
Abstract
Cancer patients receiving anthracycline-based chemotherapy are at risk to develop life-threatening chronic cardiotoxicity with the pathophysiological mechanism of action not fully understood. Besides the most common hypothesis that anthracycline-induced congestive heart failure (CHF) is mainly caused by generation of reactive oxygen species, recent data point to a critical role of topoisomerase II beta (TOP2B), which is a primary target of anthracycline poisoning, in the pathophysiology of CHF. As the use of the only clinically approved cardioprotectant dexrazoxane has been limited by the FDA in 2011, there is an urgent need for alternative cardioprotective measures. Statins are anti-inflammatory and anti-oxidative drugs that are clinically well established for the prevention of cardiovascular diseases. They exhibit pleiotropic beneficial properties beyond cholesterol-lowering effects that most likely rest on the indirect inhibition of small Ras homologous (Rho) GTPases. The Rho GTPase Rac1 has been shown to be a major factor in the regulation of the pro-oxidative NADPH oxidase as well as in the regulation of type II topoisomerase. Both are discussed to play an important role in the pathophysiology of anthracycline-induced CHF. Therefore, off-label use of statins or novel Rac1 inhibitors might represent a promising pharmacological approach to gain control over chronic cardiotoxicity by interfering with key mechanisms of anthracycline-induced cardiomyocyte cell death.
Similar content being viewed by others
Facts
-
Anthracycline-induced cardiotoxicity is an unresolved major problem in cancer therapy.
-
Rho GTPases have nuclear functions that might influence the doxorubicin-induced DNA damage response.
-
Rho GTPases interfere with two of the supposed main mechanisms of anthracycline-induced cardiotoxicity: generation of reactive oxygen species and topoisomerase II poisoning.
-
A preventive treatment with statins or specific inhibitors of Rho GTPases are promising pharmaceutical approaches to alleviate anthracycline-induced cardiotoxicity.
Open questions
-
Does topoisomerase II-mediated mtDNA damage play a role in anthracycline-induced cardiotoxicity?
-
How do Rho GTPases regulate topoisomerase II activity?
-
Are nuclear functions of Rho GTPases involved in the anthracycline-induced DNA damage response?
-
What is more relevant for chronic cardiotoxicity: the generation of reactive oxygen species or topoisomerase II beta poisoning?
-
The cardioprotective effects of statins in anthracycline-based chemotherapy needs verification in randomized prospective studies.
Anthracyclines are potent chemotherapeutics, which are used for the treatment of a broad spectrum of malignancies.1 The supposed antineoplastic mechanism is the induction of DNA damage, predominantly in the S- and G2-phase of proliferating cells.2 Anthracyclines such as epirubicin or doxorubicin inhibit type II topoisomerases, thereby causing DNA double-strand breaks (DSBs),3 which represent a strong apoptotic stimulus if left unrepaired.4, 5 In addition, anthracyclines intercalate into DNA, form bulky DNA adducts and DNA crosslinks, which interfere with DNA replication and transcription. They can damage DNA directly due to the generation of reactive oxygen species (ROS), leading to oxidized nucleotides, base mismatches, point mutations and DNA single-strand breaks. The production of ROS also causes a DNA damage-independent stimulation of cytotoxic mechanisms, resulting from oxidative protein modifications, in particular, lipid peroxidation.6, 7 Last, anthracyclines interfere with DNA helicase activity and DNA strand separation.8
Unfortunately, the geno- and cytotoxic effects evoked by anthracyclines are not limited to tumour cells. Adverse effects of anthracycline-based chemotherapy on normal tissue can be severe and dose limiting.9 Patients are at considerable risk to develop acute and chronic cardiotoxicity with the mechanism(s) involved under debate. Acute cardiotoxicity during therapy is rare, not dose-related and often associated with pre-existing cardiac diseases.10, 11 More common and by far more serious is chronic cardiotoxicity, which can occur weeks or even years after treatment. In >50% of patients who survived childhood leukaemia echocardiographic abnormalities are detectable after anthracycline-based therapeutic regimen.12 Chronic cardiotoxicity usually manifests during the first year after the end of anthracycline treatment but can also occur decades later.13, 14, 15, 16, 17, 18, 19
Breast cancer patients treated with the anthracycline-derivative doxorubicin showed decreased left ventricular ejection fraction (LVEF) when the cumulative doxorubicin dose exceeded 350 mg/m2 (refs 20, 21). In a retrospective study comprising 4000 patients, 88 developed congestive heart failure (CHF) after treatment. The incidence ranged from 0.1 to 7.0% depending on the cumulative dose (<400–550 mg/m2). In patients receiving ≥700 mg/m2 the incidence was 18%.22 In consequence of these data, reduction of the maximum cumulative dose to 550 mg/m2 was recommended, which unfortunately is accompanied by reduced anti-tumour efficiency. Notably, even when adhering to the suggested maximum doxorubicin dose, ~26% of patients are at risk to develop CHF.9 A cohort study of adult survivors of childhood leukaemia found that these patients have a twofold higher risk of developing CHF when having received a cumulative dose of <250 mg/m2 and a fivefold higher risk when >250 mg/m2 were applied (compared to patients who received a non-anthracycline-based therapy).23
Mechanisms of anthracycline-induced cardiotoxicity
A hallmark of anthracycline-induced chronic cardiotoxicity is the reduction of left ventricular wall thickness due to the loss of cardiomyocytes, resulting in restricted LVEF.24 Anthracycline-induced cardiomyocyte cell death is likely mediated through caspase-3-related apoptotic pathways activated by p53 and/or TNF-signalling.25 The trigger stimuli ultimately causing cardiomyocyte cell death are uncertain and controversially discussed. Suggested mechanisms for the development of cardiomyopathy include accumulation of toxic metabolites (e.g., doxorubinicol), autophagy, production of peroxynitrite and ROS, TOP2B inhibition, and disruption of mitochondrial homeostasis/integrity.8, 26 A detailed discussion of each of these putative factors is beyond the scope of this review. Only the most common original hypothesis and the latest most topical hypothesis, for which new evidence was found recently, are highlighted here.
The ‘ROS and iron’ hypothesis
Cardiomyocytes are described to be particular sensitive to ROS. It is believed that their anti-oxidative defence systems are saturated by endogenous oxidative metabolism.27 Additional ROS exposure could exhaust this system faster as compared to other tissues. Anthracyclines could cause such additional ROS generation because they are reductively activated to a semiquinone radical, which undergoes redox cycling, thereby producing superoxide (O2−) and hydrogen peroxide (H2O2). In the presence of iron, anthracyclines can form Fe3+–anthracycline complexes, which further catalyse the conversion of H2O2 to various ROS species, including cytotoxic hydroxyl radicals (OH−; Fenton’s reaction). Redox cycling from a single anthracycline molecule could lead to the accumulation of ROS7 (Figure 1). This so-called ‘ROS and iron’ hypothesis is highly popular to explain the cardiotoxicity of anthracyclines, but the postulated relevance for chronic cardiotoxicity lacks convincing experimental evidence. Besides, it is under debate whether ROS formation plays a predominant pathophysiological role when clinically relevant (low) doses of anthracyclines are used. Importantly, most of the in vitro and in vivo studies demonstrating ROS formation after doxorubicin treatment used supra-clinical doses that are magnitudes higher than the concentrations detected in the serum of patients during chemotherapy.6, 8, 28 In vitro studies with rat cardiomyocytes showed that anthracycline-induced ROS formation was very low or even below detection limit at clinically relevant doses (≤2 μM doxorubicin).29, 30

Model of doxorubicin-mediated generation of reactive oxygen species (ROS) by redox cycling and Fenton‘s reaction. Doxorubicin produces ROS by redox cycling at its semiquinone structure. In the Fenton‘s reaction, iron catalyses the generation of OH radicals (•OH) from H2O2. SOD, superoxide dismutase; adapted from Simunek et al.6 and Stêrba et al.7
Transgenic mice that overexpress human manganese-dependent superoxide dismutase (MnSOD) in the heart are protected from doxorubicin-induced acute cardiotoxicity, pointing to a critical role of ROS as inducer of acute cardiac damage and MnSOD as major detoxification mechanism.31 However, the doses of 10–25 mg/kg doxorubicin used in this study are equivalent to the 1–2.5-fold LD50 of mice.32 There are plenty of studies addressing acute toxic anthracycline effects after high dosage and short follow-up time that claim the relevance of ROS, anti-oxidative drugs or ROS-detoxifying enzymes.6 Yet, none of them proves a pivotal role of ROS in the pathophysiology of chronic cardiotoxicity observed under clinical relevant settings of anthracycline-based anti-cancer therapy. Furthermore, it is hard to distinguish whether ROS generation is the primary mode of cardiotoxic action of anthracyclines or a secondary phenomenon resulting from cardiotoxicity. For instance, enhanced ROS levels can also result from mitochondrial dysfunction or endoplasmatic reticulum stress caused by accumulation of misfolded proteins.33 Another point to be considered is that anthracyclines exhibit a strong auto-fluorescence.34, 35 As common ROS detection methods are fluorescence-based, this can lead to the misinterpretation of results, in particular when high doses (>2 μM) of anthracyclines are used. Down to the present day, it is unclear whether or not generation of ROS occurs in relevant amounts under standard anthracycline treatment regimens, undermining its role as main trigger of chronic cardiotoxicity.
Application of antioxidants and iron-chelating agents led to inconclusive results in animal experiments. Importantly, no protection from anthracycline-induced cardiotoxicity was found in patients treated with N-acetyl cysteine or combinations of antioxidants.6 By contrast, the iron-chelating drug dexrazoxane (ICRF-187), which is structurally related to EDTA, effectively alleviated anthracycline-induced cardiotoxicity in numerous animal and human studies. Yet, the precise cytoprotective mechanism of dexrazoxane remains unclear and studies with other iron chelators failed to show cardioprotection.6 Apart from iron chelation, which inhibits the production of hydroxyl radicals resulting from Fenton’s reaction, dexrazoxane also inhibits type II topoisomerases36, 37, 38, 39, 40, 41 by a catalytic mechanisms that is different from anthracycline poisoning and does not comprise cleavable complex formation39, 40, 42 (Figure 2). In light of this, it is feasible that dexrazoxane does not solely bind free iron, but may also lower the amount of the accessible main substrate (i.e., topoisomerase II) for anthracycline poisoning.39, 40 This could result in reduced cardiotoxicity but may also impair the anti-tumour efficiency of anthracyclines. Early clinical studies with dexrazoxane dispelled those concerns, as tumour protection was not observed.43, 44 However, two recent studies claimed that dexrazoxane bears the risk to affect anthracycline’s anti-tumour efficiency and to increase the incidence of secondary tumours.45, 46

Inhibition of type II topoisomerases by anthracyclines and dexrazoxane. To resolve tensions in supercoiled DNA, topoisomerase II induces a DNA double-strand break (DSB) to allow passage of another DNA strand, which is delivered from the open clamp (orange) (a). The DNA strand passes downwards through the DSB of the first strand and subsequently the open strand is resealed again (c). If anthracyclines bind at the region of interplay between the opened DNA and the topoisomerase II (b), the resealing of the DSB is disabled and a so-called cleavable complex consisting of anthracycline, DNA and topoisomerase persists until it is proteasomally degraded (d). Consequently, the opened DNA is left over containing a highly cytotoxic DSB that can trigger a pro-apoptotic DNA damage response. Binding of dexrazoxane to the ATPase domain of the topoisomerase keeps its clamp in a closed state (c), which prevents anthracycline binding as well as entrance into another enzymatic cycle (transition from the state illustrated under c to the state illustrated under a). A, anthracycline; DEX, dexrazoxane; DSB, DNA double-strand break; adapted from Lyu et al.54 and Vejpongsa and Yeh39
Type II topoisomerase poisoning
Proliferating cells depend on topoisomerase II alpha (TOP2A) for chromosome condensation and segregation.47, 48 The beta form (TOP2B) predominantly contributes to transcription.49 Knockout of the alpha form revealed that the beta form may substitute for its function in chromosome condensation, but cannot complement for chromosome segregation.50 Proliferating cells show an enhanced expression of the alpha form in S- and G2-phase, whereas its amount is low in G0/G1 phase cells.51 The beta isoform is constitutively expressed independent of the cell cycle.52 Type II topoisomerases cut DNA double-strands to allow strand passage for unwinding and unknotting of supercoiled DNA (Figure 2). Typically, these cuts are resealed right after strand passage.39, 40, 53 Upon poisoning of topoisomerase by anthracyclines, resealing of the transient DSB becomes impossible. The anthracycline stabilizes the temporary cleavage complex consisting of DNA and topoisomerase to a permanently stalled complex. Presumably, this complex is proteasomally degraded and DNA DSBs remain.54 Like stated above, accumulation of non-repaired DSBs represents a strong apoptotic stimulus4, 5, 55 that can trigger cardiomyocyte death.
Cardiomyocytes lack the expression of TOP2A but harbour relative high levels of TOP2B as compared to other tissues.56 This might render them particular vulnerable to topoisomerase II poisons such as anthracyclines. In line with this, Lyu et al.54 showed that knockout of TOP2B protects mouse embryonal fibroblasts from anthracycline-induced cytotoxicity. In addition, the expression level of TOP2B correlates with anthracycline-induced apoptosis in peripheral blood cells.57 Most importantly, mice with a cardiac-specific top2b knockout exhibit less cardiotoxicity following anthracyline treatment.58 The top2b knockout mice revealed lower levels of DSBs, no decline in LVEF after doxorubicin treatment and showed reduced mitochondrial dysfunction. These results support the hypothesis that the cardioprotective effects seen with dexrazoxane are at least partially dependent on its interference with topoisomerase II.
Another interesting aspect related to TOP2 poisoning by anthracyclines is the fact that both TOP2A and TOP2B were found in the mitochondria recently.59, 60 As the beta form is predominantly expressed in the heart, it is likely the relevant form in cardiac mitochondria too.60 Anthracyclines can induce mitochondrial damage due to uncoupling of the electron transport chain, disruption of mitochondrial membrane potential and production of ROS, especially in combination with the mitochondrial iron metabolism.61 As anthracyclines induce DSBs in genomic DNA via topoisomerase II poisoning, it is feasible that a similar genotoxic mechanism also occurs in mitochondrial DNA (mtDNA) and contributes to anthracycline-induced cytotoxicity. As CHF occurs at late times after anthracycline treatment, a long-lasting mitochondrial dysfunction (and concomitant sustained increase in the steady-state levels of ROS) may add to the pathophysiology of chronic cardiotoxicity. Indeed, studies in mice and rabbits demonstrated that chronic anthracycline-induced heart failure correlates with imbalances in mitochondrial mass and reduced expression of genes regulating mitochondrial homeostasis.62, 63 However, whether TOP2-dependent DSBs in mtDNA or TOP2-independent oxidative mtDNA base lesions cause depletion of mtDNA content is hard to discriminate.
Recently, Jean et al.64 showed that mice treated with a chemically modified doxorubicin derivative, which exclusively enters mitochondria but not the nucleus, lack cardiotoxicity, arguing against a dominant contribution of mtDNA damage in the development of cardiomyopathy following anthracycline treatment. The authors suggest that altered nuclear expression of genes encoding mitochondrial biogenesis factors such as NRF1 and TFAM, is more relevant for the onset of cardiotoxicity than direct mitochondrial damage. Recently, Khiati et al.65 showed that mice lacking mitochondrial type I topoisomerase (mtTOP1), are hypersensitive to anthracyclines. As mtTOP1 is required to maintain efficient mtDNA production after mitotoxic insults, this finding supports the idea of anthracycline-induced mtDNA damage being of relevance for the pathophysiology of chronic cardiotoxicity. Regardless of whether or not TOP2 poisoning of mitochondrial or genomic DNA plays a key role in anthracycline-evoked cardiotoxicity, TOP2B seems to be a promising pharmacological target for its prevention. Pharmacological inhibition of TOP2B during the time period anthracyclines are applied might protect non-proliferating cardiac cells, whereas TOP2A would still be poisoned in proliferating tumour cells.
Current treatment and prevention strategies
As the use of dexrazoxane for the prevention of anthracycline-induced CHF was restricted by the FDA in 2011, alternative cardio-preventive measures, especially for cardioprophylaxis in children, are urgently needed. Cardioprotective preventive measures used today are limitation of the cumulative anthracycline dose and application of ‘less cardiotoxic’ derivatives or liposomal encapsuled forms. The therapy of anthracycline-induced CHF matches that of anthracycline-independent CHF and includes ACE inhibitors, beta-blockers, diuretics, aldosterone blockers and cardiac glycosides.66 The therapeutic benefit closely depends on the improvement of left ventricular function.67, 68 The ACE inhibitor enalapril and the beta-blocker carvedilol are the most effective drugs in achieving normalization of anthracycline-caused decrease in LVEF.69 Due to these promising therapeutic results, a preventive study was initiated. In the OVERCOME trail, 42% of the patients showed a preservation of LVEF by prophylactic enalapril and carvedilol treatment, and 10% of patients responded partially.70 However, these cardioprotective effects are less marked than in the case of dexrazoxane-based prevention.
Statins in the prevention of cardiovascular disease
Statins (HMG-CoA reductase inhibitors) are first choice for the treatment of hypercholesterolemia since 1987.71 Elevated blood cholesterol levels are a major risk factor for atherosclerosis.72 Following cholesterol accumulation on blood vessel walls (so-called plaques) and macrophage infiltration, the constriction of blood vessels is promoted.73 Statins competitively inhibit the conversion of HMG-CoA to mevalonate, a precursor for cholesterol synthesis,74 thereby eventually reducing the cell’s intrinsic cholesterol synthesis and favouring the uptake of serum LDL cholesterol.75 As the mevalonate pathway is essential for the synthesis of numerous isoprenoids, statins have multiple so-called pleiotropic effects that are independent of cholesterol biosynthesis.76, 77
Non-lipid-lowering effects of statins
Statins prevent from cholesterol-related plaque generation, reduce already formed plaques, prevent their breakage and attenuate thrombocyte-mediated vessel blockage.78, 79 In addition, they exhibit anti-inflammatory properties and reduce deleterious heart tissue remodelling.80, 81 These beneficial effects cannot be explained solely by changes in LDL/HDL ratios. Rather, they were mostly traced back to the inhibition of small Ras homologous (Rho) GTPases.75 Rho GTPases are molecular switches and signal transducers on the inner cell membrane that transmit extracellular stimuli to mitogen-activated protein kinases and transcription factors.82, 83 Inhibition of the HMG-CoA reductase causes depletion of the cellular pool of isoprene precursors, which are also essential for C-terminal prenylation and subsequent membrane localization of Rho proteins84, 85, 86, 87 (Figure 3). Rho GTPases are famous as key regulators of the actin-cytoskeleton, but exhibit manifold functions beyond that.88, 89, 90 For instance, RhoA is involved in the regulation of endothelial cell migration, blood vessel tension and thrombocyte aggregation.91, 92, 93 The RhoA downstream effector Rho-associated coiled-coil forming protein kinase (ROCK) regulates the activity of the endothelial nitric oxide synthase (eNOS).94 RhoA/ROCK inhibition or treatment with statins stabilizes eNOS mRNA as well as AKT-mediated activation of eNOS.95, 96 The release of nitric oxide reduces endothelial tension, vascular inflammation, thrombocyte aggregation and thus works against atherosclerosis.92, 95, 97 Rac1 is part of the NADPH oxidase complex, which initiates pro-inflammatory processes.98, 99 In endothelial cells, Rac1 is a major regulator of migration, adhesion and controls vascularization.100 Taken together, attenuation of Rho GTPase signalling seems to contribute to the anti-atherosclerotic properties of statins and might also be of relevance beyond the maintenance of cardiovascular health.

Indirect inhibition of Rho GTPase signaling by HMG-CoA reductase inhibitors (statins). Non-cholesterol-related beneficial effects of statins mainly rest on the indirect inhibition of Rho GTPases. The statin-mediated depletion of the intracellular pool of isoprene precursors causes reduced anchorage of Rho proteins on the inner cell membrane, which impairs their activation by GEFs on this particular location. Depicted are selected cellular functions regulated by Rho GTPases that might be relevant for the heart. CHF, congestive heart failure; CR, cellular receptor; DDR, DNA damage response; FPP, farnesyl pyrophosphate; GAP, GTPase activating protein; GDP, guanosine diphosphate; GEF, guanine nucleotide exchange factor; GGP, geranylgeranyl pyrophosphate; GTP, guanosine triphosphate; NOS, nitric oxide synthase; Rho, Ras homologous proteins; ROS, reactive oxygen species; adapted from Fritz et al.155
A quite novel aspect is the possible role of Rho GTPases in the regulation of the DNA damage response (DDR).101, 102, 103, 104 The DDR is a complex and fine-tuned network of signalling cascades that are involved in recognition of DNA damage, DNA repair, cell cycle progression, cell death and survival.4, 5, 105 Surprisingly, the RhoA-specific guanine nucleotide exchange factor (GEF) Net1 was found in the nucleus,103 implicating nuclear functions of RhoA. GEFs are a group of proteins that mediate GTP-binding and thereby the activation of Rho GTPases106, 107 (Figure 3). For Rac1, cell cycle-dependent nuclear translocation was shown and associated with mitosis.108 Moreover, it was shown that prevention of Rac1 prenylation by statins promotes Rac1 translocation to the nucleus,108 pointing to statin-mediated loss of cytosolic function of Rac1 going along with a gain of nuclear function. Recent data suggest that nuclear Rac1 might be involved in the regulation of topoisomerase II activity and the DDR.102, 104, 109 The latter is supported by the finding that oxidized DNA bases can act as Rac1 GEFs.110 Taken together, inhibition of Rho proteins such as RhoA or Rac1 by statins also modulates mechanisms of the DDR independent of cholesterol metabolism. The pleiotropic, cardioprotective characteristics of statins and their favourable tolerability make them promising candidates for the prevention and/or treatment of anthracycline-induced cardiotoxicity.
Statins and anthracycline-induced cardiotoxicity
In 2000 Felezko et al. tested the anti-tumour potential of lovastatin if combined with doxorubicin in mouse xenograft models.111 The statin improved the anti-tumour efficiency of the anthracycline. In addition, co-treated mice showed less troponin T release, pointing to cardioprotective effects of the statin. This finding implies that statins foster a win–winsituation in anthracycline-based chemotherapy: sensitizing tumour cells while protecting the heart. Indeed, statins are described to sensitize different tumour entities against various chemotherapeutics in rodent models112, 113, 114, 115, 116, 117 and are useful in chemoprevention of colorectal cancer.118
In rats, rosuvastatin was more potent to protect from doxorubicin-induced delayed cardiotoxicity than carvedilol.119 The beneficial effects seemed to be independent of the anti-oxidative properties of the statin. By contrast, Riad et al. suggested both anti-oxidative and anti-inflammatory effects of statins to contribute to cardioprotection. The statin enhanced SOD2 levels, reduced caspase-3-mediated apoptosis and mitigated cardiac inflammation following doxorubicin treatment.120 However, their experimental set-up was designed for the molecular analysis of acute toxic effects shortly after high doses of doxorubicin and statin. Transferring these results to the chronic situation is complicated. Noteworthy, certain statins show anti-oxidative capacity (e.g., atorvastatin, simvastatin or fluvastatin)121 and activate anti-oxidative Keap1/Nrf2 signalling.122 Nrf2 is responsible for the expression of genes coding for anti-oxidative factors (i.e., SOD, GST or GSH).123 Like stated above, the small Rho GTPase Rac1 is a key regulator of the NADPH oxidase complex. This complex is a major factor in the defence against endo-parasites but is also involved in the regulation of endothelial functions and vascular tone.124, 125 Immune cells such as macrophages use this complex to produce superoxide anions that are released to the intercellular space. The resulting oxidative stress can be a driver of chronic inflammation with deregulated macrophage activity.126, 127 Mice with impaired Nox2 NADPH oxidase complex are less sensitive to anthracycline-induced cardiotoxicity, pointing to a crucial role of this Rac1-regulated enzyme in the pathology of cardiotoxicity evoked by anthracyclines.128 Moreover, a Rac1 knockout in cardiomyocytes of mice prevents angiotensin II-induced cardiac hypertrophy, which also involves NADPH oxidase activation.129 It is tempting to speculate that statins may counteract cardiomyocyte injury by inhibition of Rac1-driven pro-oxidative mechanisms. Yet, one should keep in mind that the cardioprotective effect of prototypical antioxidants against anthracycline-induced injury as observed in animal models was inconsistent and, moreover, antioxidants were ineffective in clinical trials.7
Regarding their anti-inflammatory properties, statins are described to inhibit nuclear translocation of Nf-kappaB by RhoA/ROCK inhibition, in vitro.130, 131 RhoA, Cdc42 and Rac1 stimulate the transcriptional activity of Nf-kappaB by favouring phosphorylation of IkappaB alpha.132 Accordingly, specific inhibition of these Rho GTPases by clostridial toxins attenuates LPS-stimulated induction of IL6/TNF-alpha as well as Nf-kappaB translocation.133 As Nf-kappaB functions as pro-inflammatory transcription factor and is regulated in a Rho-dependent manner, it is feasible that statins interfere with anthracycline-induced cardiac inflammation by inhibition of Nf-kappaB signalling as well as interference with NADPH oxidase. In addition, statins are known to influence TGFbeta/SMAD/CTGF signalling as well as STAT signalling, which both play important roles in fibrosis and cancer development.134, 135, 136, 137 Taken together, statins hold the potential to ameliorate anthracycline-induced pro-oxidative, inflammatory and/or fibrotic cardiac responses due to inhibition of Rho-GTPase signalling.
In vitro experiments revealed geno- and cytoprotective effects of lovastatin following treatment of non-transformed immortalized and primary cells with doxorubicin or etoposide.102, 113, 130, 138 Both drugs have in common that they poison type II topoisomerases, leading to the induction of DSBs. This geno-protective nature of lovastatin points to a link between isoprene precursor depletion (which attenuates Rho GTPase signalling) and the activity of type II topoisomerases. This might be of particular relevance, as catalytic inhibition of TOP2B with dexrazoxane, sobuzoxane (MST-16) and merbarone protects cardiomyocytes from anthracycline-induced cytotoxicity.41 In some of these studies, the protective effects of the statin (lowered DSBs levels, attenuated DDR and reduced apoptotic cell death) were mimicked by specific inhibition of Rac1, employing small-molecule inhibitors or clostridial toxins.102, 113 The predominant role of Rac1 in the promotion of doxorubicin-induced DSB formation was confirmed in experiments using the Rac1-specific inhibitor EHT1864 and employing a set of immortalized cell lines.139 Rac1 inhibition reduced doxorubicin-induced DNA damage formation and response in a cell-type-specific manner, as measured on the basis of S139 phosphorylation of histone H2AX (γH2AX, a well-established surrogate marker of DSBs) and using the comet assay (detection of DNA single-strand breaks and DSBs on single cell level). The geno-protection occurred in a p53-independent manner. This is worth mentioning as the role of p53 in anthracycline-induced cardiomyocyte apoptosis is still contradictory and enigmatic.26, 140, 141, 142, 143, 144, 145 Extensive analyses with inhibitors of different Rac1-regulated protein kinases pointed to JNK, ERK, PI3K, PAK and CK1 to be involved in doxorubicin-induced DDR. In addition, Rac1 inhibition reduced phosphorylation of TOP2A on positions S1106 and S1247, pointing to a possible role of Rac1 in the regulation of type II topoisomerase. This finding can be seen in context with the experiments of Lyu et al. and Zhang et al. who showed cardioprotection from doxorubicin exposure, when type II topoisomerases were inhibited or knocked out in vitro and in vivo.54, 58 The connection between Rac1 and type II topoisomerases is still elusive but underpinned by findings of Sandrock et al.109 who showed nuclear Rac1 to co-precipitate with type II topoisomerases.
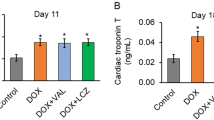
Huelsenbeck et al.113 demonstrated that a statin co-treatment attenuates acute anthracycline-induced cardiotoxicity in BALB/c mice as mirrored by reduced mRNA levels of pro-fibrotic and pro-inflammatory cytokines. It also protected from doxorubicin-induced sub-acute cardiac damage. In a similar study, atorvastatin protected mice from doxorubicin-induced DNA damage, lipid peroxidation and glutathione depletion.146 Using wistar rats, the cardioprotective potential of rosuvastatin (0.5 and 2.0 mg/kg per day; i.p.) was compared to that of carvedilol (1 mg/kg per day; i.p.).147 The rats were pre-treated with each drug for 1 month, received a single high dose of doxorubicin (30 mg/kg; i.p.) and were analysed 24 h later. Both drugs similarly prevented doxorubicin-induced increase in blood pressure and caspase-3 protein levels in cardiomyocytes. Like mentioned above, a similar study comparing the protective effects of rosuvastatin and carvedilol 4 weeks after multiple low doses of doxorubicin identified the statin as more potent cardioprotectant than carvedilol.119
Yoshida et al.142 suggested that doxorubicin-induced cardiotoxicity is mediated by oxidative DNA damage followed by ATM/p53-regulated apoptosis. In this study, mice were treated with doxorubicin (6 mg/kg; i.p.) once a week for 4 weeks and co-treated with pitavastatin (3 mg/kg per day). In line with the aforementioned in vitro studies of Huelsenbeck et al.,113 where rat cardiomyocytes (H9c2) were employed, Yoshida et al. also found protective effects of a Rac1 inhibition following anthracycline treatment in H9c2 cells. They assigned the protective effect of Rac1 inhibition to its role in NADPH oxidase regulation and oxidative stress and, in line with other data,148 suggested that statin-mediated effects are mainly related to inhibition of Rac1 signalling. Accordingly, a cardiac rac1 knockout protected mice from acute anthracycline-induced cardiotoxicity in a ROS-dependent as well as -independent manner.149 Moreover, mice with liver-specific rac1 knockout are protected from acute anthracycline-triggered DSB formation in hepatocytes too.150
Most studies investigating anthracycline effects under situation of inhibition of Rho GTPases point to Rac1-regulated oxidative stress or Rac1-regulated topoisomerase II functions as major factors in acute or sub-acute anthracycline-induced cardiotoxicity. Yet, protection from chronic cardiotoxicity would be clinically more relevant than prevention of acute adverse effects of anthracyclines. Only few animal experiments addressed the possible usefulness of statins in chronic anthracycline-evoked cardiotoxicity so far. In one of those studies, an increase in cardiac pro-inflammatory and pro-fibrotic mRNA levels as well as mitochondrial hyperproliferation was found following anthracycline treatment.63 Echocardiographical analyses showed a decrease in the left ventricular posterior wall diameter in doxorubicin-treated mice. These adverse cardiac effects were diminished in animals co-treated with lovastatin. Taken together, statins have the potential to alleviate acute, sub-acute and chronic cardiotoxicity following anthracycline treatment in vivo. Due to their pleiotropic effects, statins might counteract the complex dose- and time-dependent mechanisms underlying the cardiac toxicity of anthracyclines. Whereas specific RhoA and/or Rac1 inhibitory drugs are not clinically approved, statins represent Rho inhibitory drugs that are clinically well established, well tolerated and characterized by a wide therapeutic window.
A retrospective clinical cohort study from 2012 found a significant lower risk of heart failure in breast cancer patients receiving statins during the therapy.151 Furthermore, recent meta-study demonstrated that statins are at least equally potent as dexrazoxane, beta-blockers or angiotensin antagonists in the prevention of anthracycline-induced cardiotoxicity.152 Unfortunately, there are only a few small-scaled prospective studies available that focused on cytoprotective effects of statins in patients receiving anthracycline-based therapies. In one of these studies, 20 patients undergoing anthracycline-based chemotherapy were compared to 20 patients who were co-treated with high doses of atorvastatin (40 mg/kg) during chemotherapy with doxorubicin or idarubicin for up to 6 months.153 In the statin co-treated group, no significant change in LVEF was observed, whereas the group that did not receive the statin showed a significant decrease in LVEF (~63% baseline versus ~ 55% after therapy). In another study that included 14 patients who received statins during an anthracycline-based chemotherapy and 37 patients who did not,154 the statin-treated group showed no significant change in LVEF, whereas those not receiving the statin had a mean LVEF decline of ~7%. Certainly, there is a need of additional randomized prospective studies comprising higher number of patients to validate these cardioprotective effects of statins in anthracycline-based chemotherapy in humans.
Conclusion and perspective
Prevention and treatment of anthracycline-induced cardiotoxicity with enalapril, carvedilol and statins are conceivable strategies to reduce the cardiovascular risk. Statins exhibit anti-inflammatory as well as anti-fibrotic properties and interfere with at least two of the suggested main mechanisms involved in anthracycline-caused cardiotoxicity, (i) the generation of ROS and (ii) topoisomerase II inhibition (Figure 4). In animal studies, statins exhibited stronger cardioprotective potential than carvedilol and small-scaled clinical studies showed improvement of cardiac function by statins in anthracycline-treated cancer patients. In addition, statins may sensitize certain tumour entities to chemotherapeutics, which might even further enhance the anti-cancer efficiency of anthracycline-based therapeutic regimen while concomitantly protecting normal tissue, thus widening the therapeutic window. Many of the beneficial effects of a statin treatment could be broken down to the damping of RhoA or Rac1 signalling, making these Rho GTPases preferable targets for future chemo-preventive strategies. Animal studies addressing anthracycline-induced chronic cardiotoxicity in mice lacking Rac1 specifically in cardiomyocytes will clarify the relevance of Rac1 signalling in anthracycline-mediated cardiomyocyte death. Altogether, the pre-clinical and clinical data available so far encourage forthcoming larger phase II studies that address the clinical efficacy of statins for normal tissue protection in anthracycline-based therapeutic regimens.

Rho GTPases are involved in the regulation of main factors in anthracycline-induced cardiotoxicity. Anthracyclines induce reactive oxygen species (ROS) by redox cycling as well as Fenton’s reaction. ROS induce oxidative DNA damage and are a driver of inflammation. In addition, anthracyclines inhibit type II topoisomerases (TOP2), causing highly cytotoxic DNA double-strand breaks (DSBs), which forcefully trigger a pro-apoptotic DNA damage response and can ultimately result in cardiomyocyte cell death. Rho GTPases such as RhoA or Rac1 are known to play a role in the regulation of cell death and survival. Rac1 is described to regulate ROS production via the NADPH oxidase complex, RhoA and Rac1 participate in the regulation of inflammatory processes by (among others) altering NF-κB signaling after genotoxic insults. Oxidized guanine can act as GEF for nuclear Rac1, making this Rho GTPase a possible novel factor in the regulation of the DNA damage response. In addition, Rac1 is described to interact with type II topoisomerases
References
Weiss RB . The anthracyclines: will we ever find a better doxorubicin? Semin Oncol 1992; 19: 670–686.
Ling YH, el-Naggar AK, Priebe W, Perez-Soler R . Cell cycle-dependent cytotoxicity, G2/M phase arrest, and disruption of p34cdc2/cyclin B1 activity induced by doxorubicin in synchronized P388 cells. Mol Pharmacol 1996; 49: 832–841.
Nitiss JL . DNA topoisomerases in cancer chemotherapy: using enzymes to generate selective DNA damage. Curr Opin Investig Drugs 2002; 3: 1512–1516.
Roos WP, Kaina B . DNA damage-induced cell death: from specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett 2013; 332: 237–248.
Roos WP, Thomas AD, Kaina B . DNA damage and the balance between survival and death in cancer biology. Nat Rev Cancer 2016; 16: 20–33.
Simunek T, Sterba M, Popelova O, Adamcova M, Hrdina R, Gersl V . Anthracycline-induced cardiotoxicity: overview of studies examining the roles of oxidative stress and free cellular iron. Pharmacol Rep 2009; 61: 154–171.
Sterba M, Popelova O, Vavrova A, Jirkovsky E, Kovarikova P, Gersl V et al. Oxidative stress, redox signaling, and metal chelation in anthracycline cardiotoxicity and pharmacological cardioprotection. Antioxid Redox Signal 2013; 18: 899–929.
Gewirtz DA . A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem Pharmacol 1999; 57: 727–741.
Swain SM, Whaley FS, Ewer MS . Congestive heart failure in patients treated with doxorubicin: a retrospective analysis of three trials. Cancer 2003; 97: 2869–2879.
Kremer LC, van Dalen EC, Offringa M, Ottenkamp J, Voute PA . Anthracycline-induced clinical heart failure in a cohort of 607 children: long-term follow-up study. J Clin Oncol 2001; 19: 191–196.
Giantris A, Abdurrahman L, Hinkle A, Asselin B, Lipshultz SE . Anthracycline-induced cardiotoxicity in children and young adults. Crit Rev Oncol Hematol 1998; 27: 53–68.
Lipshultz SE, Colan SD, Gelber RD, Perez-Atayde AR, Sallan SE, Sanders SP . Late cardiac effects of doxorubicin therapy for acute lymphoblastic leukemia in childhood. N Engl J Med 1991; 324: 808–815.
Sorensen K, Levitt GA, Bull C, Dorup I, Sullivan ID . Late anthracycline cardiotoxicity after childhood cancer: a prospective longitudinal study. Cancer 2003; 97: 1991–1998.
Lipshultz SE, Lipsitz SR, Sallan SE, Dalton VM, Mone SM, Gelber RD et al. Chronic progressive cardiac dysfunction years after doxorubicin therapy for childhood acute lymphoblastic leukemia. J Clin Oncol 2005; 23: 2629–2636.
Elbl L, Hrstkova H, Chaloupka V . The late consequences of anthracycline treatment on left ventricular function after treatment for childhood cancer. Eur J Pediatr 2003; 162: 690–696.
Rathe M, Carlsen NL, Oxhoj H . Late cardiac effects of anthracycline containing therapy for childhood acute lymphoblastic leukemia. Pediatr Blood Cancer 2007; 48: 663–667.
Ganame J, Claus P, Uyttebroeck A, Renard M, D'Hooge J, Bijnens B et al. Myocardial dysfunction late after low-dose anthracycline treatment in asymptomatic pediatric patients. J Am Soc Echocardiogr 2007; 20: 1351–1358.
Pein F, Sakiroglu O, Dahan M, Lebidois J, Merlet P, Shamsaldin A et al. Cardiac abnormalities 15 years and more after adriamycin therapy in 229 childhood survivors of a solid tumour at the Institut Gustave Roussy. Br J Cancer 2004; 91: 37–44.
van Dalen EC, van der Pal HJ, Kok WE, Caron HN, Kremer LC . Clinical heart failure in a cohort of children treated with anthracyclines: a long-term follow-up study. Eur J Cancer 2006; 42: 3191–3198.
Alexander J, Dainiak N, Berger HJ, Goldman L, Johnstone D, Reduto L et al. Serial assessment of doxorubicin cardiotoxicity with quantitative radionuclide angiocardiography. N Engl J Med 1979; 300: 278–283.
Buzdar AU, Marcus C, Smith TL, Blumenschein GR . Early and delayed clinical cardiotoxicity of doxorubicin. Cancer 1985; 55: 2761–2765.
Von Hoff DD, Layard MW, Basa P, Davis HL Jr, Von Hoff AL, Rozencweig M et al. Risk factors for doxorubicin-induced congestive heart failure. Ann Intern Med 1979; 91: 710–717.
Mulrooney DA, Yeazel MW, Kawashima T, Mertens AC, Mitby P, Stovall M et al. Cardiac outcomes in a cohort of adult survivors of childhood and adolescent cancer: retrospective analysis of the Childhood Cancer Survivor Study cohort. BMJ 2009; 339: b4606.
Lenzhofer R, Dudczak R, Gumhold G, Graninger W, Moser K, Spitzy KH . Noninvasive methods for the early detection of doxorubicin-induced cardiomyopathy. J Cancer Res Clin Oncol 1983; 106: 136–142.
Thorburn A, Frankel AE . Apoptosis and anthracycline cardiotoxicity. Mol Cancer Ther 2006; 5: 197–199.
Zhang YW, Shi J, Li YJ, Wei L . Cardiomyocyte death in doxorubicin-induced cardiotoxicity. Arch Immunol Ther Exp 2009; 57: 435–445.
Thayer WS . Adriamycin stimulated superoxide formation in submitochondrial particles. Chem Biol Interact 1977; 19: 265–278.
Gianni L, Vigano L, Locatelli A, Capri G, Giani A, Tarenzi E et al. Human pharmacokinetic characterization and in vitro study of the interaction between doxorubicin and paclitaxel in patients with breast cancer. J Clin Oncol 1997; 15: 1906–1915.
Bernuzzi F, Recalcati S, Alberghini A, Cairo G . Reactive oxygen species-independent apoptosis in doxorubicin-treated H9c2 cardiomyocytes: role for heme oxygenase-1 down-modulation. Chem Biol Interact 2009; 177: 12–20.
Rharass T, Gbankoto A, Canal C, Kursunluoglu G, Bijoux A, Panakova D et al. Oxidative stress does not play a primary role in the toxicity induced with clinical doses of doxorubicin in myocardial H9c2 cells. Mol Cell Biochem 2016; 413: 199–215.
Yen HC, Oberley TD, Vichitbandha S, Ho YS St, Clair DK . The protective role of manganese superoxide dismutase against adriamycin-induced acute cardiac toxicity in transgenic mice. J Clin Invest 1996; 98: 1253–1260.
Lewis RJ, Sax NI In: Lewis RJ(ed.). Sax's Dangerous Properties of Industrial Materials. 11th edn, vol. 11. Wiley-Interscience, Wiley & Sons Inc.: Hoboken, NJ, USA, 2004, p 88.
Murphy MP . Mitochondrial dysfunction indirectly elevates ROS production by the endoplasmic reticulum. Cell Metab 2013; 18: 145–146.
Shen F, Chu S, Bence AK, Bailey B, Xue X, Erickson PA et al. Quantitation of doxorubicin uptake, efflux, and modulation of multidrug resistance (MDR) in MDR human cancer cells. J Pharmacol Exp Ther 2008; 324: 95–102.
Karukstis KK, Thompson EH, Whiles JA, Rosenfeld RJ . Deciphering the fluorescence signature of daunomycin and doxorubicin. Biophys Chem 1998; 73: 249–263.
Fortune JM, Osheroff N . Topoisomerase II as a target for anticancer drugs: when enzymes stop being nice. Prog Nucleic Acid Res Mol Biol 2000; 64: 221–253.
Hasinoff BB, Kuschak TI, Yalowich JC, Creighton AM . A QSAR study comparing the cytotoxicity and DNA topoisomerase II inhibitory effects of bisdioxopiperazine analogs of ICRF-187 (dexrazoxane). Biochem Pharmacol 1995; 50: 953–958.
Ishida R, Miki T, Narita T, Yui R, Sato M, Utsumi KR et al. Inhibition of intracellular topoisomerase II by antitumor bis(2,6-dioxopiperazine) derivatives: mode of cell growth inhibition distinct from that of cleavable complex-forming type inhibitors. Cancer Res 1991; 51: 4909–4916.
Vejpongsa P, Yeh ET . Topoisomerase 2beta: a promising molecular target for primary prevention of anthracycline-induced cardiotoxicity. Clin Pharmacol Ther 2014; 95: 45–52.
Roti Roti EC, Salih SM . Dexrazoxane ameliorates doxorubicin-induced injury in mouse ovarian cells. Biol Reprod 2012; 86: 96.
Vavrova A, Jansova H, Mackova E, Machacek M, Haskova P, Tichotova L et al. Catalytic inhibitors of topoisomerase II differently modulate the toxicity of anthracyclines in cardiac and cancer cells. PLoS One 2013; 8: e76676.
Larsen AK, Escargueil AE, Skladanowski A . Catalytic topoisomerase II inhibitors in cancer therapy. Pharmacol Ther 2003; 99: 167–181.
Swain SM, Vici P . The current and future role of dexrazoxane as a cardioprotectant in anthracycline treatment: expert panel review. J Cancer Res Clin Oncol 2004; 130: 1–7.
van Dalen EC, Caron HN, Dickinson HO, Kremer LC . Cardioprotective interventions for cancer patients receiving anthracyclines. Cochrane Database Syst Rev 2011; 6: CD003917.
Tebbi CK, London WB, Friedman D, Villaluna D, De Alarcon PA, Constine LS et al. Dexrazoxane-associated risk for acute myeloid leukemia/myelodysplastic syndrome and other secondary malignancies in pediatric Hodgkin's disease. J Clin Oncol 2007; 25: 493–500.
Salzer WL, Devidas M, Carroll WL, Winick N, Pullen J, Hunger SP et al. Long-term results of the pediatric oncology group studies for childhood acute lymphoblastic leukemia 1984-2001: a report from the children's oncology group. Leukemia 2010; 24: 355–370.
Grue P, Grasser A, Sehested M, Jensen PB, Uhse A, Straub T et al. Essential mitotic functions of DNA topoisomerase IIalpha are not adopted by topoisomerase IIbeta in human H69 cells. J Biol Chem 1998; 273: 33660–33666.
Linka RM, Porter AC, Volkov A, Mielke C, Boege F, Christensen MO . C-terminal regions of topoisomerase IIalpha and IIbeta determine isoform-specific functioning of the enzymes in vivo. Nucleic Acids Res 2007; 35: 3810–3822.
Ju BG, Lunyak VV, Perissi V, Garcia-Bassets I, Rose DW, Glass CK et al. A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science 2006; 312: 1798–1802.
Carpenter AJ, Porter AC . Construction, characterization, and complementation of a conditional-lethal DNA topoisomerase IIalpha mutant human cell line. Mol Biol Cell 2004; 15: 5700–5711.
Villman K, Stahl E, Liljegren G, Tidefelt U, Karlsson MG . Topoisomerase II-alpha expression in different cell cycle phases in fresh human breast carcinomas. Mod Pathol 2002; 15: 486–491.
Turley H, Comley M, Houlbrook S, Nozaki N, Kikuchi A, Hickson ID et al. The distribution and expression of the two isoforms of DNA topoisomerase II in normal and neoplastic human tissues. Br J Cancer 1997; 75: 1340–1346.
Liu LF, Rowe TC, Yang L, Tewey KM, Chen GL . Cleavage of DNA by mammalian DNA topoisomerase II. J Biol Chem 1983; 258: 15365–15370.
Lyu YL, Kerrigan JE, Lin CP, Azarova AM, Tsai YC, Ban Y et al. Topoisomerase IIbeta mediated DNA double-strand breaks: implications in doxorubicin cardiotoxicity and prevention by dexrazoxane. Cancer Res 2007; 67: 8839–8846.
Lips J, Kaina B . DNA double-strand breaks trigger apoptosis in p53-deficient fibroblasts. Carcinogenesis 2001; 22: 579–585.
Capranico G, Tinelli S, Austin CA, Fisher ML, Zunino F . Different patterns of gene expression of topoisomerase II isoforms in differentiated tissues during murine development. Biochim Biophys Acta 1992; 1132: 43–48.
Kersting G, Tzvetkov MV, Huse K, Kulle B, Hafner V, Brockmoller J et al. Topoisomerase II beta expression level correlates with doxorubicin-induced apoptosis in peripheral blood cells. Naunyn Schmiedebergs Arch Pharmacol 2006; 374: 21–30.
Zhang S, Liu X, Bawa-Khalfe T, Lu LS, Lyu YL, Liu LF et al. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med 2012; 18: 1639–1642.
Low RL, Orton S, Friedman DB . A truncated form of DNA topoisomerase IIbeta associates with the mtDNA genome in mammalian mitochondria. Eur J Biochem 2003; 270: 4173–4186.
Zhang H, Zhang YW, Yasukawa T, Dalla Rosa I, Khiati S, Pommier Y . Increased negative supercoiling of mtDNA in TOP1mt knockout mice and presence of topoisomerases IIalpha and IIbeta in vertebrate mitochondria. Nucleic Acids Res 2014; 42: 7259–7267.
Mordente A, Meucci E, Silvestrini A, Martorana GE, Giardina B . Anthracyclines and mitochondria. Adv Exp Med Biol 2012; 942: 385–419.
Jirkovsky E, Popelova O, Krivakova-Stankova P, Vavrova A, Hroch M, Haskova P et al. Chronic anthracycline cardiotoxicity: molecular and functional analysis with focus on nuclear factor erythroid 2-related factor 2 and mitochondrial biogenesis pathways. J Pharmacol Exp Ther 2012; 343: 468–478.
Henninger C, Huelsenbeck S, Wenzel P, Brand M, Huelsenbeck J, Schad A et al. Chronic heart damage following doxorubicin treatment is alleviated by lovastatin. Pharmacol Res 2015; 91: 47–56.
Jean SR, Tulumello DV, Riganti C, Liyanage SU, Schimmer AD, Kelley SO . Mitochondrial targeting of doxorubicin eliminates nuclear effects associated with cardiotoxicity. ACS Chem Biol 2015; 10: 2007–2015.
Khiati S, Dalla Rosa I, Sourbier C, Ma X, Rao VA, Neckers LM et al. Mitochondrial topoisomerase I (top1mt) is a novel limiting factor of doxorubicin cardiotoxicity. Clin Cancer Res 2014; 20: 4873–4881.
Volkova M, Russell R 3rd . Anthracycline cardiotoxicity: prevalence, pathogenesis and treatment. Curr Cardiol Rev 2011; 7: 214–220.
Jensen BV, Skovsgaard T, Nielsen SL . Functional monitoring of anthracycline cardiotoxicity: a prospective, blinded, long-term observational study of outcome in 120 patients. Ann Oncol 2002; 13: 699–709.
Mousavi N, Tan TC, Ali M, Halpern EF, Wang L, Scherrer-Crosbie M . Echocardiographic parameters of left ventricular size and function as predictors of symptomatic heart failure in patients with a left ventricular ejection fraction of 50-59% treated with anthracyclines. Eur Heart J Cardiovasc Imaging 2015; 16: 977–984.
Cardinale D, Colombo A, Lamantia G, Colombo N, Civelli M, De Giacomi G et al. Anthracycline-induced cardiomyopathy: clinical relevance and response to pharmacologic therapy. J Am Coll Cardiol 2010; 55: 213–220.
Bosch X, Rovira M, Sitges M, Domenech A, Ortiz-Perez JT, de Caralt TM et al. Enalapril and carvedilol for preventing chemotherapy-induced left ventricular systolic dysfunction in patients with malignant hemopathies: the OVERCOME trial (prevention of left ventricular dysfunction with enalapril and carvedilol in patients submitted to intensive chemotherapy for the treatment of malignant hemopathies). J Am Coll Cardiol 2013; 61: 2355–2362.
Endo A . A historical perspective on the discovery of statins. Proc Jpn Acad Ser B Phys Biol Sci 2010; 86: 484–493.
Vaughan CJ, Gotto AM Jr, Basson CT . The evolving role of statins in the management of atherosclerosis. J Am Coll Cardiol 2000; 35: 1–10.
Vaughan CJ, Murphy MB, Buckley BM . Statins do more than just lower cholesterol. Lancet 1996; 348: 1079–1082.
Kreisberg RA . Reductase inhibitor therapy of hypercholesterolemia. Trans Am Clin Climatol Assoc 1991; 102: 153–163.
Zhou Q, Liao JK . Pleiotropic effects of statins. - Basic research and clinical perspectives. Circ J 2010; 74: 818–826.
Futterman LG, Lemberg L . Statin pleiotropy: fact or fiction? Am J Crit Care 2004; 13: 244–249.
Waldman A, Kritharides L . The pleiotropic effects of HMG-CoA reductase inhibitors: their role in osteoporosis and dementia. Drugs 2003; 63: 139–152.
Bellosta S, Ferri N, Bernini F, Paoletti R, Corsini A . Non-lipid-related effects of statins. Ann Med 2000; 32: 164–176.
Palinski W . New evidence for beneficial effects of statins unrelated to lipid lowering. Arterioscler Thromb Vasc Biol 2001; 21: 3–5.
Endres M . Statins: potential new indications in inflammatory conditions. Atheroscler Suppl 2006; 7: 31–35.
Reddy R, Chahoud G, Mehta JL . Modulation of cardiovascular remodeling with statins: fact or fiction? Curr Vasc Pharmacol 2005; 3: 69–79.
Bokoch GM, Der CJ . Emerging concepts in the Ras superfamily of GTP-binding proteins. FASEB J 1993; 7: 750–759.
Etienne-Manneville S, Hall A . Rho GTPases in cell biology. Nature 2002; 420: 629–635.
Adamson P, Marshall CJ, Hall A, Tilbrook PA . Post-translational modifications of p21rho proteins. J Biol Chem 1992; 267: 20033–20038.
Adamson P, Paterson HF, Hall A . Intracellular localization of the P21rho proteins. J Cell Biol 1992; 119: 617–627.
Casey PJ . Protein lipidation in cell signaling. Science 1995; 268: 221–225.
Goldstein JL, Brown MS . Regulation of the mevalonate pathway. Nature 1990; 343: 425–430.
Hall A . Ras-related GTPases and the cytoskeleton. Mol Biol Cell 1992; 3: 475–479.
Hall A, Paterson HF, Adamson P, Ridley AJ . Cellular responses regulated by rho-related small GTP-binding proteins. Philos Trans R Soc Lond B Biol Sci 1993; 340: 267–271.
Heasman SJ, Ridley AJ . Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol 2008; 9: 690–701.
Wolfrum S, Jensen KS, Liao JK . Endothelium-dependent effects of statins. Arterioscler Thromb Vasc Biol 2003; 23: 729–736.
Yao L, Romero MJ, Toque HA, Yang G, Caldwell RB, Caldwell RW . The role of RhoA/Rho kinase pathway in endothelial dysfunction. J Cardiovasc Dis Res 2010; 1: 165–170.
Aslan JE, McCarty OJ . Rho GTPases in platelet function. J Thromb Haemost 2013; 11: 35–46.
Tousoulis D, Kampoli AM, Tentolouris C, Papageorgiou N, Stefanadis C . The role of nitric oxide on endothelial function. Curr Vasc Pharmacol 2012; 10: 4–18.
Rikitake Y, Liao JK . Rho GTPases, statins, and nitric oxide. Circ Res 2005; 97: 1232–1235.
Ming XF, Viswambharan H, Barandier C, Ruffieux J, Kaibuchi K, Rusconi S et al. Rho GTPase/Rho kinase negatively regulates endothelial nitric oxide synthase phosphorylation through the inhibition of protein kinase B/Akt in human endothelial cells. Mol Cell Biol 2002; 22: 8467–8477.
Zhou Q, Liao JK . Rho kinase: an important mediator of atherosclerosis and vascular disease. Curr Pharm Des 2009; 15: 3108–3115.
Heyworth PG, Knaus UG, Settleman J, Curnutte JT, Bokoch GM . Regulation of NADPH oxidase activity by Rac GTPase activating protein(s). Mol Biol Cell 1993; 4: 1217–1223.
Mizuno T, Kaibuchi K, Ando S, Musha T, Hiraoka K, Takaishi K et al. Regulation of the superoxide-generating NADPH oxidase by a small GTP-binding protein and its stimulatory and inhibitory GDP/GTP exchange proteins. J Biol Chem 1992; 267: 10215–10218.
Tan W, Palmby TR, Gavard J, Amornphimoltham P, Zheng Y, Gutkind JS . An essential role for Rac1 in endothelial cell function and vascular development. FASEB J 2008; 22: 1829–1838.
Fritz G, Kaina B . Rac1 GTPase, a multifunctional player in the regulation of genotoxic stress response. Cell Cycle 2013; 12: 2521–2522.
Huelsenbeck SC, Schorr A, Roos WP, Huelsenbeck J, Henninger C, Kaina B et al. Rac1 protein signaling is required for DNA damage response stimulated by topoisomerase II poisons. J Biol Chem 2012; 287: 38590–38599.
Dubash AD, Guilluy C, Srougi MC, Boulter E, Burridge K, Garcia-Mata R . The small GTPase RhoA localizes to the nucleus and is activated by Net1 and DNA damage signals. PLoS One 2011; 6: e17380.
Fritz G, Henninger C . Rho GTPases: novel players in the regulation of the DNA damage response? Biomolecules 2015; 5: 2417–2434.
Ciccia A, Elledge SJ . The DNA damage response: making it safe to play with knives. Mol Cell 2010; 40: 179–204.
Boissier P, Huynh-Do U . The guanine nucleotide exchange factor Tiam1: a Janus-faced molecule in cellular signaling. Cell Signal 2014; 26: 483–491.
Quilliam LA, Khosravi-Far R, Huff SY, Der CJ . Guanine nucleotide exchange factors: activators of the Ras superfamily of proteins. Bioessays 1995; 17: 395–404.
Michaelson D, Abidi W, Guardavaccaro D, Zhou M, Ahearn I, Pagano M et al. Rac1 accumulates in the nucleus during the G2 phase of the cell cycle and promotes cell division. J Cell Biol 2008; 181: 485–496.
Sandrock K, Bielek H, Schradi K, Schmidt G, Klugbauer N . The nuclear import of the small GTPase Rac1 is mediated by the direct interaction with karyopherin alpha2. Traffic 2010; 11: 198–209.
Hajas G, Bacsi A, Aguilera-Aguirre L, Hegde ML, Tapas KH, Sur S et al. 8-Oxoguanine DNA glycosylase-1 links DNA repair to cellular signaling via the activation of the small GTPase Rac1. Free Radic Biol Med 2013; 61: 384–394.
Feleszko W, Mlynarczuk I, Balkowiec-Iskra EZ, Czajka A, Switaj T, Stoklosa T et al. Lovastatin potentiates antitumor activity and attenuates cardiotoxicity of doxorubicin in three tumor models in mice. Clin Cancer Res 2000; 6: 2044–2052.
Fritz G, Kaina B . Rho GTPases: promising cellular targets for novel anticancer drugs. Curr Cancer Drug Targets 2006; 6: 1–14.
Huelsenbeck J, Henninger C, Schad A, Lackner KJ, Kaina B, Fritz G . Inhibition of Rac1 signaling by lovastatin protects against anthracycline-induced cardiac toxicity. Cell Death Dis 2011; 2: e190.
Cho SJ, Kim JS, Kim JM, Lee JY, Jung HC, Song IS . Simvastatin induces apoptosis in human colon cancer cells and in tumor xenografts, and attenuates colitis-associated colon cancer in mice. Int J Cancer 2008; 123: 951–957.
Kochuparambil ST, Al-Husein B, Goc A, Soliman S, Somanath PR . Anticancer efficacy of simvastatin on prostate cancer cells and tumor xenografts is associated with inhibition of Akt and reduced prostate-specific antigen expression. J Pharmacol Exp Ther 2011; 336: 496–505.
Gao J, Jia WD, Li JS, Wang W, Xu GL, Ma JL et al. Combined inhibitory effects of celecoxib and fluvastatin on the growth of human hepatocellular carcinoma xenografts in nude mice. J Int Med Res 2010; 38: 1413–1427.
Yu X, Luo Y, Zhou Y, Zhang Q, Wang J, Wei N et al. BRCA1 overexpression sensitizes cancer cells to lovastatin via regulation of cyclin D1-CDK4-p21WAF1/CIP1 pathway: analyses using a breast cancer cell line and tumoral xenograft model. Int J Oncol 2008; 33: 555–563.
Lochhead P, Chan AT . Statins and colorectal cancer. Clin Gastroenterol Hepatol 2013; 11: 109–118.
Kim YH, Park SM, Kim M, Kim SH, Lim SY, Ahn JC et al. Cardioprotective effects of rosuvastatin and carvedilol on delayed cardiotoxicity of doxorubicin in rats. Toxicol Mech Methods 2012; 22: 488–498.
Riad A, Bien S, Westermann D, Becher PM, Loya K, Landmesser U et al. Pretreatment with statin attenuates the cardiotoxicity of Doxorubicin in mice. Cancer Res 2009; 69: 695–699.
Franzoni F, Quinones-Galvan A, Regoli F, Ferrannini E, Galetta F . A comparative study of the in vitro antioxidant activity of statins. Int J Cardiol 2003; 90: 317–321.
Habeos IG, Ziros PG, Chartoumpekis D, Psyrogiannis A, Kyriazopoulou V, Papavassiliou AG . Simvastatin activates Keap1/Nrf2 signaling in rat liver. J Mol Med (Berl) 2008; 86: 1279–1285.
Zhu H, Jia Z, Misra BR, Zhang L, Cao Z, Yamamoto M et al. Nuclear factor E2-related factor 2-dependent myocardiac cytoprotection against oxidative and electrophilic stress. Cardiovasc Toxicol 2008; 8: 71–85.
Kleniewska P, Piechota A, Skibska B, Goraca A . The NADPH oxidase family and its inhibitors. Arch Immunol Ther Exp 2012; 60: 277–294.
Segal BH, Grimm MJ, Khan AN, Han W, Blackwell TS . Regulation of innate immunity by NADPH oxidase. Free Radic Biol Med 2012; 53: 72–80.
Forman HJ, Torres M . Reactive oxygen species and cell signaling: respiratory burst in macrophage signaling. Am J Respir Crit Care Med 2002; 166 ((12 Pt 2)): S4–S8.
Cathcart MK . Regulation of superoxide anion production by NADPH oxidase in monocytes/macrophages: contributions to atherosclerosis. Arterioscler Thromb Vasc Biol 2004; 24: 23–28.
Zhao Y, McLaughlin D, Robinson E, Harvey AP, Hookham MB, Shah AM et al. Nox2 NADPH oxidase promotes pathologic cardiac remodeling associated with Doxorubicin chemotherapy. Cancer Res 2010; 70: 9287–9297.
Satoh M, Ogita H, Takeshita K, Mukai Y, Kwiatkowski DJ, Liao JK . Requirement of Rac1 in the development of cardiac hypertrophy. Proc Natl Acad Sci USA 2006; 103: 7432–7437.
Gnad R, Kaina B, Fritz G . Rho GTPases are involved in the regulation of NF-kappaB by genotoxic stress. Exp Cell Res 2001; 264: 244–249.
Wei CY, Huang KC, Chou YH, Hsieh PF, Lin KH, Lin WW . The role of Rho-associated kinase in differential regulation by statins of interleukin-1beta- and lipopolysaccharide-mediated nuclear factor kappaB activation and inducible nitric-oxide synthase gene expression in vascular smooth muscle cells. Mol Pharmacol 2006; 69: 960–967.
Perona R, Montaner S, Saniger L, Sanchez-Perez I, Bravo R, Lacal JC . Activation of the nuclear factor-kappaB by Rho, CDC42, and Rac-1 proteins. Genes Dev 1997; 11: 463–475.
Guo F, Xing Y, Zhou Z, Dou Y, Tang J, Gao C et al. Guanine-nucleotide exchange factor H1 mediates lipopolysaccharide-induced interleukin 6 and tumor necrosis factor alpha expression in endothelial cells via activation of nuclear factor kappaB. Shock 2012; 37: 531–538.
Rodriguez-Vita J, Sanchez-Galan E, Santamaria B, Sanchez-Lopez E, Rodrigues-Diez R, Blanco-Colio LM et al. Essential role of TGF-beta/Smad pathway on statin dependent vascular smooth muscle cell regulation. PLoS One 2008; 3: e3959.
Rodrigues Diez R, Rodrigues-Diez R, Lavoz C, Rayego-Mateos S, Civantos E, Rodriguez-Vita J et al. Statins inhibit angiotensin II/Smad pathway and related vascular fibrosis, by a TGF-beta-independent process. PLoS One 2010; 5: e14145.
Iwamoto T, Oshima K, Seng T, Feng X, Oo ML, Hamaguchi M et al. STAT and SMAD signaling in cancer. Histol Histopathol 2002; 17: 887–895.
Eberlein M, Heusinger-Ribeiro J, Goppelt-Struebe M . Rho-dependent inhibition of the induction of connective tissue growth factor (CTGF) by HMG CoA reductase inhibitors (statins). Br J Pharmacol 2001; 133: 1172–1180.
Damrot J, Nubel T, Epe B, Roos WP, Kaina B, Fritz G . Lovastatin protects human endothelial cells from the genotoxic and cytotoxic effects of the anticancer drugs doxorubicin and etoposide. Br J Pharmacol 2006; 149: 988–997.
Wartlick F, Bopp A, Henninger C, Fritz G . DNA damage response (DDR) induced by topoisomerase II poisons requires nuclear function of the small GTPase Rac. Biochim Biophys Acta 2013; 1833: 3093–3103.
Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L . Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev 2004; 56: 185–229.
Zhu W, Zhang W, Shou W, Field LJ . P53 inhibition exacerbates late-stage anthracycline cardiotoxicity. Cardiovasc Res 2014; 103: 81–89.
Yoshida M, Shiojima I, Ikeda H, Komuro I . Chronic doxorubicin cardiotoxicity is mediated by oxidative DNA damage-ATM-p53-apoptosis pathway and attenuated by pitavastatin through the inhibition of Rac1 activity. J Mol Cell Cardiol 2009; 47: 698–705.
Wang S, Konorev EA, Kotamraju S, Joseph J, Kalivendi S, Kalyanaraman B . Doxorubicin induces apoptosis in normal and tumor cells via distinctly different mechanisms. intermediacy of H(2)O(2)- and p53-dependent pathways. J Biol Chem 2004; 279: 25535–25543.
Nithipongvanitch R, Ittarat W, Velez JM, Zhao R St, Clair DK, Oberley TD . Evidence for p53 as guardian of the cardiomyocyte mitochondrial genome following acute adriamycin treatment. J Histochem Cytochem 2007; 55: 629–639.
Perego P, Corna E, De Cesare M, Gatti L, Polizzi D, Pratesi G et al. Role of apoptosis and apoptosis-related genes in cellular response and antitumor efficacy of anthracyclines. Curr Med Chem 2001; 8: 31–37.
Ramanjaneyulu SV, Trivedi PP, Kushwaha S, Vikram A, Jena GB . Protective role of atorvastatin against doxorubicin-induced cardiotoxicity and testicular toxicity in mice. J Physiol Biochem 2013; 69: 513–525.
Sharma H, Pathan RA, Kumar V, Javed S, Bhandari U . Anti-apoptotic potential of rosuvastatin pretreatment in murine model of cardiomyopathy. Int J Cardiol 2011; 150: 193–200.
Rashid M, Tawara S, Fukumoto Y, Seto M, Yano K, Shimokawa H . Importance of Rac1 signaling pathway inhibition in the pleiotropic effects of HMG-CoA reductase inhibitors. Circ J 2009; 73: 361–370.
Ma J, Wang Y, Zheng D, Wei M, Xu H, Peng T . Rac1 signalling mediates doxorubicin-induced cardiotoxicity through both reactive oxygen species-dependent and -independent pathways. Cardiovasc Res 2013; 97: 77–87.
Bopp A, Wartlick F, Henninger C, Kaina B, Fritz G . Rac1 modulates acute and subacute genotoxin-induced hepatic stress responses, fibrosis and liver aging. Cell Death Dis 2013; 4: e558.
Seicean S, Seicean A, Plana JC, Budd GT, Marwick TH . Effect of statin therapy on the risk for incident heart failure in patients with breast cancer receiving anthracycline chemotherapy: an observational clinical cohort study. J Am Coll Cardiol 2012; 60: 2384–2390.
Kalam K, Marwick TH . Role of cardioprotective therapy for prevention of cardiotoxicity with chemotherapy: a systematic review and meta-analysis. Eur J Cancer 2013; 49: 2900–2909.
Acar Z, Kale A, Turgut M, Demircan S, Durna K, Demir S et al. Efficiency of atorvastatin in the protection of anthracycline-induced cardiomyopathy. J Am Coll Cardiol 2011; 58: 988–989.
Chotenimitkhun R, D'Agostino R Jr, Lawrence JA, Hamilton CA, Jordan JH, Vasu S et al. Chronic statin administration may attenuate early anthracycline-associated declines in left ventricular ejection function. Can J Cardiol 2015; 31: 302–307.
Fritz G, Henninger C, Huelsenbeck J . Potential use of HMG-CoA reductase inhibitors (statins) as radioprotective agents. Br Med Bull 2011; 97: 17–26.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by J Chipuk
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Henninger, C., Fritz, G. Statins in anthracycline-induced cardiotoxicity: Rac and Rho, and the heartbreakers. Cell Death Dis 8, e2564 (2018). https://doi.org/10.1038/cddis.2016.418
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2016.418
This article is cited by
-
Moving Beyond Cardiotoxicity Detection to Prevention: A Pharmacologic Review
Current Treatment Options in Cardiovascular Medicine (2024)
-
Efficacy and safety of cardioprotective drugs in chemotherapy-induced cardiotoxicity: an updated systematic review & network meta-analysis
Cardio-Oncology (2023)
-
Ultrastructural Myocardial Reorganization during Experimental Treatment with Doxorubicin and Atorvastatin
Bulletin of Experimental Biology and Medicine (2022)
-
In Vivo Murine Models of Cardiotoxicity Due to Anticancer Drugs: Challenges and Opportunities for Clinical Translation
Journal of Cardiovascular Translational Research (2022)
-
Dose-Dependent Cardioprotective Effect of Hemin in Doxorubicin-Induced Cardiotoxicity Via Nrf-2/HO-1 and TLR-5/NF-κB/TNF-α Signaling Pathways
Cardiovascular Toxicology (2021)
