A protein expression is also regulated by the eukaryotic initiation factor 2α (eIF2α), which is essential for translation attenuation.12 The function of eIF2α is regulated by phosphorylation on Ser51, which results in eIF2α sequestration and loss of function.13 Thus, eIF2α hyperphosphorylation results in a marked decrease of CDC25A protein expression. Silencing of Smad7 in both HCT-116 and DLD-1 cells increased the Ser51 phosphorylation of eIF2α and this preceded CDC25A protein downregulation (Figure 4d and not shown). Smad7 antisense oligonucleotide-treated CRC cells also showed reduced protein synthesis compared with control cells (Figure 4e). To confirm the link between eIF2α and CDC25A in CRC cells, eIF2α function was specifically inhibited in HCT-116 and DLD-1 by salubrinal, which interferes with Ser51 dephosphorylation. This resulted in an increase of p-eIF2α and a decrease of CDC25A protein level (Supplementary Figure 3).
Blockade of Smad7 reduces the proliferation of neoplastic cells in human CRC explants and the in vivo growth of colon tumors in mice
To mechanistically link the elevated levels of Smad7 with CRC cell growth, we used two different models of colon carcinogenesis. First, Smad7 antisense was added to organ cultures of human CRC explants, and cell growth and cell cycle-related proteins were analyzed after 24 h. Immunohistochemistry showed that Smad7 antisense reduced the fraction of Smad7-, PCNA- and CDC25A-expressing CRC cells and increased the percentage of cells expressing p-eIF2α (Figure 5). Second, we tested the ability of Smad7 antisense to inhibit intestinal tumorigenicity in vivo using a spontaneous model of CRC in multiple intestinal neoplasia (MIN) mice carrying a mutation in the adenomatous polyposis coli (Apc) gene. Treatment of Apcmin/+ mice with the carcinogen azoxymethane (AOM) increases tumor incidence, number and size, particularly in the colon.14 Mice were treated intraperitoneally with AOM (10 mg/kg) once a week for 2 weeks and monitored for tumor formation. One week after the last AOM injection, mice received oral gavage of either Smad7 antisense or sense oligonucleotide (125 μg/mouse) three times a week, until killing. Some mice were given oral FITC-conjugated Smad7 antisense (125 μg/mouse) also, and killed 8 h later in order to assess intestinal uptake of the oligonucleotide (Figure 6a).

Inhibition of Smad7 with the specific Smad7 antisense oligonucleotide (AS) reduces the proliferation of neoplastic cells in human CRC explants. Representative pictures of SMAD7-, PCNA-, p-eIF2α Ser51- and CDC25A-stained sections of freshly obtained CRC explants treated with either Smad7 sense oligonucleotide (S) or AS for 24 h. Isotype control stainings are also indicated. The scale bars are 40 μm. The scale bars in the insets are 10 μm. One of five representative experiments in which similar results were obtained is shown

Smad7 downregulation significantly reduces colonic tumorigenesis in Apcmin/+ mice. (a) Orally administered fluorescent-labeled Smad7 antisense oligonucleotide (AS) in Apcmin/+ mice is taken up by both epithelial and lamina propria mononuclear cells in the colon after 8 h of administration. No relevant staining is seen in mice treated with PBS (CTR). The scale bars are 40 μm. The scale bar in the inset is 10 μm. One of three separate experiments is shown. (b) Left panels show representative endoscopic pictures and H&E-stained sections of colon tumors developed in mice treated with either Smad7 sense oligonucleotide (S) or AS. The scale bars are 100 μm. Graphs show the number of lesions and the endoscopic scoring of tumors developed in mice treated with either Smad7 S or AS. Data indicate mean±S.E.M. of three independent experiments in which five mice per group were considered (n=15). (c–e) Representative images showing Smad7- (c), CDC25A- (d) and p-eIF2α Ser51-positive cells (e) in colonic sections taken from Apcmin/+ mice treated with either Smad7 S or AS. The scale bars are 20 μm. One of five representative experiments in which similar results were obtained is shown. NT, nontumoral area; T, tumoral area
Endoscopy on day 78 showed that control mice developed multiple large tumors, whereas the number and size of tumors were reduced in the colon of Smad7 antisense oligonucleotide-treated mice (Figure 6b). Immunohistochemistry of tumor and nontumor sections showed that Smad7 was upregulated in the neoplastic areas and inhibited by antisense treatment (Figure 6c). Moreover, antisense oligonucleotide treatment reduced the levels of CDC25A (Figure 6d) and upregulated p-eIF2α (Figure 6e) in the tumors themselves.
Discussion
The development of CRC is a multifactorial process with environmental (e.g., diet, cigarette smoke exposure and microbial infection), epigenetic and genetic factors being implicated.15 Compelling evidence indicates that CRC cells are marked by enhanced activation of various intracellular signals, which ultimately promote the expression of molecules involved in cell survival/anti-apoptosis and cell cycle progression. In recent years, a better understanding of these deregulated pathways (e.g., the EGFR pathway) led to the development of therapeutic strategies, which have been employed with some success in CRC patients.16 Here, we have identified a significant increase of Smad7 in human CRC relative to surrounding nontumor tissues and showed that silencing of Smad7 suppresses CRC cell growth. This later finding was seen in human CRC cell lines, organ cultures of human CRC explants and in vivo in Apcmin/+ mice, which spontaneously develop CRC. Smad7 antisense oligonucleotide inhibited proliferation of both HCT-116 and DLD-1 cells, which are known to be unresponsive to TGF-β1. In these cultures, neither TGF-β1 nor a neutralizing TGF-β1 antibody changed the percentages of proliferating CRC cells following Smad7 inhibition, thus suggesting that the anti-mitogenic effect of Smad7 antisense oligonucleotide, at least in these cell types, is not linked to the Smad7-mediated suppression of TGF-β1/Smad2/3-associated signaling. This result is in line with previous studies showing the ability of Smad7 to either function as a TGF-β1-independent transcriptional modulator17, 18 or functionally inactivate retinoblastoma protein without blocking the activation of TGF-β type I receptor and the nuclear translocation of Smad2/3.19
Inhibition of Smad7 was associated with arrest of CRC cells in S phase of the cell cycle and a marked and specific downregulation of CDC25A, a phosphatase that controls progression of the cell cycle through the S phase via the dephosphorylation CDK2 and inhibition of CDK2–cyclin complex activity. Interestingly, inhibition of CDC25A by the Smad7 antisense oligonucleotide occurred at protein and not at RNA level, thus implying a posttranscriptional control of CDC25A by Smad7. In this context, it is noteworthy that CDC25 RNA expression was even more pronounced in Smad7-deficient cells in comparison with control cells, probably as a result of a compensatory mechanism, which enhances CDC25 gene transcription following CDC25 protein downregulation. Treatment of CRC cells with the proteasome pathway inhibitors did not alter the effect of Smad7 antisense oligonucleotide on CDC25A protein inhibition, arguing against the possibility that reduction of CDC25A in Smad7-deficient cells is secondary to enhanced protein degradation. As inhibition of Smad7 in CRC cell lines was associated with reduced protein synthesis, we hypothesized that CDC25 protein downregulation could reflect changes in the function of molecules involved in CDC25 protein synthesis. So, we focused the next experiments on eIF2α, because it has a critical role in regulating translation attenuation. Indeed, it has been reported that, under various stressful conditions, hyperphosphorylation of eIF2α determines a sustained shutoff of protein synthesis and suppression of CDC25A protein expression.12 Time–course studies showed that inhibition of Smad7 enhanced phosphorylation of eIF2α and this preceded CDC25A protein downregulation. Studies are now ongoing to ascertain the basic mechanism by which knockdown of Smad7 leads to eIF2α hyperphosphorylation as well as to determine the factors/mechanisms implicated in the control of Smad7 expression in CRC cells.
The demonstration that Smad7 is overexpressed in the cancer cells within the CRC mass and that silencing of Smad7 abrogates CRC cell growth confirms and expands on earlier reports of the role of Smad7 in negatively influencing prognosis of patients with oesophageal, gastric, pancreatic and intestinal malignancies.8 These findings, together with the demonstration that the Smad7 antisense oligonucleotide used for inhibiting Smad7 in CRC cells was safe and well-tolerated in patients with active Crohn’s disease,20 pave the way for CRC to be examined as a potential clinical target indication for anti-Smad7-based therapy.
Materials and Methods
Patients and samples
Paired tissue samples were taken from the tumoral area and the macroscopically unaffected, adjacent, colonic mucosa of 14 patients who underwent colon resection for sporadic CRC at the Tor Vergata University Hospital (Rome, Italy) and used for Smad7 evaluation by western blotting. Additional samples were taken from the non tumoral area of three CRC patients and used for isolating primary intestinal epithelial cells. Moreover, samples taken from the tumoral area of five CRC patients were used for organ culture experiments. Finally, samples taken from the tumoral area and the macroscopically unaffected, adjacent, colonic mucosa of six CRC patients were used for assessing Smad7 by immunohistochemistry. All patients received neither radiotherapy nor chemotherapy prior to undergoing surgery. The human studies were approved by the local Ethics committee and each patient gave written informed consent.
Animals
Rag1-deficient (Rag1−/−) and Apcmin/+ mice were obtained from The Jackson Laboratory (Bar Harbor, ME, USA) and maintained in filter-topped cages on autoclaved food and water at the Plaisant animal facility (Rome, Italy). Mice were routinely tested (every 3 months) for health status and infections according to the Federation of European Laboratory Animal Science Associations (FELASA) guidelines. Mice were negative for all pathogens included in this protocol. Mice were also negative for the presence of Helicobacter hepaticus and Helicobacter bilis. All animal experiments were approved by the local Institutional Animal Care and Use Committee.
Inhibition of Smad7 by Smad7 antisense oligonucleotide
Details concerning the Smad7 antisense oligonucleotide and the Smad7 sense oligonucleotide used in these studies have been previously reported.21 Because human and murine Smad7 genes are homologous, mouse Smad7 was inhibited using the same Smad7 antisense oligonucleotide shown to be effective in inhibiting human Smad7.
Cell culture
All reagents were from Sigma-Aldrich (Milan, Italy) unless specified. The human CRC cell lines HCT-116, DLD-1 and HT-29, and the human hepatocellular carcinoma cell line HepG2 were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). The human CRC cell line HT-115 was obtained from the European Collection of Cell Cultures (ECACC, Salisbury, UK). Cells were maintained in McCoy’s 5A (HCT-116, HT-29), DMEM (HT-115, HepG2) and RPMI1640 (DLD-1) medium, all supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin (P/S) (all from Lonza, Verviers, Belgium) and 50 μg/ml gentamycin in a 37 °C, 5% CO2, fully humidified incubator. Cell lines were recently authenticated by STR DNA fingerprinting using the PowerPlex 18D System kit according to the manufacturer’s instructions (Promega, Milan, Italy). The STR profiles of all the cell lines matched the known DNA fingerprints. To investigate the role of Smad7 in CRC cell growth and death, HCT-116 and DLD-1 cells were either left untreated or transfected with either Smad7 sense oligonucleotide or Smad7 antisense oligonucleotide for 24 h using Opti-MEM medium and Lipofectamine 2000 reagent (both from Life Technologies, Milan, Italy) according to the manufacturer’s instructions. Cells were then washed with PBS and recultured with the respective fresh medium containing 0.05% bovine serum albumin (BSA) for further 12–48 h. The efficiency of the transfection was determined by flow cytometry using a fluorescein-labeled Smad7 antisense oligonucleotide. To evaluate the role of the proteasome pathway in the Smad7 antisense oligonucleotide-mediated regulation of CDC25A, cells were transfected with either Smad7 sense oligonucleotide or Smad7 antisense oligonucleotide (both used at 1 μg/ml) for 24 h and cultured in the presence of the proteasome inhibitors MG115 and MG132 (used at 25 μM, both from EMD Millipore, Milan, Italy) or DMSO (vehicle) for further 24 h. To test the effect of the eIF2α inhibitor Salubrinal (EMD Millipore) on p-eIF2α and CDC25A expression, CRC cells were cultured in the presence of either 25 μM Salubrinal or DMSO (vehicle) for 6 and 24 h, respectively. To assess whether Smad7 knockdown modulates translation intensity, Smad7 sense oligonucleotide and Smad7 antisense oligonucleotide-transfected cells were pulsed with 10 μg/ml puromycin (P8833, diluted in PBS) 10 min before harvesting at 37° and 5% CO2. Cells were harvested 24 h after the end of transfection. Total proteins were extracted and subjected to immunoblot analysis using an anti-puromycin antibody (clone 12D10, EMD Millipore). To evaluate the role of TGF-β1 signaling pathway in the Smad7 antisense oligonucleotide-mediated CRC cell growth inhibition, cells were transfected with either Smad7 sense oligonucleotide or Smad7 antisense oligonucleotide (both used at 1 μg/ml) for 24 h and then cultured in the presence or absence of TGF-β1 (2 ng/ml, R&D Systems, Minneapolis, MN, USA) and anti-TGF-β1 (1 μg/ml, R&D Systems) for further 24 h. The percentage of proliferating cells was assessed by flow cytometry. To rule out the possibility that the Smad7 antisense oligonucleotide-mediated CRC cell growth inhibition was secondary to culture medium-related stress signals, CRC cells were transfected with either Smad7 sense oligonucleotide or Smad7 antisense oligonucleotide for 24 h and cultured with fresh medium supplemented with 2% FBS for further 24 h. The percentage of proliferating cells was then assessed by flow cytometry.
Flow cytometry evaluation of Smad7 in CRC cells
HCT-116 and DLD-1 cells were harvested, washed in PBS and fixed in IC fixation buffer (eBioscience, San Diego, CA, USA) at 4 °C for at least 3 h. Cells were then washed twice in permeabilization buffer 1 × (eBioscience) and stained with either Smad7 (Biorbyt, Cambridge, UK) or rabbit IgG control antibody (R&D Systems) for 1 h at 4 °C. Cells were then washed and labeled with a FITC-conjugated anti-rabbit secondary antibody (Abcam, Cambridge, UK) for 30 min at 4 °C. Smad7-positive cells were evaluated by flow cytometry (FACSCalibur, Becton Dickinson, Milan, Italy).
Analysis and quantification of cell proliferation, cycle and death
Cell proliferation was assessed using carboxyfluorescein diacetate succinimidyl ester (CFSE; Molecular Probes, Eugene, OR, USA), which covalently binds cell components to yield a fluorescence that is divided equally between daughter cells at each division. Briefly, cells were either left untreated or transfected with either Smad7 sense oligonucleotide or Smad7 antisense oligonucleotide (both used at 1 μg/ml). After 24 h, cells were washed with PBS and incubated with CFSE according to the manufacturer’s instructions. After 30 min, the medium was removed and fresh medium containing 0.05% BSA was added for further 24 h. At the end, cells were collected, washed twice with PBS, and then incubated with 5 μg/ml of PI for 15 min at 4 °C in the dark. CFSE- and/or PI-positive cells were determined by flow cytometry and the data were analyzed using ModFit LT 2.0 (Verity Software House, Inc., Topsham, ME, USA).
For analysis of cell cycle distribution, cells were either left untreated or transfected with either Smad7 sense oligonucleotide or Smad7 antisense oligonucleotide (both used at 1 μg/ml). After 24 h, cells were washed with PBS and recultured with fresh medium containing 0.05% BSA for further 24 h. At the end, cells were collected, washed twice with PBS, fixed in 70% cold ethanol, and stored at −20 °C for at least 3 h. Samples were washed twice with PBS, treated with 20 μg/ml RNase A and 100 μg/ml PI, and analyzed by flow cytometry using ModFit LT 2.0.
To score cell death, cells were either left untreated or transfected with either Smad7 sense oligonucleotide or Smad7 antisense oligonucleotide (both used at 1 μg/ml). After 24 h, cells were washed with PBS and re-cultured with fresh medium containing 0.05% BSA for further 24–48 h. Cells were then collected, washed twice in PBS, stained with FITC–annexin V (AV, 1 : 100 final dilution, Immunotools, Friesoyte, Germany) according to the manufacturer’s instructions and incubated with 5 μg/ml PI for 30 min at 4 °C, and their fluorescence was measured using FL-1 and FL-2 channels of FACSCalibur using Cell Quest Pro software. Viable cells were considered as AV−/PI− cells, apoptotic cells as AV+/PI− cells, whereas secondary necrotic cells were characterized by AV+/PI+ staining.
Isolation of primary colonic epithelial cells
Freshly obtained colonic mucosal samples were washed with Hank’s balanced salt solution (HBSS) Ca+-and Mg+-free (Lonza) containing dithiothreitol (DTT) to remove mucus, finely minced and incubated in HBSS containing 5 mM EDTA, 1% P/S and 50 μg/ml gentamycin at 37 °C for 30 min. Primary colonic epithelial cells were obtained by collecting the supernatants from HBSS/EDTA washing and then purified by negative selection using CD45 magnetic beads (Miltenyi Biotec, Calderara di Reno, Italy).
Western blotting
Protein extracts were prepared and run as previously described.22 Blots were incubated with Smad7 (1 μg/ml, R&D Systems), p-CDK2 (Thr-14/Tyr-15) (sc-28435-R), CDK2 (sc-6248), CDC25A (sc-7389), CDC25B (sc-5619), CDC25C (sc-13138), eIF2-α antibodies (1 : 500 final dilution, all from Santa Cruz Biotechnology, Santa Cruz, CA, USA), p-eIF2α Ser51 (1 : 1000 final dilution, Cell Signaling Technology, Danvers, MA, USA), followed by a secondary antibody conjugated to horseradish peroxidase (Dako, Milan, Italy). After analysis, each blot was stripped and incubated with a mouse–anti-human monoclonal β-actin antibody (1 : 5000 final dilution) to ascertain equivalent loading of the lanes. Computer-assisted scanning densitometry (Total Lab, AB.EL Science-Ware Srl, Rome, Italy) was used to analyze the intensity of the immunoreactive bands.
RNA extraction, cDNA preparation and Real-time PCR
Total RNA was extracted from cells by using TRIzol reagent (Life Technologies). A constant amount of RNA (1 μg/sample) was retrotranscribed into complementary DNA (cDNA), and 1 μl of cDNA/sample was then amplified using the following conditions: denaturation 1 min at 95 °C, annealing 30 s at 58 °C for CDC25A or 30 s at 60 °C for β-actin, followed by 30 s of extension at 72 °C. Human CDC25A primers were as follows: FWD: 5′-GTA CAA AGA GGA GGA AGA GC-3′; REV: 5′-GAT GCC AGG GAT AAA GAC TG-3′. Real-time-PCR was performed using the IQ SYBR Green Supermix (Bio-Rad Laboratories, Milan, Italy) and RNA expression was calculated relative to the housekeeping β-actin gene on the base of the ΔΔCt algorithm.
Organ culture
CRC explants were placed on Millicell inserts (EMD Millipore) in six-well plates containing RPMI 1640 medium supplemented with 10% FBS, 1% P/S and 50 μg/ml gentamycin in the presence of either Smad7 sense oligonucleotide or Smad7 antisense oligonucleotide (both used at 1 μg/ml) for 24 h. The culture was performed in an organ culture chamber at 37 °C in a 5% CO2/95% O2 atmosphere.
Effect of Smad7 antisense oligonucleotide on the in vivo formation of HCT-116-derived tumors
HCT-116 (5 × 105 cells) were resuspended in 500 μl BD Matrigel Basement Membrane Matrix High Concentration (BD Biosciences, Milan, Italy) and injected subcutaneously into the flank of 8-week-old female Rag1−/− mice, whose fur was shaved and depilated. After 1 week, mice with similar tumor volume, determined by caliper measurement, were divided into two groups and received daily intraperitoneal injection of either Smad7 sense oligonucleotide or Smad7 antisense oligonucleotide (both at 100 μg/mouse resuspended in 300 μl PBS). Mice were sacrificed at day 21. Tumors were excised, photographed, their volume calculated according to the following formula: 1/2 × (short diameter) × (long diameter) × (height) and finally included in OCT (Thermo Scientific, Rockford, IL, USA) for histochemistry experiments. To assess the effective uptake of the Smad7 antisense oligonucleotide in the growing tumors, in preliminary experiments mice received a single intraperitoneal injection of either FITC-conjugated Smad7 antisense oligonucleotide (used at 100 μg/mouse) or PBS (control). Twenty-four hours later mice were killed, tumors excised and FITC-conjugated Smad7 antisense oligonucleotide distribution assessed by immunofluorescence.
Experimental model of sporadic CRC
Cohoused 5-week-old female Apcmin/+ mice received intraperitoneal injections of 10 mg/kg AOM once a week for 2 weeks in order to selectively increase colon tumorigenesis as previously reported.14 One week after the last AOM injection, mice were randomly divided in two groups and treated with either Smad7 sense or antisense oligonucleotide (both used at 125 μg/mouse in 100 μl of pH 9.5 bicarbonate solution) by oral gavage every other day until killing (day 80). Mice were endoscopically screened 2 days before being killed using the high-resolution endoscopic system and protocols described by Becker et al.23 To assess the in vivo colonic uptake of the orally administered Smad7 antisense oligonucleotide, in preliminary experiments mice were given a single administration of either FITC-conjugated Smad7 antisense oligonucleotide (used at 125 μg/mouse) or PBS (control) by oral gavage. Eight hours later, mice were killed, colon collected, cut in sections and FITC-conjugated Smad7 antisense oligonucleotide distribution assessed by immunofluorescence.
Endoscopic procedures
Colonoscopy was performed in a blinded manner for monitoring of tumorigenesis using the Coloview high-resolution mouse endoscopic system (Karl-Storz, Tuttlingen, Germany). Tumors observed during endoscopy were counted to obtain the overall number of lesions. Furthermore, tumor sizes of all tumors in a given mouse were scored, yielding the tumor score as described by Becker et al.23 Tumor sizes were graded as follows: grade 1 (very small but detectable tumor), grade 2 (tumor covering up to one-eighth of the colonic circumference), grade 3 (tumor covering up to a quarter of the colonic circumference), grade 4 (tumor covering up to half of the colonic circumference) and grade 5 (tumor covering more than half of the colonic circumference).23
Immunofluorescence
Cryosections were prepared from HCT-116-derived xenografts developed in Rag1−/− mice that were intraperitoneally injected with either FITC-conjugated Smad7 antisense oligonucleotide or PBS (control) and from colonic samples of Apcmin/+ mice that were given either FITC-conjugated Smad7 antisense oligonucleotide or PBS (control) by oral gavage. Sections were counterstained with DAPI (Life Technologies) and evaluated using a fluorescence microscope (Olympus, Milan, Italy).
Immunohistochemistry
Tissue sections of surgical specimens taken from patients with sporadic CRC were cut, deparaffinized, dehydrated through xylene and ethanol, and incubated with a mouse anti-human Smad7 antibody (R&D Systems) for 1 h at room temperature. Immunoreactive cells were visualized using MACH4 Universal HRP-Polymer kit with DAB (Biocare Medical, Concord, CA, USA), according to the manufacturer’s instructions, and lightly counterstained with hematoxylin. Isotype control sections were prepared under identical immunohistochemical conditions, as described above, replacing the primary antibody with a purified, normal mouse IgG2b control antibody (R&D Systems).
Cryosections prepared from HCT-116-derived xenografts and human CRC explants were stained with Smad7 (R&D Systems), CDC25A (Biorbyt) p-eIF2α Ser51 (Cell Signaling Technology). Isotype control-stained sections were prepared under identical immunohistochemical conditions replacing the primary antibody with either a mouse or rabbit normal IgG control antibody (R&D Systems). Proliferating cells were evaluated using a PCNA staining kit (ZYMED Laboratories, Carlsbad, CA, USA) according to the manufacturer’s instructions. Negative control-stained sections were prepared under identical immunohistochemical conditions omitting the primary antibody. Colonic cryosections of Apcmin/+ mice were stained either with hematoxylin and eosin (H&E) or with Smad7 (Biorbyt), CDC25A (Biorbyt) and p-eIF2α Ser51 (Cell Signaling Technology). Proliferating cells were evaluated using a PCNA staining kit (ZYMED Laboratories) according to the manufacturer’s instructions.
Statistical analysis
Differences between groups were compared using the two-tailed Student’s t-test. P-values <0.05 were considered statistically significant.
Abbreviations
- TGF:
-
transforming growth factor
- CRC:
-
colorectal cancer
- CDK:
-
cyclin-dependent kinase
- eIF2:
-
eukaryotic initiation factor 2
- AV:
-
annexin V
- PI:
-
propidium iodide
- PBS:
-
phosphate-buffered saline
- FITC:
-
fluorescein isothiocyanate
- PCNA:
-
proliferating cell nuclear antigen
- APC:
-
adenomatous polyposis coli
- MIN:
-
multiple intestinal neoplasia
- AOM:
-
azoxymethane
- EGFR:
-
epidermal growth factor receptor
- FBS:
-
fetal bovine serum
- BSA:
-
bovine serum albumin
- DMSO:
-
dimethyl sulfoxide
- CFSE:
-
carboxyfluorescein diacetate succinimidyl ester
- DAPI:
-
4′,6-diamidino-2-phenylindole
- DAB:
-
3,3′-diaminobenzidine
- DTT:
-
dithiothreitol
References
Center MM, Jemal A, Smith RA, Ward E . Worldwide variations in colorectal cancer. CA Cancer J Clin 2009; 59: 366–378.
Vainio H, Miller AB . Primary and secondary prevention in colorectal cancer. Acta Oncol 2003; 42: 809–815.
Fearon ER, Vogelstein B . A genetic model for colorectal tumorigenesis. Cell 1990; 61: 759–767.
Slattery ML, Herrick J, Curtin K, Samowitz W, Wolff RK, Caan BJ et al. Increased risk of colon cancer associated with a genetic polymorphism of SMAD7. Cancer Res 2010; 70: 1479–1485.
Thompson CL, Plummer SJ, Acheson LS, Tucker TC, Casey G, Li L . Association of common genetic variants in SMAD7 and risk of colon cancer. Carcinogenesis 2009; 30: 982–986.
Boulay JL, Mild G, Lowy A, Reuter J, Lagrange M, Terracciano L et al. SMAD7 is a prognostic marker in patients with colorectal cancer. Int J Cancer 2003; 104: 446–449.
Nakao A, Afrakhte M, Moren A, Nakayama T, Christian JL, Heuchel R et al. Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling. Nature 1997; 389: 631–635.
Briones-Orta MA, Tecalco-Cruz AC, Sosa-Garrocho M, Caligaris C, Macias-Silva M . Inhibitory Smad7: emerging roles in health and disease. Curr Mol Pharmacol 2011; 4: 141–153.
Shapiro GI . Cyclin-dependent kinase pathways as targets for cancer treatment. J Clin Oncol 2006; 24: 1770–1783.
Kristjansdottir K, Rudolph J . Cdc25 phosphatases and cancer. Chem Biol 2004; 11: 1043–1051.
Busino L, Chiesa M, Draetta GF, Donzelli M . Cdc25A phosphatase: combinatorial phosphorylation, ubiquitylation and proteolysis. Oncogene 2004; 23: 2050–2056.
Tomko RJ Jr, Lazo JS . Multimodal control of Cdc25A by nitrosative stress. Cancer Res 2008; 68: 7457–7465.
Wek RC, Jiang HY, Anthony TG . Coping with stress: eIF2 kinases and translational control. Biochem Soc Trans 2006; 34: 7–11.
Issa AY, Volate SR, Muga SJ, Nitcheva D, Smith T, Wargovich MJ . Green tea selectively targets initial stages of intestinal carcinogenesis in the AOM-ApcMin mouse model. Carcinogenesis 2007; 28: 1978–1984.
Fearon ER . Molecular genetics of colorectal cancer. Annu Rev Pathol 2011; 6: 479–507.
Waldner MJ, Neurath MF . The molecular therapy of colorectal cancer. Mol Aspects Med 2010; 31: 171–178.
Pulaski L, Landstrom M, Heldin CH, Souchelnytskyi S . Phosphorylation of Smad7 at Ser-249 does not interfere with its inhibitory role in transforming growth factor-beta-dependent signaling but affects Smad7-dependent transcriptional activation. J Biol Chem 2001; 276: 14344–14349.
Gronroos E, Hellman U, Heldin CH, Ericsson J . Control of Smad7 stability by competition between acetylation and ubiquitination. Mol Cell 2002; 10: 483–493.
Boyer Arnold N, Korc M . Smad7 abrogates transforming growth factor-beta1-mediated growth inhibition in COLO-357 cells through functional inactivation of the retinoblastoma protein. J Biol Chem 2005; 280: 21858–21866.
Monteleone G, Fantini MC, Onali S, Zorzi F, Sancesario G, Bernardini S et al. Phase I clinical trial of Smad7 knockdown using antisense oligonucleotide in patients with active Crohn's disease. Mol Ther 2012; 20: 870–876.
Monteleone G, Kumberova A, Croft NM, McKenzie C, Steer HW, MacDonald TT . Blocking Smad7 restores TGF-beta1 signaling in chronic inflammatory bowel disease. J Clin Invest 2001; 108: 601–609.
Monteleone G, Franchi L, Fina D, Caruso R, Vavassori P, Monteleone I et al. Silencing of SH-PTP2 defines a crucial role in the inactivation of epidermal growth factor receptor by 5-aminosalicylic acid in colon cancer cells. Cell Death Differ 2006; 13: 202–211.
Becker C, Fantini MC, Wirtz S, Nikolaev A, Kiesslich R, Lehr HA et al. In vivo imaging of colitis and colon cancer development in mice using high resolution chromoendoscopy. Gut 2005; 54: 950–954.
Acknowledgements
The study was supported by the Fondazione ‘Umberto Di Mario ONLUS’, Rome, Associazione Italiana per la Ricerca sul Cancro (IG-13049 to GM and MFAG-12108 to CS) and Giuliani SpA, Milan, Italy.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
GM has filed a patent related to the treatment of inflammatory bowel diseases with Smad7 antisense oligonucleotides. The remaining authors declare no conflict of interest.
Additional information
Edited by G Melino
Supplementary Information accompanies this paper on Cell Death and Disease website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Stolfi, C., De Simone, V., Colantoni, A. et al. A functional role for Smad7 in sustaining colon cancer cell growth and survival. Cell Death Dis 5, e1073 (2014). https://doi.org/10.1038/cddis.2014.49
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2014.49
Keywords
This article is cited by
-
Chimeric TβRII-SE/Fc overexpression by a lentiviral vector exerts strong antitumoral activity on colorectal cancer-derived cell lines in vitro and on xenografts
Cancer Gene Therapy (2024)
-
PEACOCK: a machine learning approach to assess the validity of cell type-specific enhancer-gene regulatory relationships
npj Systems Biology and Applications (2023)
-
Use of patient-derived explants as a preclinical model for precision medicine in colorectal cancer: A scoping review
Langenbeck's Archives of Surgery (2023)
-
Identification and characterization of colorectal-cancer-associated SNPs on the SMAD7 locus
Journal of Cancer Research and Clinical Oncology (2023)
-
Exploiting gene dependency to inform drug development for multiple myeloma
Scientific Reports (2022)

{kind=link}
{kind=link}
{kind=link}