
Abstract
Chimeric antigen receptor (CAR) gene-engineered T cell therapy holds the potential to make a meaningful difference in the lives of patients with terminal cancers. For decades, cancer therapy was based on biophysical parameters, with surgical resection to debulk, followed by radiation and chemotherapy to target the rapidly growing tumor cells, while mostly sparing quiescent normal tissues. One breakthrough occurred with allogeneic bone-marrow transplant for patients with leukemia, which provided a sometimes curative therapy. The field of adoptive cell therapy for solid tumors was established with the discovery that tumor-infiltrating lymphocytes could be expanded and used to treat and even cure patients with metastatic melanoma. Tumor-specific T-cell receptors (TCRs) were identified and engineered into patient peripheral blood lymphocytes, which were also found to treat tumors. However, these were limited by patient HLA-restriction. Close behind came generation of CAR, combining the exquisite recognition of an antibody with the effector function of a T cell. The advent of CD19-targeted CARs for treating patients with multiple forms of advanced B-cell malignancies met with great success, with up to 95% response rates. Applying CAR treatment to solid tumors, however, has just begun, but already certain factors have been made clear: the tumor target is of utmost importance for clinicians to do no harm; and solid tumors respond differently to CAR therapy compared with hematologic ones. Here we review the state of clinical gene-engineered T cell immunotherapy, its successes, challenges, and future.
Similar content being viewed by others

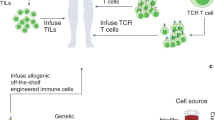
Introduction: fantastic voyage
In recent years, T-cell-mediated immunotherapy of cancer has become a celebrated therapy, essentially a revolution in what have otherwise been multiple levels of non-specific, sometimes highly toxic treatments involving invasive surgeries, debilitating radiation and chemotherapy1,2. What has been less clear, is the long-standing role that T cells, and adoptive cell therapies have had in treating this disease. The first successful adoptive cell therapy of cancer took place over half a century ago3. In 1956 in New York, Dr E Donnall Thomas performed lethal irradiation on a patient with leukemia, followed by a bone-marrow transplant (BMT) from the patient's identical twin. The result was a complete regression of the leukemia via graft-versus-tumor (GVT) activity, a finding that earned Thomas a Nobel Prize in 1990. Attempts to transfer cells from non-related donors resulted in severe damage to the patient from graft-versus-host disease (GVHD), and often mortality of the patient, so these BMTs were limited to identical twin donors for another decade, until non-twin sibling donors were used to reduce potential HLA mismatch. It took another decade until BMT involving a partially HLA-matched non-related donor was able to treat a patient with leukemia in 1979 in Seattle, WA. The practice of non-related donor BMT continues today as the only curative option for many types of leukemia, but carries a heavy price, with many patients developing GVHD, which may be severe and lethal in some cases.
T-cell therapy of cancer
Fast forward to the 21st Century, the first published report out of Steven Rosenberg's group at the National Cancer Institute (NCI) Surgery Branch used patient T cells extracted from tumor (or tumor-infiltrating lymphocytes, TIL), expanded ex vivo and re-infused to successfully treat metastatic melanoma4. Though exciting news, this approach proved to have its own challenges. Not all patients have resectable tumor; of those with resectable disease, not all tumors grew lymphocytes; of those that grew lymphocytes, not all demonstrated anti-tumor activity; of those that demonstrated anti-tumor function, many patients would not survive the eight-plus weeks required to grow and expand their T cells to therapeutic levels for reinfusion. Adding to this the challenge is that many tumor types do not give rise to TIL, and the 'boutique' nature of high-cost facilities and experienced personnel required to grow and test such cells; thus, even with a 50% objective response (OR) rate5,6, this treatment strategy remains largely un-utilized.
This quandary brought about the observed need for a 'universal' T cell that could recognize tumors in different patients, without the costly and resource-heavy dependence on growing each patient's TIL. Over several years of treating hundreds of melanoma patients with TIL, it was noted that T cells grown from many different patients all showed recognition of the highly expressed melanoma antigens MART-1 and gp100. Nicholas Restifo's group at NCI generated preclinical mouse models of T cell-mediated melanoma immunotherapy that supported these observations7,8,9,10,11, and introduced additional methods to both augment clinical therapies, and help explain the mechanisms of T-cell anti-tumor function.
TCR-engineered lymphocytes
Based on the theory that T cells targeting these shared melanoma antigens could be used to target similar tumors from different patients sharing the same HLA, one predominant T-cell clone (DMF4) from a patient whose TIL consisted of only 2 tumor-reactive cell types (DMF4 and DMF5), both MART-1/HLA-A*02(A2)-reactive, was selected12. Patient peripheral blood T cells were transduced with a retroviral construct expressing the T-cell receptor (TCR) of DMF4, and reinfused back to the patient intravenously (diagrammed in Figure 1). In 2006 Richard Morgan in Steven Rosenberg's group published these first successful gene-engineered MART-1 TCR clinical trial results (Table 1)13. Two out of seventeen (12%) patients with metastatic melanoma experienced an anti-tumor response, which although far from a cure and lower than the rate observed for TIL, provided the first proof-of-concept that gene-engineered peripheral T cells could actually work in patients with advanced metastatic cancer.

Patient-centered gene-engineered T-cell therapy of cancer. (1) Patient blood is collected by peripheral blood draw, or leukapheresis. (2) Ex vivo T cells are transduced or transfected with T-cell receptor (TCR) or chimeric antigen receptor (CAR) genes via gamma-retrovirus, lentivirus, or non-viral (transposon) gene transfer. (3) Gene-transferred T cells are expanded ex vivo. (4) TCR- or CAR-engineered T-cell product is readministered back to patient intravenously. (5) Receptor-engineered T cells circulate through the patient's bloodstream to encounter tumor, where they bind cognate antigen, activate, and destroy the tumor.
Searching for ways to improve this approach, two questions were: (1) is MART-1 a 'good' target, or would another one work better? (2) does TCR affinity matter, or is it entirely a result of the host T cell function? In considering these questions, it is important to frame our definition of avidity and affinity. The affinity of a TCR is a biophysical measure of the force required to separate the antigenic target (major histocompatibility complex (MHC) + peptide) from the TCR protein, and is typically determined by surface plasmon resonance. The avidity of a T cell for its target cell is determined by a number of contributing factors, including the number of TCR on the surface, the density of cognate antigen on the target cell, the presence of co-receptors such as cellular differentiation (CD) 4 or CD8, as well as the affinity of the TCR.
To answer the first question about antigen targets, T cells from TIL of responding patients were cloned out and found to contain high numbers of naturally occurring MART-1 (27-35)- and gp100-reactive cells14. Three epitopes of gp100 were evaluated, gp100 (209-217), (280-288) and (154-162). Of 576 individual T-cell clones, 7% had reactivity for MART-1, 5% for gp100 (280-288), 0.5% for gp100 (209-217) and 0.3% for gp100 (154-162). MART-1-reactive clones were segregated based upon their relative avidity for cognate antigen on target cells, and their relative TCR affinity for targets, based on modified tetramer-binding assays. TCRs from high-, medium-, and low-reactivity clones were isolated and expressed in donor T cells. The resulting avidity followed the hierarchy observed in the original T cell clones, with TCRs from higher-avidity clones conferring higher avidity to transduced T cell progeny. At the highest avidity TCRs were able to confer functional recognition even in CD4+ T cells, albeit to a lesser extent than CD8+ counterparts.
Affinity matters
The fallout of this work was that the MART-1 TCR being used clinically (DMF4) fell in the mid- to below-average range of avidity/affinity compared with the other MART-1-reactive clones. In fact, the single highest-avidity, high-functioning TCR belonged to the second MART-1-reactive T-cell clone from the same patient as DMF4, i.e., the DMF5 clone. Based on these results, a second clinical trial was designed, replacing the lower-affinity TCR with the high-affinity DMF5 TCR. Upon treatment of patients with metastatic melanoma, it became apparent that something very different was taking place, as most patients developed a robust skin rash five days after cell infusion, often followed by depigmentation and vitiligo15. Several patients complained about sight or hearing troubles, and upon examination, they were found to have these MART-1 TCR T cells infiltrating their skin, ears, and even their eyes, damaging the pigmented cells within. Fortunately, all of these toxicities receded upon local administration of steroids, with the exception of the skin rash, which abated naturally following depletion of the pigmented melanocytes contained within. While these pigmented cells may seem like natural targets in retrospect, at the time no one expected this, based upon the lack of any such signs in patients in the prior low-affinity MART-1 TCR clinical trial. In the DMF5 TCR trial, 6/20 treated patients (30%) experienced an objective tumor response (Table 1), a percentage higher than that in the previous trial, but still too low to statistically determine whether this improved efficacy.
At the same time, a murine-derived high-affinity TCR against human gp100 (154-162) was generated by injecting an HLA-A2 transgenic mouse with peptide and used to treat 16 patients with metastatic melanoma (Table 1)15. The results were similar to those observed with the high-affinity MART-1 TCR, namely a high incidence of depigmenting autotoxicity and/or ocular and auricular toxicity (80%), and similar if slightly decreased anti-tumor efficacy (3/16 OR, 19%)15. What these combined TCR trials made abundantly clear is that receptor affinity matters, and engineered T cells can be a very powerful therapy. With great power comes great responsibility, thus future targets would need to be chosen with extreme care.
Moving beyond melanoma
On the heels of this work came the first TCR clinical trials for non-melanoma malignancies, with TCRs targeting the cancer-testis antigen NY-ESO-1, the onco-fetal carcinoembryonic antigen CEA, or melanoma antigen-encoding genes (MAGEs). Originally described by Li et al.16 in 2005, and further affinity-tuned by Robbins et al.17, a high-avidity NY-ESO-1 HLA-A2-restricted 1G4 TCR was the first gene-engineered TCR to treat patients with non-melanoma tumors. The first clinical study, using gamma-retrovirus to deliver the NY-ESO-1 TCR to T cells, was published by the Rosenberg group in 201118. Objective clinical responses were seen in 4/6 patients with synovial cell sarcoma and 5/11 patients with melanoma. This was followed up in 2015, expanding the numbers to 11/18 (61%) patients with synovial cell sarcoma, and 11/20 (55%) with melanoma (Table 1)19. Patients on both trials received a lymphodepleting preparative chemotherapy with cyclophosphamide (Cy) and fludarabine (Flu). Long-term follow-up demonstrated 3- and 5-year OS rates were 38% and 14% respectively for sarcoma, and 33% (both) for melanoma. At UPENN, Carl June's group extended this TCR therapy using lentivirus delivery to treat patients with multiple myeloma, reporting clinical responses in 16/20 (80%) in a phase I/II trial20, with notably high levels of long-term persistence of TCR positive cells in patient blood (Table 1).
To date in 2016, there are nine clinical trials listed using the NY-ESO-1 TCR to treat patients with bladder carcinoma, breast cancer, esophagus carcinoma, lung cancer (unspecified type), melanoma, multiple myeloma, neuroblastoma, ovarian cancer, synovial cell sarcoma, non-small cell lung cancer (NSCLC), and other metastatic cancers, as listed on www.ClinicalTrials.gov and in Table 2.
A fine balance
HLA-A2 transgenic mice were used to isolate a high-affinity TCR against human CEA for treating patients with metastatic colorectal cancer at NCI21. After treating three patients, although anti-tumor activity was observed in the form of 74%-99% reduction in serum CEA levels, and one out of three patients with objective tumor regressions, all three developed acute and severe autoimmune inflammatory colitis in the colon, presumably in response to normal CEA expression (Table 1)22. The trial was discontinued due to toxicity.
Three independent clinical trials using TCRs targeting MAGE genes have been reported (Table 1): MAGE-A3 in the context of HLA-A2 was targeted in patients with metastatic melanoma, synovial cell sarcoma, or esophageal cancer by Steven Rosenberg's group at NCI23; Carl June's group at UPENN targeted MAGE-A3/HLA-A1 to treat two patients with myeloma and melanoma24; Hiroshi Shiku's group in Japan targeted MAGE-A4 on HLA-A24:02 in patients with recurrent esophageal cancer25.
The NCI trial (MAGE-A3/HLA-A2) treated nine patients23. Five experienced clinical objective responses. However, 3 additional patients experienced severe mental status changes within 2 days of treatment, and 2 of these lapsed into coma and died. Autopsies showed extensive necrotizing leukoencephalopathy associated with influx of T cells. It was determined that both patient and normal brain samples showed neuronal staining with an antibody that recognizes multiple MAGE-A family members. The TCR used in the trial was generated in response to MAGE-A3 peptide priming, however, it was also known to cross-react with MAGE-A9 and -A1226. Further evaluation determined that MAGE-A12 is normally expressed in human brain and this was concluded to be the ultimate cause of toxicity.
Danger of supraphysiological affinity-tuning of TCR
The UPENN trial treated only two patients, one with multiple myeloma, and one with melanoma24. The MAGE-A3/HLA-A1 TCR was originally obtained from a vaccinated patient27,28, and subjected to affinity modulation to increase affinity for target antigen (similarly to17). These first two patients developed cardiogenic shock and died within two weeks of T-cell infusion. Upon autopsy investigation, extensive T-cell infiltrate and diffuse myocyte necrosis were observed in heart tissues of both patients, and death was attributed to this toxicity. However, no expression of MAGE-A3 could be detected in the heart tissues. It was only upon a thorough evaluation using live, beating myocytes in culture that cross-reactivity with the MAGE-A3 TCR was detected. The culprit was determined to be an unrelated muscle-specific protein, Titin, that yielded a similar peptide in the HLA-A1 cleft, in a form of molecular mimicry24,29,30. This observation brings forth the question of whether this would have happened with a naturally selected TCR, or if increasing TCR affinity may contribute to off-target recognition of other similar peptide targets? The answer remains unknown, but is worth considering moving forward.
In a trial with less dramatic results targeting MAGE-A4 on HLA-A24:02, ten patients with esophageal cancer were treated with the TCR25. In this trial, although persistence of transferred cells could be detected in the blood by PCR up to five months after treatment, no objective responses were observed, and there was minimal toxicity. The results of these published TCR oncology clinical trials are compared in Table 1.
Moving beyond self
Most recently, investigations of non-self antigens are being pursued using TCRs targeting viral antigen from the human papillomavirus (HPV). Currently, HPV E6 TCR immunotherapy is being investigated for use against HPV-associated vaginal, cervical, anal, penile, and oropharyngeal cancers in a trial led by Christian Hinrichs at NCI31. Current clinical trials involving gene-engineered TCRs to treat cancers are listed in Table 2.
CARs: driving therapy beyond haplotype restriction
While TCR gene-engineered T-cell therapy has certainly proven to be potent, and efficacious in some cases, it faces a number of caveats. First and foremost is that it is necessarily restricted to a subset of patients. Not only do patients' tumors have to express the protein of interest, they must also match the haplotype restriction of the TCR, and both the HLA molecule and the intracellularly processed peptide must be presented together on the surface of the tumor cell. By necessity, this limits the therapy to a small subset of patients.
First generation chimeric antigen receptors (CARs) — back in the day
CARs were originally conceptualized by Zelig Eshhar and colleagues in 198932. Their findings, published in Proceedings of the National Academy of Sciences, described the generation of an “immunoglobulin-T-cell receptor chimeric molecule” by splicing the heavy and light chain variable regions of a monoclonal antibody (mAb) followed by transfection of these two fragments along with the constant region of a TCR into a T lymphocyte cell line. They later modified their approach by generating a single-chain fragment (scFv) encoding both heavy and light variable regions joined by a linker sequence, negating the need for multiple gene transfers to achieve antibody-like receptor specificity33. Upon scFv binding to a cognate antigen, the CAR signals through the CD3ζ chain to activate the receptor-bearing T lymphocyte (Figure 2). These first generation constructs, defined by the inclusion of a single intracellular signaling domain, were evaluated in early clinical cancer trials using CARs targeting folate receptor in patients with ovarian cancer34, carbonic anhydrase IX (CAIX) in patients with renal cancer35, and CD171/L1-CAM in pediatric patients with neuroblastoma36. While these early trials did not demonstrate long-term cell engraftment nor anti-tumor efficacy, the CAIX trial did lead to reports of unexpected biliary tract toxicity, illustrating the potential risk for acute in vivo on-target, off-tumor effects in patients (Table 3)35.

T-cell receptor (TCR) and chimeric antigen receptor (CAR) structure. T-cell receptors are composed of two separate proteins, the alpha (α) and beta (β) chains. TCR antigen-binding sites are located in the membrane-distal variable regions, which are attached to the membrane-proximal constant region. CARs are composed of a membrane-distal single-chain variable region (scFv) made of the variable heavy and light chains joined by a linker molecule. Upon encountering cognate antigen, T-cell activation by both TCR and CAR occurs through intracellular TCR zeta (ζ) signaling.
The first indication of positive clinical outcome using CARs came in a report published in 2008 by Malcolm Brenner and colleagues at Baylor College of Medicine in Houston, TX. A first generation CAR targeting disialoganglioside GD2 in pediatric patients with neuroblastoma induced a complete remission in three out of eleven patients37,38 (Table 3), even though there were less than 0.1% of transferred cells detectable in the blood by 24 hours after treatment. These results suggest that CAR T-cell therapy has the potential to induce effective antitumor responses, however, an emerging theme was the challenge of promoting long-term CAR T-cell engraftment in vivo in patients.
These early first generation CARs consisted of the variable regions of an antibody joined together by a linker sequence to form a scFv, molecularly fused to the intracellular CD3ζ TCR signaling chain. Subsequent improvements to clinical CARs included adding additional co-stimulatory signals, including 4-1BB to prolong T cell survival via upregulation of the anti-apoptotic factor Bcl-xL, and CD28 to increase the potency of the T cell response39,40,41,42,43,44. These improved CARs were termed 'second' or 'third' generation, depending on the presence of one (second) or more than one (third) co-stimulatory region (Figure 3)45,46,47,48. Additional CARs are now incorporating surface-expressed costimulatory receptor ligands such as 4-1BBL in addition to internal costimulation and CD3ζ signaling motifs used in the second and third generation CARs48.

Chimeric antigen receptor (CAR) generations. First generation CARs were composed of the single-chain variable region (scFv) linked to intracellular CD3 zeta (ζ). Second generation CARs incorporated an intracellular signaling motif from a T-cell costimulatory molecule followed by CD3ζ, while third generation CARs include more than one costimulatory molecule in addition to CD3ζ.
The first clinical trial using third generation CARs to treat patients with metastatic melanoma targeted a well-documented tumor-associated antigen, receptor tyrosine-protein kinase erbB-2 (ERBB2)/human epidermal growth factor receptor 2 (HER2)49 (Table 3). In recent years, HER2 had been successfully exploited as a target for tumor immunotherapy, primarily using a HER2-targeting antibody (Herceptin®) in the adjuvant setting for patients with resected breast cancer. Over 400 000 women have received Herceptin treatment, with a low rate of reported toxicity. Based on this safety profile, the Herceptin mAb scFv was chosen to develop into a CAR to treat patients with HER2-positive metastatic melanoma at NCI50. The first patient treated was given a high number of cells, 1 × 1011, with intensive prior lymphodepletion. Within minutes of the cell infusion, the patient experienced severe distress, followed rapidly by coma with X-ray evidence of high levels of pulmonary infiltrate, and died 5 days later, despite medical intervention including high-dose steroids49. Upon autopsy, transferred CAR T cells were found throughout multiple regions of the body, with high accumulations in the lung, and associated tissue destruction. Post-mortem tissue evaluation demonstrated expression of HER2 antigen in lung epithelium that was concluded to be the cause of death. The results of published CAR solid tumor clinical trials are listed in Table 3.
CD19 CARs for hematologic malignancies — a success story
The patient death in the HER2 CAR trial made a big impact in the ongoing selection of antigenic targets. It became prudent to look for antigens that are either entirely tumor-specific, which are few and far-between, or shared between tumor and non-crucial tissues. In the field of hematologic oncology, this presented an opportunity to target multiple B-cell-related malignancies. CD19 and CD20 are cell differentiation antigens expressed on the surface of B lymphocytes. While B lymphocytes are required biologically as precursors of plasma cells for antibody secretion, individuals with congenital absence of B cells have been documented, and can live essentially normal lives with prophylactic infusions of gamma-globulin. One of the earliest CARs used to treat blood cancers targeted CD2051,52, without much impact and with negligible CAR T cell persistence after infusion.
CD19 CAR was proposed to target leukemia in preclinical publications in 2007, 2008 and 2009 by Michel Sadelain53, Carl June41,54, and Steven Rosenberg55, respectively. Things really changed for the field however, in 2010, when the first results of their respective clinical CAR trials targeting CD19 in various leukemias were released. James Kochenderfer in Steven Rosenberg's group published initial results of a clinical trial using a second generation (CD28 costimulation) gamma-retroviral vector encoded CD19 CAR to treat a patient with advanced follicular lymphoma (FL)56. Both the cancerous cells and the patient's B lymphocytes were eliminated and remained so throughout the 39-week follow-up of the study. This work was followed up in 2012 with the results of eight patients treated on the trial, with 6/8 experiencing clinical remissions, and 4/8 also experiencing long-term depletion of their normal B lymphocytes57. Along with these results, 5/8 patients had more than 1% of peripheral blood mononuclear cells (PBMCs) as CAR T cells in their blood, peaking around 10 days after infusion, with two patients attaining more than 10%. However, all patients' blood CAR T cell levels returned to < 0.01% by one month after treatment.
In 2011, Carl June's group at UPENN published the case report of a patient with chronic lymphocytic leukemia (CLL) who was treated with a second generation (4-1BB costimulation) CD19 CAR encoded in a lentiviral vector58. This patient showed a complete remission of the cancer, an elimination of all normal B lymphocytes, as well as a notable immune-related fever attributed to cytokine release by a high number of CAR T cells in the body. The clinical anti-tumor response was maintained through the 8-month follow-up of the study. What was particularly intriguing about this study was that the patient was treated with a very low number of CAR T cells, only 1.42 × 107. Other clinical trials typically used 2-3 logs higher numbers, closer to 109-1011.
Also in 2011, Renier Brentjens and Michel Sadelain reported results of a second generation (CD28 costimulation) CD19 CAR clinical trial treating nine patients with CLL or acute lymphocytic leukemia (ALL) at New York's Memorial Sloan-Kettering59 with approximately 107 cells/kg of patient body weight, with or without Cy-mediated lymphodepletion preconditioning. In the first cohort of 3 patients treated without Cy, all patients experienced rapid disease progression. Moving to a lymphodepleting regimen, they reported that most patients experienced fevers within 24 hours of cell infusion. All were transient, with the exception for one patient with CLL, whose fever continued until he died within 48 hours60. The ultimate cause of death was unknown, but the authors speculated that it was likely due to a prior existing sepsis. Following an initial increase in tumor size, one patient with CLL experienced a partial reduction in tumor that lasted until 6 months after treatment59. A second patient was treated while in remission with ALL, and experienced a reduction in circulating normal B lymphocytes that lasted until 8 weeks later, when he received an allogeneic BMT and was removed from the study. Notably, transferred CAR T cells disappeared from the circulation in all patients by two weeks post-infusion, and the authors found the rapidity of elimination was proportional to the patient's tumor load.
Since these initial reports of CD19 CAR success, follow-up trials have been conducted in higher numbers of patients with FL, CLL, and ALL, with longer follow-up, as well as for patients with additional B-cell malignancies61,62,63,64,65,66,67,68,69. Of these, one of the most notable is the successful treatment of a patient with multiple myeloma (MM).
Although MM may be derived from B-lineage cells, it is normally considered to be a disease of fully differentiated, CD19-negative plasma cells. In 2015, Alfred Garfall and Edward Stadtmauer from Carl June's group at UPENN reported CD19-targeted, 4-1BB-costimulated, CAR treatment of a patient with MM70. The patient was a 43-year-old woman who had previously received high-dose chemotherapy and an autologous stem-cell transplant (SCT), and experienced a transient partial remission lasting less than 6 months. Since then she had undergone nine separate lines of chemotherapy without success. At the time of CD19 CAR treatment, her immune-globulin (Ig) A serum tumor marker level was 6310 mg/dL, her bone marrow was 95% MM, and her cancer was 99.95% CD19-negative. She received myeloablative chemotherapy with Cy and melphalan that served to keep her IgA counts below 300-6 000 mg/dL, and then received a second auto-SCT followed by the infusion of a small number (5 × 107) of CD19 CAR T cells. By 100 days after CAR treatment, her serum IgA and IgM spike dropped to 0, and her bone marrow was cleared of tumor. The CAR T cells were detectable at low levels in blood and bone marrow after 2 months, but had disappeared by day 100, when a rebound in normal B lymphocytes was observed. The patient remained in full remission through the duration of follow-up, which was 1 year after CAR treatment. This result was quite unexpected, as the patient's disease was almost entirely CD19-negative (0.05% CD19+), and has led to the speculation that CD19 CARs may target the cancer stem cell-like population that expressed CD19, leading to elimination of the tumor-regenerating population. Alternatively, as CD19 CARs eliminated the entire B cell repertoire, this may also have depleted a potential reservoir of pro-tumor, immune-suppressing cell type71,72,73. Ten MM patients were treated in this clinical trial, five of whom (in addition to the patient described in the case report) remained progression-free with a range of 35-222 days at the time of publication70.
Hematologic malignancies beyond CD19
With the notable successes of CD19-targeting CAR in lymphocytic leukemias, there has been a surge in CAR research targeting additional hematologic markers. Although the high response rate with CD19 targeting has been remarkable, there have been reports of subsequent CD19 loss in responding patients with recurrent disease, and more obviously, not all hematologic cancers occur in B cells. Clinicaltrials.gov currently lists open immunotherapy trials for myeloma using CD138 and B-cell maturation antigen (BCMA) in China, and at UPENN/NIH, respectively; and targeting CD33 and CD123 to treat acute myelogenous leukemia (AML) in China and at UPENN, respectively. CARs for hematologic cancers have been described extensively in several reviews74,75, and hence will not be discussed in great detail here.
Double-edged sword
One important fact about cancer therapy of any sort is summed up in the adage, “That which doesn't kill us, makes us stronger”. When targeting 'self gone bad', there is likely to be some collateral damage to the rest. This is clearly seen in traditional radiation and chemotherapy, whereby rapidly proliferating cells are preferentially eliminated; but this is not limited to tumor cells, causing damage throughout the rest of the body as well. In particular, cells in the gut, skin, hair follicles, and reproductive organs are also in a continual state of turnover. The same basic premise seems to hold for adoptive T-cell immunotherapy: the harder it hits, the better it works. In this case, the target is specified, but sometimes, the side-effects are systemic. Activated T cells produce high levels of inflammatory cytokines including tumor-necrosis factor (TNF)-α, multiple interleukins (ILs) such as IL-1, 2, 6, and 8, and interferon (IFN)-γ. Each of these has intense effects on the body including development of vascular leak syndrome, resulting in low blood pressure and rapid oedema into tissues; fever and malaise; recruitment and activation of additional innate and adaptive immune cells, and cascades of events that can themselves lead to serious harm or even death57,58,67,76. The generalized term for this has come to be called 'cytokine release syndrome' (CRS). The traditional way that clinicians would use to treat this presentation would be to deliver high doses of systemic corticosteroids; the problem in the context of CAR therapy is that steroids work by shutting down the immune system in general, and T cells in particular. So the conundrum presents itself in CD19 CAR clinical trials: at what point does the risk/benefit of treating the CRS override the potential therapeutic benefit of anti-tumor function?
In many leukemia patients treated with CD19 CARs, CRS has become a correlate of clinical anti-tumor response67. So much so, that clinicians and patients alike become excited when they detect a fever or a rash coming on after T-cell infusions. The diagnosis and treatment of CRS is an evolving science, but focusing on reducing one cytokine in particular, IL-6, appears to be effective. The discovery of IL-6 blockade in treating CRS falls squarely in the category of serendipity. Early on in the initial CD19 CAR clinical trial at UPENN, a young girl, seven years old with terminal leukemia, had been one of the first patients treated with CARs. Four days after receiving CD19 CARs, she developed a rapidly rising fever, and a life-threatening loss of blood pressure, necessitating hospitalization in the intensive care unit by day five. She did not respond to steroid treatment, and no one knew how else to treat her, as no one knew what was causing the problem. The clinicians worked together through the night with the laboratory scientists, and ran a multiple-cytokine/chemokine assay (Luminex) on her blood to see if anything came up. While many of the factors we now understand to be elevated in CRS were highly expressed (IFNγ, TNFα, IL-1, IL-2, IL-8, etc.), one cytokine in particular raised alarm bells, IL-6. It so happened that one of the clinical team had a family member with rheumatoid arthritis, a clinically approved treatment for which is the IL-6-receptor inhibitor, Tocilizumab. With the patient at death's door, the clinical team obtained Tocilizumab from the hospital dispensary and administered it to the patient. They then watched as her fever almost immediately subsided, her blood pressure increased, and all her other symptoms steadily returned to normal62.
Recently, the UPENN group identified 24 predictive biomarkers for CRS in patients following CD19 CAR-T therapy77. Further analyses identified three specific markers that could be used prospectively to identify which patients would experience severe CRS, early on during treatment. Intriguingly, though three specific markers were found to predict CRS in both adult and pediatric patients, the specificities of these markers differed between the two groups. In adult patients, soluble gp130 (sgp130), IFNγ and IL1RA could predict CRS, while for pediatric patients, the markers were IFNγ, IL13 and MIP1α. The treatment consensus was that interruption of the IL-6/IL-6R signaling by administering Tocilizumab generally resulted in alleviation of symptoms within 24-36 hours. Unfortunately, even with Tocilizumab treatment, 2 out of 14 patients died of CRS, underlining the need for early identification and potentially more effective intervention.
A further complication of CD19 CAR treatments has become apparent, most notably by a recent US Federal Drug Administration (FDA)-mandated hold on a phase I trial following three patient deaths. The deaths were attributed to cerebral oedema, and occurred in a specific patient cohort receiving intensified lymphodepleting chemotherapy with fludarabine prior to CAR T-cell delivery. As this trial was sponsored by a large company, Juno Therapeutics, news of the hold was widely and rapidly disseminated, resulting in much speculation over the safety and future of CAR-T therapy. However, the research team was granted approval to resume the clinical trial, without the intensified chemotherapy regimen, after only three days. This cerebral oedema, along with other neurologic toxicities that have also been observed in other CD19 CAR trials66,67,78, is associated with CD19 CAR T-cell detection in cerebral spinal fluid, and was not able to be blocked by treatment with Tocililzumab66. Unfortunately the removal of the chemopreparatory regimen was not sufficient, as two more patient deaths were reported shortly thereafter in the same trial. Further investigation into the cause, prevention and treatment of these cerebral toxicities is needed, will be ongoing, and will likely require intervention that is not limited by the blood-brain barrier (BBB), such as systemic administration of steroid, or intra-thecal delivery of mAb-based therapeutic agents such as Tocilizumab.
Boldly going where no one has gone before
The impressive results seen in blood cancers fueled resurgence in the CAR field. For the first time, large pharmaceutical companies started showing interest, and competing to pair with academic centers to put staggering sums of money (as regards the average academic research budget) in the tens to hundreds of millions of dollars, towards research and development of CAR-T immunotherapy. In exchange, industry would own intellectual property rights, or licensing rights, or some combination thereof, of the resulting research products. The first such major announcement was Novartis Pharmaceuticals pairing with UPENN, followed rapidly by the newly formed Kite Pharma entering sponsored research agreements with NCI. Others involved in CAR research, including Michel Sadelain, Renier Brentjens and Isabelle Rivière from New York's MSKCC, and Stanley Riddell, Philip Greenberg, and Michael Jensen from the Hutchinson Cancer Center/Seattle Children's Hospital, came together to form their own company, Juno Pharmaceuticals, using private investment and publicly traded stock to fund their research. Other big pharmas have invested in platforms to utilize CAR technologies, including Eli Lily, GlaxoSmithKline, Janssen, and Merck. As well, an entire field of new companies were formed to capitalize on this technology, including Bluebird Technologies, Bellicum, Cellectis, and Intrexon-Ziopharm.
Moving away from CD19-targeting of CARs into treatment of solid tumors has met with more resistance. Unfortunately, a common refrain from newcomers to the field is, “But CARs don't work in solid tumors!” Where this opinion originates is unknown, and perhaps it is inferred deductively by the lack of positive reports of CARs in solid tumors. To date, there have been few completed clinical trials published utilizing second and third generation CARs in solid tumor malignancies (Table 3). Exceptions include two case reports and results of a 19-patient trial. The first case, out of Steven Rosenberg's group in the NCI Surgery Branch in 2010, reported on the first in human use of a next (third) generation CAR in solid tumors to treat a patient with refractory metastatic melanoma. Unfortunately, this patient experienced an immediate lethal severe adverse event (SAE) following infusion of a large number (1011) of HER2 CAR T cells, attributed to off-tumor, on-target toxicity in normal tissues49. Another report of using the next (second) generation CAR in solid tumors came recently from Baylor, targeting the same HER2 antigen, in patients with refractory metastatic sarcomas (16 of 19 with osteosarcoma)79. Understandably, this trial started at a much lower cell dose, with as few as 104 T cells, working up to a maximum dose of 108 cells, still 3 logs below the dosage given in the NCI trial. While this trial showed no overt toxicity, it did have some indication of efficacy, with one patient experiencing a partial tumor response revealed by positron emission tomography (PET). The first report on second generation CARs targeting a different antigen, mesothelin (meso), came out of UPENN and described two patients, one with malignant pleural mesothelioma (MPM) and the other with metastatic pancreatic cancer, who were treated with meso-targeting RNA CARs. While neither experienced toxicity, the MPM patient experienced a partial, though transient anti-tumor response80. To date, other published solid-tumor CAR clinical trials utilized pre-CD19 CAR-success era 'first in human' first generation constructs. While original trials with first-generation GD2- and interleukin-13 receptor-α2 (IL13Rα2)-targeting CARs had little or no impact on disease38,81, they also suffered from a lack of engraftment and persistence of the CAR T cells. In fact, these trials were the impetus behind developing the costimulatory molecule-inclusive constructs comprising later generation CARs. An additional barrier that early trials faced was inefficient vector delivery and gene expression in T cells81,82. The methods used suffered from low efficiency of gene transfer, requiring long-term ex vivo expansion and repeated stimulations of transgene T cells81, something that in itself has been shown to be detrimental to in vivo therapy83.
When does a rose not smell so sweet? Shared self-antigens
With this resurgence in research, publications on preclinical CARs have abounded; however, an emerging theme predominates: efficacy versus toxicity. Since cancer starts from an otherwise normal cell, the difficulty lies in selecting targets that upon being attacked will result in tumor regression, without damaging normal tissue. The first HER2 CAR tragedy was a harsh lesson on how potent these T cells can be, and the importance of choosing a 'safe' target. The problem is there are very few known surface proteins that are absolutely tumor-specific, or even shared with non-essential tissues. As discovered with MART-115, gp10015, and CEA22, in targeted T cell therapies, even so-called 'tumor-associated' or 'embryonic' antigens may still turn out to be expressed in essential adult tissues (Table 1).
Low-hanging fruit
With a dismal prognosis of 15-month survival, and a third of tumors expressing a mutated tumor-specific surface protein, CAR targeting of epidermal growth factor receptor variant three (EGFRvIII) in glioblastoma multiforme (GBM) was an ideal choice. Several groups, including ours, have published preclinical mouse model studies targeting EGFRvIII, with human and murine CARs84,85,86,87,88,89. As EGFRvIII is a tumor-specific mutant protein, normal tissue toxicity is unlikely to be induced by EGFRvIII CARs; however, the possibility of cross-reactivity cannot be completely excluded given the structural similarity of EGFRvIII to normal EGFR.
As a prerequisite to clinical translation and in an effort to rule out cross-reactivity to wild-type EGFR protein, we evaluated potential binding of normal EGFR by anti-EGFRvIII scFv at the protein:protein interaction level, and then evaluated EGFRvIII CARs in vitro against primary skin cells (keratinocytes) known to express high levels of EGFR. As a final test, we evaluated the CARs in vivo in a human skin-grafted murine model, using EGFR CARs as a positive control, and found no toxicity or cross-recognition of normal EGFR by EGFRvIII CARs84. As a result, UPENN is currently treating GBM patients with EGFRvIII CARs (scFv 2173, a humanized version of murine-derived 3C10), costimulated by 4-1BB, at two sites: UPENN and UCSF. Additionally, Steven Rosenberg's group at NCI also has a clinical trial open for patients with high-grade gliomas, using an EGFRvIII CAR (human scFv 139) with CD28 and 4-1BB costimulatory signals. And most recently, Duke University also has an EGFRvIII CAR GBM Clinical Trial pending in ClinicalTrials.gov (Table 4 and Figure 4).

Coggle diagram of completed and ongoing TCR and CAR gene-engineered T-cell immunotherapy clinical trials (per ClinicalTrials.gov).
There is over a decade of preclinical research using chimeric T cells to target IL13Rα2, highly expressed in GBM but with low to absent expression reported in normal tissues90. Michael Jensen's previous group at City of Hope in Duarte, CA has done much work developing a non scFv-based CAR using a recombinant IL13 “zetakine” molecule with increased affinity for IL13Rα2 over IL13Rα190. In 2015, they published results of a clinical trial treating 3 patients with recurrent GBM with up to 12 intracavitary doses of CD8+ T cells expressing first-generation zetakine CARs along with HyTK drug selection/suicide gene fusion proteins81. Unfortunately, in this trial gene transfer into patient T cells was achieved by DNA plasmid electroporation and drug selection for positive cells, followed by extensive ex vivo expansions; as a result, each individual product took 3-4 months to prepare, and thus although 10 patients were enrolled on the trial, only 3 survived to receive CAR treatment. The CAR T cells did not persist in the patients, although this is unsurprising, given the lack of costimulatory signaling, and as published by the authors' previous work, the immunogenicity of the HyTK protein51. All patients underwent tumor resection prior to cell infusion, hence no anti-tumor efficacy could be determined, and all 3 patients died of their disease less than 1 year after treatment.
Shared self-antigens, revisited
For many tumor-associated shared antigens (TAAs), cancers have super-physiologic levels of expression when compared with expression levels on normal tissues. This is the rationale behind the continued pursuit of several TAAs as targets for CARs. These types of targets can be highly expressed on numerous different types of cancers, where they either vary from normal tissues by expression level, or geographic location. A large number of these are currently being evaluated for safety in phase I clinical trials in addition to HER2, including mesothelin, cMet, MUC1, GD2, EphA2, EGFR, CD171, and CD133 (Table 4 and Figure 4). Additionally, there are shared antigens being targeted that are not directly displayed on tumors, but are present in the specific tumor microenvironment, such as fibroblast activation protein (FAP) and vascular endothelial growth factor receptor (VEGFR)-291,92,93,94.
The Goldilocks theorem
The possibility of a 'treatment window' has long been discussed, whereby CARs might target only those tissues (i.e., tumors) expressing abnormally high levels of target antigen. This has been observed in the TCR field, whereby the avidity of a T-cell is judged by the minimal level of surface peptide required to trigger activation95, however it is less well-known in the CAR field. The reason for this is likely due to the distinct biology of TCR and CAR. TCR:MHC-peptide interactions are typically in the micromolar (μM) affinity range, while mAbs and scFvs generally interact with their target epitopes in the nanomolar (nM) affinity range. We have observed that affinity matters in MART-1 TCR gene-engineered T-cell therapy clinical trials at NCI. Use of the high-affinity DMF5 TCR (KD of 43 μM) caused major toxicity to melanocytes in skin, eye, and inner ear, as well as an anti-tumor effect of 30% objective response (OR) rate15, while the low-affinity DMF4 TCR (KD of 170 μM)14,96,97 showed no melanocyte toxicity and a 13% OR rate13. In contrast, the affinity range of scFvs used in clinical CARs varies, but is generally 3 or more logs higher than that of TCR. For example, the humanized EGFRvIII 2173 CAR in our current GBM clinical trial at UPENN has an affinity of 101 nM84, and the affinity of the previous NCI HER2 CAR scFv was 0.58 nM98.
It has only recently been shown how changes in the affinity levels of scFv can impact CAR function and antigen recognition. In 2015, two publications were released in the same issue of Cancer Res, one from Yangbing Zhao's group at UPENN98 and another from Laurence Cooper's group at MD Anderson Cancer Center99, where the authors introduced mutations to cause changes in affinity in EGFR- and/or HER2-targeting scFv, and evaluated the impact on CAR function against tumors and normal cells. Intriguingly, both groups were able to find a 'sweet spot' of affinity, where tumors (with high antigen expression levels) were recognized by CAR T cells in vitro and in vivo in mouse models, but cells with normal levels of antigen were not. This is supported by the clinical trial results out of Baylor, showing that HER2 CAR T cells (up to 108 cells) with a different scFv, targeting a different HER2 epitope, caused no observed toxicity in 19 treated patients79.
Limiting toxicity
RNA CARs In light of the unfortunate fatality in the first HER2 CAR clinical trial, investigators are considering this shared-antigen route with caution. One approach to limiting potential toxicity of CARs targeting these antigens is to limit the duration of exposure of treatment. Currently, this method is being investigated by the June group at UPENN in the form of using meso-targeting CAR RNA-electroporated T cells to treat patients with meso-expressing solid tumors including pancreatic and ovarian cancer, and mesothelioma (Table 3)80. CAR RNA is generated in vitro, and then electroporated into patient T cells prior to reinfusion. This method has a high gene transfer rate, with > 90% of cells expressing the CAR transgene, however, a cost is the loss of approximately 50% of cells entering the electroporation process. In addition, once translated the CAR protein is only expressed for about seven days on the cell surface, thus requiring the T cell to encounter target antigen on tumor cells during that time. As such, these cells are a 'one and done' hit, and thus cell infusions will likely need to be repeated several times to see any effect. While there were no target-related toxicities observed in this trial, there was some anti-tumor impact observed in one patient. With this initial safety data, the meso-targeting CAR T-cell clinical trial has now transitioned to using lentivirally transduced T cells at UPENN; there are also meso-targeting CAR T-cell trials underway at NCI and MSKCC, and in China (Table 4).
There is another caveat to repeated CAR T cell infusions, which became abundantly clear in one patient at UPENN who received multiple meso-targeting RNA CAR treatments temporally separated by several weeks. The scFv for the UPENN meso-targeting CAR is of murine origin, and thus initiated an anti-mouse immune response in the recipient patient. Upon re-treatment, the patient experienced an immediate and severe anaphylactic reaction to the murine protein (a human anti-mouse antibody (HAMA) humoral response)100, requiring steroid intervention that fortunately subdued the reaction.
Route of administration A second method intended to minimize toxicity is the use of localized intra-tumoral (IT) CAR T cell injection. The theory behind this is that the T cells should remain localized at the site of the tumor, rather than spreading through the body via the blood stream as they would upon intravenous injection. While not practical for widely disseminated or metastatic disease, this approach may be effective in the setting of localized cancer, such as GBM, which kills without ever leaving the brain. This idea has been employed by Michael Jensen's group in their IL13Rα2 CAR T cell therapy81, and is a delivery method proposed by the Gottschalk/Ahmed group at Baylor in Texas, to treat patients with GBM with HER2 CARs (per ClinicalTrials.gov NCT02442297) (Table 4).
Reducing dosage Another way the Baylor group is attempting to attenuate potential toxicity is by dramatically reducing the dose of CAR T cells given. In the original NCI HER2 CAR clinical trial, the first and only patient treated was given an extremely high dose of cells (1011)49. In the current trial at Baylor, not only are the cells being delivered via the IT route, but the trial has also employed a dose-escalation regimen, starting with 1 × 107 cells, with a maximum dose cohort at 1 × 108 cells, delivered in 2 infusions of 5 × 107 cells each on successive days (per ClinicalTrials.gov NCT02442297). At Baylor there are two additional HER2 CAR trials listed in ClinicalTrials.gov, one for GBM and one so-called 'bucket trial' treating any HER2-expressing malignancy. Both trials administer CAR T cells intravenously, however, they start at an even lower dose followed by a dose escalation. The bucket trial, which opened in 2009, but is currently listed as not recruiting patients, calls for a starting dose of 104 cells/m2 body surface (or approximately 104 cells total, for typical adults).
Baylor's first HER2 GBM trial opened in 2010 (also not currently recruiting patients per ClinicalTrials.gov) started with a higher, though still low, dose of 106 cells/m2 followed by a dose escalation, and utilized highly selected cytomegalovirus (CMV)-specific memory T cells for CAR transduction and expansion. There are differing views on what makes the 'best' cell for adoptive cell therapy38,83,101,102,103,104,105,106; results from the Baylor group suggest starting with virus-specific memory cells may provide the best treatment38. While this selection allows use of a well-defined subset of cells as the starting material, a drawback is the number of ex vivo stimulations and expansions, and consequently the length of time required to reach sufficient numbers of cells for treatment. When targeting a shared antigen like HER2 however, use of these cells may provide an added level of safety, as the repeated stimulations of these cells ex vivo may limit their overall potential for activation in vivo before they become exhausted, senescent, or are eliminated.
Cell suicide systems One mechanism to limit toxicity may have occurred serendipitously — endogenous immune rejection of murine scFv protein. The potential for induction of an immune response against the murine portion of mouse-derived mAb scFv had been speculated prior to any CAR clinical trials. Retrospective studies on patient serum from the NCI clinical trial for patients with melanoma who had received autologous T cells expressing the gp100 (154-162)-specific TCR generated in an HLA-A2 transgenic mouse, showed development of HAMA107. Intriguingly, this mechanism to remove TCR- and CAR-engineered T cells from the body was not a factor in the cell persistence, or success of the CD19 CAR clinical trials, as all normal B lymphocytes were eliminated, in addition to tumors, and thus no antibody-based response could be elicited. In treating patients with solid tumors or CD19-negative leukemic malignancies however, this immunogenicity could be an obstacle to treatment, or a boon to limiting off-tumor toxicity in the long-term. More recently, for CARs based on murine-derived scFv, efforts have been made to 'humanize' the foreign sequences in attempts to reduce HAMA immunogenicity84.
In the wake of the high immunogenicity demonstrated by earlier cell suicide genes like HSV/TK51,108,109,110, there has been a prolonged search for a non-immunogenic, effective system to regulate CAR T cell activity in vivo in patients. A traditional method to shut down T-cell function is systemic administration of steroids, which can reduce symptoms of inflammation and T cell activation within hours. However, as seen with the patient who succumbed rapidly after administration of the first HER2 CARs, steroids alone were not sufficient to reverse the damage done. A new method would require almost instantaneous action, with immediate T-cell shutdown, or more likely, destruction of the CAR T cell itself.
Currently MSKCC is running a bucket-style clinical trial treating patients with meso-expressing solid tumors with CD28-costimulated meso-targeting CARs with an engineered safety 'off' switch (Table 4). These cells include an inducible Caspase 9 (iCasp9) protein, rapidly activated in cells upon delivery of a drug to the patient111,112,113. This rapid activation is possible, since the system does not depend upon transcription of new genes or translation of proteins, rather, a modified version of caspase 9 is continually made. The protein is only active as a dimer, and dimerization is induced selectively through exposure to a specific drug. The active dimerized protein rapidly sends the cell into apoptotic cell death. Originally described as a shutoff switch in adoptive-transfer T cell therapy, this inducible Caspase 9 system has been adapted for use in the gene-engineered TCR and CAR field111,114,115, and may prove particularly useful in the case of any off-tumor toxicity.
To be continued
Currently there are 39 clinical trials listed on ClinicalTrials.gov using CAR T cells to treat solid tumor malignancies, as listed in Table 4, and depicted in Figure 4. Most target tumor-associated self-antigens, including HER2, GD2, cMET, EGFR, MUC1, EphA2, GPC3, CD133 and CD171. A few target non-tumor targets that are regarded as essential for tumor growth and survival in vivo, such as VEGFR or FAP. Recently, a new class of tumor targets have been identified, that slipped through the cracks of the typical mutated or over-expressed tumor antigens, namely those resulting from post-translational modifications. We recently reported on a tumor-specific CAR target, presented by glycosylation of MUC1 surface proteins, that was uniform across several types of cancer, but undetected on normal tissues116. Presumably, it will not be long before the results of these trials are published, providing insight into routes to success or challenges faced using this platform to treat solid cancers.
Future directions
In parallel with the development of engineered T cell immunotherapy, the field of cancer immunotherapy as a whole has made great leaps recently, particularly in the form of checkpoint blockade antibodies. In direct relation to a T cell's ability to achieve anti-tumor function in vivo, is a series of checkpoint molecules involved in turning T cells 'on' or 'off'. The arm of T cell-activating proteins has been utilized thoroughly in CAR design, incorporating stimulating molecules such as CD28, 4-1BB, OX-40, ICOS, CD27 and others to potentiate function. However, there is another arm that is more insidious, and is exploited by many cancers, particularly in solid tumor malignancies. These insidious proteins have been called checkpoint modulators, and act to shut down T cells by triggering the expression of PD1, CTLA4, TIM3, LAG3, and VISTA, among others, on the T cell surface. Administration of antibodies that block PD1- and CTLA4-mediated T cell inhibition has met with great success in the clinic, with reports of tumor regressions in patients with melanoma, lung cancer, spindle-cell carcinoma, and renal cell cancer117,118,119,120,121,122,123,124,125.
It has been thought that the mechanism of action of these checkpoint inhibitor antibodies is to free endogenous anti-tumor T cells to destroy their tumor targets126, and the same principle likely applies to gene-engineered TCR and CAR T cells upon tumor encounter in patients. In early correlative studies of patient blood after treatment with TCR-engineered cells, the only difference found between responding and non-responding patients was the ability of the engineered cells to function upon tumor encounter immediately ex vivo. All patients, responding or not, had high levels of engineered T cells in circulation, with an average of 20% of circulating T cells one month after treatment. However, only T cells from patients who had a clinical response were able to respond to tumor exposure ex vivo by IFNγ and IL2 production, while TCR-positive peripheral blood lymphocytes (PBLs) from non-responding patients lost the ability to respond to tumor15. In a parallel study in patients' naturally occurring TIL, there was a similar observation of MART-1 TCR-positive T cells that were unable to produce type 1 cytokines in response to stimulation; in this case, the suppressed T cells were found to have selective expression of PD1, in contrast with MART-1 TCR-positive cells that did produce type 1 cytokines and were PD1-negative127.
The recent discovery that many tumor antigens targeted by endogenous T cells are actually derived from the non-synonymous passenger mutations that develop in tumor cells, and not necessarily those posed by driver mutations may direct a shift back towards using TCR to identify intracellular mutated proteins as presented on MHC128,129. The choice of CAR over TCR has both benefits (no MHC restriction) and drawbacks (shared surface proteins only), and both face similar problems (self-antigen toxicity). Table 5 summarizes some of these considerations.
Discussion
Part of the problem in applying gene-engineered T cells to clinical treatment of patients with cancer, is that the field is so new that researchers and clinicians are still learning the biology of the system, and the 'rules'. Choice of target alone may not be sufficient to predict toxicity or efficacy; additional attributes include method of gene delivery into T cells to sustain transgene expression (DNA/RNA/plasmid, gamma retrovirus, lentivirus, non-viral transposon recombination), method of stimulating (mAbs versus beads versus cells versus antigen) and growing T cells ex vivo (cytokine mixtures, duration of culture, and bulk versus selected cells), and T cell subsets (naïve versus effector versus memory). All of these minor differences can impact T cell engraftment, survival, and function in patients. The length of construct, choice of hinge region, location of target epitope, all contribute and differ in each case. Additionally, the choice of costimulatory molecule is a contentious issue. The two most commonly used molecules, CD28 and 4-1BB, appear to have very different impacts in vivo, in contrast to their similar functions in vitro. CD28 appears to pack a more powerful up-front punch, which may aid in initial tumor destruction, but result in faster CAR T cell elimination65,130. 4-1BB on the other hand, has a more moderate initial impact, but confers long-term survival and engraftment to the CAR T cells, allowing them to persist for months or even years in the patient66,131,132. Beyond use of canonical T cell genes, a recent publication replaced CAR T cell modules (CD3ζ and costimulatory domain) with NK cell signaling proteins 2DS and DAP12, which resulted in substantial augmentation of CAR anti-tumor function in vivo against solid tumors in preclinical mouse models133.
Even without the differences already noted, it appears that targeting the exact same antigen may have different results, depending on either the antigenic epitope targeted, or the affinity of the mAb scFv. Two preclinical papers were recently published, both targeting tumor stroma-associated FAP. Both used syngeneic preclinical mouse models and showed similar in vitro function. One group (Rosenberg, NCI) found that the FAP CAR T cells were acutely toxic in their mouse models with minimal anti-tumor activity, and counseled against FAP as a clinical target94. The other group (Puré, UPENN) found the opposite; they saw no in vivo toxicity, and observed anti-tumor efficacy, encouraging the use of FAP CARs in the clinic93. Upon close inspection, the major differences were 4-1BB signaling in the latter study, versus CD28 signaling in the former, and each used a different mAb scFv to target FAP. It turns out that both results are probably correct; each individual construct, target, and treatment need empirical testing for evaluation.
Conclusion
100 different types of solid tumor cancers seem to be more like 100 different diseases each with their own barriers to treatment. Likely a return to basic science will be required to elucidate the suppressive barriers encountered in each. Pancreatic cancer for example, builds a physical barrier of stromal tissue against T cells, while many GBM secrete transforming growth factor (TGF) β and express T-cell inhibitory ligands. Happily, with the recent influx of capital and interest in developing immunotherapies for the clinic, there are more tools than ever available to use alone, or increasingly in combination to treat cancer. Age-old treatments including chemotherapies and radiation are being rediscovered for their ability to synergize with newly discovered immunotherapies like TCRs, CARs and checkpoint blockade antibodies. An entire class of new high-impact journals has been formed in response to the rapid influx of discoveries, including Sci Transl Med, Cancer Discov, and Cancer Immunology Research. While no one can predict exactly where the research will lead, one thing is certain, the future of cancer immunotherapy is bright indeed.
References
Fesnak AD, June CH, Levine BL . Engineered T cells: the promise and challenges of cancer immunotherapy. Nat Rev Cancer 2016; 16:566–581.
Topalian SL, Wolchok JD, Chan TA, et al. Immunotherapy: The path to win the war on cancer? Cell 2015; 161:185–186.
DeVita VT Jr, Rosenberg SA . Two hundred years of Cancer Research. N Engl J Med 2012; 366:2207–2214.
Dudley ME, Wunderlich JR, Robbins PF, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 2002; 298:850–854.
Dudley ME, Gross CA, Somerville RP, et al. Randomized selection design trial evaluating CD8+-enriched versus unselected tumor-infiltrating lymphocytes for adoptive cell therapy for patients with melanoma. J Clin Oncol 2013; 31:2152–2159.
Hinrichs CS, Rosenberg SA . Exploiting the curative potential of adoptive T-cell therapy for cancer. Immunol Rev 2014; 257:56–71.
Overwijk WW, Tsung A, Irvine KR, et al. gp100/pmel 17 is a murine tumor rejection antigen: induction of “self”-reactive, tumoricidal T cells using high-affinity, altered peptide ligand. J Exp Med 1998; 188:277–286.
Overwijk WW, Restifo NP . B16 as a mouse model for human melanoma. Curr Protoc Immunol 2001; Chapter 20:Unit 20.1.
Muranski P, Boni A, Antony PA, et al. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood 2008; 112:362–373.
Gattinoni L, Klebanoff CA, Restifo NP . Paths to stemness: building the ultimate antitumour T cell. Nat Rev Cancer 2012; 12:671–684.
Wrzesinski C, Paulos CM, Kaiser A, et al. Increased intensity lymphodepletion enhances tumor treatment efficacy of adoptively transferred tumor-specific T cells. J Immunother 2010; 33:1–7.
Hughes MS, Yu YY, Dudley ME, et al. Transfer of a TCR gene derived from a patient with a marked antitumor response conveys highly active T-cell effector functions. Hum Gene Ther 2005; 16:457–472.
Morgan RA, Dudley ME, Wunderlich JR, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 2006; 314:126–129.
Johnson LA, Heemskerk B . Powell DJ Jr, et al. Gene transfer of tumor-reactive TCR confers both high avidity and tumor reactivity to nonreactive peripheral blood mononuclear cells and tumor-infiltrating lymphocytes. J Immunol 2006; 177:6548–6559.
Johnson LA, Morgan RA, Dudley ME, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 2009; 114:535–546.
Li Y, Moysey R, Molloy PE, et al. Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nat Biotechnol 2005; 23:349–354.
Robbins PF, Li YF, El-Gamil M, et al. Single and dual amino acid substitutions in TCR CDRs can enhance antigen-specific T cell functions. J Immunol 2008; 180:6116–6131.
Robbins PF, Morgan RA, Feldman SA, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol 2011; 29:917–924.
Robbins PF, Kassim SH, Tran TL, et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: long-term follow-up and correlates with response. Clin Cancer Res 2015; 21:1019–1027.
Rapoport AP, Stadtmauer EA, Binder-Scholl GK, et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat Med 2015; 21:914–921.
Parkhurst MR, Joo J, Riley JP, et al. Characterization of genetically modified T-cell receptors that recognize the CEA:691-699 peptide in the context of HLA-A2.1 on human colorectal cancer cells. Clin Cancer Res 2009; 15:169–180.
Parkhurst MR, Yang JC, Langan RC, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther 2011; 19:620–626.
Morgan RA, Chinnasamy N, Abate-Daga D, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother 2013; 36:133–151.
Linette GP, Stadtmauer EA, Maus MV, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 2013; 122:863–871.
Kageyama S, Ikeda H, Miyahara Y, et al. Adoptive Transfer of MAGE-A4 T-cell receptor gene-transduced lymphocytes in patients with recurrent esophageal cancer. Clin Cancer Res 2015; 21:2268–2277.
Chinnasamy N, Wargo JA, Yu Z, et al. A TCR targeting the HLA-A*0201-restricted epitope of MAGE-A3 recognizes multiple epitopes of the MAGE-A antigen superfamily in several types of cancer. J Immunol 2011; 186:685–696.
Karanikas V, Lurquin C, Colau D, et al. Monoclonal anti-MAGE-3 CTL responses in melanoma patients displaying tumor regression after vaccination with a recombinant canarypox virus. J Immunol 2003; 171:4898–4904.
Connerotte T, Van Pel A, Godelaine D, et al. Functions of anti-MAGE T-cells induced in melanoma patients under different vaccination modalities. Cancer Res 2008; 68:3931–3940.
Cameron BJ, Gerry AB, Dukes J, et al. Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci Transl Med 2013; 5:197ra103.
Raman MC, Rizkallah PJ, Simmons R, et al. Direct molecular mimicry enables off-target cardiovascular toxicity by an enhanced affinity TCR designed for cancer immunotherapy. Sci Rep 2016; 6:18851.
Draper LM, Kwong ML, Gros A, et al. Targeting of HPV-16+ epithelial cancer cells by TCR gene engineered T cells directed against E6. Clin Cancer Res 2015; 21:4431–4439.
Gross G, Waks T, Eshhar Z . Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci USA 1989; 86:10024–10028.
Eshhar Z, Bach N, Fitzer-Attas CJ, et al. The T-body approach: potential for cancer immunotherapy. Springer Semin Immunopathol 1996; 18:199–209.
Kershaw MH, Westwood JA, Parker LL, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res 2006; 12:6106–6115.
Lamers CH, Sleijfer S, Vulto AG, et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol 2006; 24:e20–e22.
Park JR, Digiusto DL, Slovak M, et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol Ther 2007; 15:825–833.
Louis CU, Savoldo B, Dotti G, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 2011; 118:6050–6056.
Pule MA, Savoldo B, Myers GD, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med 2008; 14:1264–1270.
Finney HM, Akbar AN, Lawson AD . Activation of resting human primary T cells with chimeric receptors: costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCR zeta chain. J Immunol 2004; 172:104–113.
Imai C, Mihara K, Andreansky M, et al. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 2004; 18:676–684.
Milone MC, Fish JD, Carpenito C, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther 2009; 17:1453–1464.
Savoldo B, Ramos CA, Liu E, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest 2011; 121:1822–1826.
Lee HW, Park SJ, Choi BK, Kim HH, Nam KO, Kwon BS . 4-1BB promotes the survival of CD8+ T lymphocytes by increasing expression of Bcl-xL and Bfl-1. J Immunol 2002; 169:4882–4888.
Kowolik CM, Topp MS, Gonzalez S, et al. CD28 costimulation provided through a CD19-specific chimeric antigen receptor enhances in vivo persistence and antitumor efficacy of adoptively transferred T cells. Cancer Res 2006; 66:10995–11004.
Sadelain M, Brentjens R, Riviere I . The basic principles of chimeric antigen receptor design. Cancer Discov 2013; 3:388–398.
Zhong XS, Matsushita M, Plotkin J, Riviere I, Sadelain M . Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol Ther 2010; 18:413–420.
Carpenito C, Milone MC, Hassan R, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci USA 2009; 106:3360–3365.
Zhao Z, Condomines M, van der Stegen SJ, et al. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell 2015; 28:415–428.
Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA . Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 2010; 18:843–851.
Zhao Y, Wang QJ, Yang S, et al. A herceptin-based chimeric antigen receptor with modified signaling domains leads to enhanced survival of transduced T lymphocytes and antitumor activity. J Immunol 2009; 183:5563–5574.
Jensen MC, Popplewell L, Cooper LJ, et al. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transplant 2010; 16:1245–1256.
Till BG, Jensen MC, Wang J, et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: pilot clinical trial results. Blood 2012; 119:3940–3950.
Brentjens RJ, Santos E, Nikhamin Y, et al. Genetically targeted T cells eradicate systemic acute lymphoblastic leukemia xenografts. Clin Cancer Res 2007; 13:5426–5435.
Huang X, Guo H, Kang J, et al. Sleeping Beauty transposon-mediated engineering of human primary T cells for therapy of CD19+ lymphoid malignancies. Mol Ther 2008; 16:580–589.
Kochenderfer JN, Feldman SA, Zhao Y, et al. Construction and preclinical evaluation of an anti-CD19 chimeric antigen receptor. J Immunother 2009; 32:689–702.
Kochenderfer JN, Wilson WH, Janik JE, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 2010; 116:4099–4102.
Kochenderfer JN, Dudley ME, Feldman SA, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 2012; 119:2709–2720.
Porter DL, Levine BL, Kalos M, Bagg A, June CH . Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med 2011; 365:725–733.
Brentjens RJ, Riviere I, Park JH, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood 2011; 118:4817–4828.
Brentjens R, Yeh R, Bernal Y, Riviere I, Sadelain M . Treatment of chronic lymphocytic leukemia with genetically targeted autologous T cells: case report of an unforeseen adverse event in a phase I clinical trial. Mol Ther 2010; 18:666–668.
Porter DL, Hwang WT, Frey NV, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med 2015; 7:303ra139.
Grupp SA, Kalos M, Barrett D, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med 2013; 368:1509–1518.
Kochenderfer JN, Dudley ME, Carpenter RO, et al. Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood 2013; 122:4129–4139.
Cruz CR, Micklethwaite KP, Savoldo B, et al. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: a phase 1 study. Blood 2013; 122:2965–2973.
Brentjens RJ, Davila ML, Riviere I, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med 2013; 5:177ra138.
Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 2014; 371:1507–1517.
Davila ML, Riviere I, Wang X, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med 2014; 6:224ra225.
Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 2015; 385:517–528.
Kochenderfer JN, Dudley ME, Kassim SH, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol 2015; 33:540–549.
Garfall AL, Maus MV, Hwang WT, et al. Chimeric Antigen receptor T cells against CD19 for multiple myeloma. N Engl J Med 2015; 373:1040–1047.
de Visser KE, Korets LV, Coussens LM . De novo carcinogenesis promoted by chronic inflammation is B lymphocyte dependent. Cancer Cell 2005; 7:411–423.
Ammirante M, Luo JL, Grivennikov S, Nedospasov S, Karin M . B-cell-derived lymphotoxin promotes castration-resistant prostate cancer. Nature 2010; 464:302–305.
Balkwill F, Montfort A, Capasso M . B regulatory cells in cancer. Trends Immunol 2013; 34:169–173.
Gill S, June CH . Going viral: chimeric antigen receptor T-cell therapy for hematological malignancies. Immunol Rev 2015; 263:68–89.
Sadelain M . CAR therapy: the CD19 paradigm. J Clin Invest 2015; 125:3392–3400.
Lee DW, Gardner R, Porter DL, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014; 124:188–195.
Teachey DT, Lacey SF, Shaw PA, et al. Identification of predictive biomarkers for cytokine release syndrome after chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Cancer Discov 2016; 6:664–679.
Hu Y, Sun J, Wu Z, et al. Predominant cerebral cytokine release syndrome in CD19-directed chimeric antigen receptor-modified T cell therapy. J Hematol Oncol 2016; 9:70.
Ahmed N, Brawley VS, Hegde M, et al. Human epidermal growth factor receptor 2 (HER2)-specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J Clin Oncol 2015; 33:1688–1696.
Beatty GL, Haas AR, Maus MV, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res 2014; 2:112–120.
Brown CE, Badie B, Barish ME, et al. Bioactivity and safety of IL13Ralpha2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res 2015; 21:4062–4072.
Cooper LJ, Topp MS, Pinzon C, et al. Enhanced transgene expression in quiescent and activated human CD8+ T cells. Hum Gene Ther 2004; 15:648–658.
Klebanoff CA, Scott CD, Leonardi AJ, et al. Memory T cell-driven differentiation of naive cells impairs adoptive immunotherapy. J Clin Invest 2016; 126:318–334.
Johnson LA, Scholler J, Ohkuri T, et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci Transl Med 2015; 7:275ra222.
Sampson JH, Choi BD, Sanchez-Perez L, et al. EGFRvIII mCAR-modified T-cell therapy cures mice with established intracerebral glioma and generates host immunity against tumor-antigen loss. Clin Cancer Res 2014; 20:972–984.
Morgan RA, Johnson LA, Davis JL, et al. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum Gene Ther 2012; 23:1043–1053.
Ohno M, Ohkuri T, Kosaka A, et al. Expression of miR-17-92 enhances anti-tumor activity of T-cells transduced with the anti-EGFRvIII chimeric antigen receptor in mice bearing human GBM xenografts. J Immunother Cancer 2013; 1:21.
Kuramitsu S, Ohno M, Ohka F, et al. Lenalidomide enhances the function of chimeric antigen receptor T cells against the epidermal growth factor receptor variant III by enhancing immune synapses. Cancer Gene Ther 2015; 22:487–495.
Han J, Chu J, Keung Chan W, et al. CAR-engineered NK cells targeting wild-type EGFR and EGFRvIII enhance killing of glioblastoma and patient-derived glioblastoma stem cells. Sci Rep 2015; 5:11483.
Kahlon KS, Brown C, Cooper LJ, Raubitschek A, Forman SJ, Jensen MC . Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Res 2004; 64:9160–9166.
Chinnasamy D, Tran E, Yu Z, Morgan RA, Restifo NP, Rosenberg SA . Simultaneous targeting of tumor antigens and the tumor vasculature using T lymphocyte transfer synergize to induce regression of established tumors in mice. Cancer Res 2013; 73:3371–3380.
Chinnasamy D, Yu Z, Theoret MR, et al. Gene therapy using genetically modified lymphocytes targeting VEGFR-2 inhibits the growth of vascularized syngenic tumors in mice. J Clin Invest 2010; 120:3953–3968.
Wang LC, Lo A, Scholler J, et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res 2014; 2:154–166.
Tran E, Chinnasamy D, Yu Z, et al. Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. J Exp Med 2013; 210:1125–1135.
Kalergis AM, Boucheron N, Doucey MA, et al. Efficient T cell activation requires an optimal dwell-time of interaction between the TCR and the pMHC complex. Nat Immunol 2001; 2:229–234.
Pierce BG, Hellman LM, Hossain M, et al. Computational design of the affinity and specificity of a therapeutic T cell receptor. PLoS Comput Biol 2014; 10:e1003478.
Borbulevych OY, Santhanagopolan SM, Hossain M, Baker BM . TCRs used in Cancer Gene Ther cross-react with MART-1/Melan-A tumor antigens via distinct mechanisms. J Immunol 2011; 187:2453–2463.
Liu X, Jiang S, Fang C, et al. Affinity-tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Res 2015; 75:3596–3607.
Caruso HG, Hurton LV, Najjar A, et al. Tuning sensitivity of CAR to EGFR density limits recognition of normal tissue while maintaining potent antitumor activity. Cancer Res 2015; 75:3505–3518.
Maus MV, Haas AR, Beatty GL, et al. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol Res 2013; 1:26–31.
Sukumar M, Liu J, Ji Y, et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest 2013; 123:4479–4488.
Lugli E, Dominguez MH, Gattinoni L, et al. Superior T memory stem cell persistence supports long-lived T cell memory. J Clin Invest 2013; 123:594–599.
Gattinoni L, Lugli E, Ji Y, et al. A human memory T cell subset with stem cell-like properties. Nat Med 2011; 17:1290–1297.
Klebanoff CA, Gattinoni L, Palmer DC, et al. Determinants of successful CD8+ T-cell adoptive immunotherapy for large established tumors in mice. Clin Cancer Res 2011; 17:5343–5352.
Sommermeyer D, Hudecek M, Kosasih PL, et al. Chimeric antigen receptor-modified T cells derived from defined CD8(+) and CD4(+) subsets confer superior antitumor reactivity in vivo. Leukemia 2016; 30:492–500.
Berger C, Jensen MC, Lansdorp PM, Gough M, Elliott C, Riddell SR . Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest 2008; 118:294–305.
Davis JL, Theoret MR, Zheng Z, Lamers CH, Rosenberg SA, Morgan RA . Development of human anti-murine T-cell receptor antibodies in both responding and nonresponding patients enrolled in TCR gene therapy trials. Clin Cancer Res 2010; 16:5852–5861.
Berger C, Flowers ME, Warren EH, Riddell SR . Analysis of transgene-specific immune responses that limit the in vivo persistence of adoptively transferred HSV-TK-modified donor T cells after allogeneic hematopoietic cell transplantation. Blood 2006; 107:2294–2302.
Recchia A, Bonini C, Magnani Z, et al. Retroviral vector integration deregulates gene expression but has no consequence on the biology and function of transplanted T cells. Proc Natl Acad Sci USA 2006; 103:1457–1462.
Ciceri F, Bonini C, Stanghellini MT, et al. Infusion of suicide-gene-engineered donor lymphocytes after family haploidentical haemopoietic stem-cell transplantation for leukaemia (the TK007 trial): a non-randomised phase I-II study. Lancet Oncol 2009; 10:489–500.
Straathof KC, Pule MA, Yotnda P, et al. An inducible caspase 9 safety switch for T-cell therapy. Blood 2005; 105:4247–4254.
Yagyu S, Hoyos V, Del Bufalo F, Brenner MK . An inducible caspase-9 suicide gene to improve the safety of therapy using human induced pluripotent stem cells. Mol Ther 2015; 23:1475–1485.
Clackson T, Yang W, Rozamus LW, et al. Redesigning an FKBP-ligand interface to generate chemical dimerizers with novel specificity. Proc Natl Acad Sci USA 1998; 95:10437–10442.
Budde LE, Berger C, Lin Y, et al. Combining a CD20 chimeric antigen receptor and an inducible caspase 9 suicide switch to improve the efficacy and safety of T cell adoptive immunotherapy for lymphoma. PLoS One 2013; 8:e82742.
Hoyos V, Savoldo B, Quintarelli C, et al. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia 2010; 24:1160–1170.
Posey AD. Jr., Schwab RD, Boesteanu AC, et al. Engineered CAR T cells targeting the cancer-associated Tn-glycoform of the membrane mucin MUC1 control adenocarcinoma. Immunity 2016; 44:1444–1454.
Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012; 366:2455–2465.
Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012; 366:2443–2454.
Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med 2013; 369:134–144.
Hodi FS, O'Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363:711–723.
Hodi FS, Oble DA, Drappatz J, et al. CTLA-4 blockade with ipilimumab induces significant clinical benefit in a female with melanoma metastases to the CNS. Nat Clin Pract Oncol 2008; 5:557–561.
Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined Nivolumab and Ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015; 373:23–34.
Robert C, Schachter J, Long GV, et al. Pembrolizumab versus Ipilimumab in advanced melanoma. N Engl J Med 2015; 372:2521–2532.
Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 2011; 364:2517–2526.
Weber JS, D'Angelo SP, Minor D, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol 2015; 16:375–384.
Tumeh PC, Harview CL, Yearley JH, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014; 515:568–571.
Ahmadzadeh M, Johnson LA, Heemskerk B, et al. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009; 114:1537–1544.
Tran E, Turcotte S, Gros A, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014; 344:641–645.
Linnemann C, van Buuren MM, Bies L, et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat Med 2015; 21:81–85.
Long AH, Haso WM, Shern JF, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med 2015; 21:581–590.
Zhang H, Snyder KM, Suhoski MM, et al. 4-1BB is superior to CD28 costimulation for generating CD8+ cytotoxic lymphocytes for adoptive immunotherapy. J Immunol 2007; 179:4910–4918.
Kawalekar OU, O'Connor RS, Fraietta JA, et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016; 44:380–390.
Wang E, Wang LC, Tsai CY, et al. Generation of Potent T-cell Immunotherapy for Cancer Using DAP12-Based, Multichain, Chimeric Immunoreceptors. Cancer Immunol Res 2015; 3:815–826.
Acknowledgements
This work was supported by the NIH Director's New Innovator Award DP2CA174502 to LAJ.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Johnson, L., June, C. Driving gene-engineered T cell immunotherapy of cancer. Cell Res 27, 38–58 (2017). https://doi.org/10.1038/cr.2016.154
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2016.154
Keywords
This article is cited by
-
SMAD7 expression in CAR-T cells improves persistence and safety for solid tumors
Cellular & Molecular Immunology (2024)
-
CAR T therapies in multiple myeloma: unleashing the future
Cancer Gene Therapy (2024)
-
The CUL5 E3 ligase complex negatively regulates central signaling pathways in CD8+ T cells
Nature Communications (2024)
-
Complete spectrum of adverse events associated with chimeric antigen receptor (CAR)-T cell therapies
Journal of Biomedical Science (2023)
-
Challenges and new technologies in adoptive cell therapy
Journal of Hematology & Oncology (2023)
