
Abstract
Herein, we investigate whether single-nucleotide polymorphisms (SNPs) across the PARK10 locus are associated with susceptibility to Parkinson's disease (PD) or age at onset (AAO) of disease. One hundred and eighty-eight SNPs were genotyped across the PARK10 locus in 180 PD patients and 180 controls from central Norway (stage 1). We then used the linkage disequilibrium (LD) structure from stage 1 to select 75 SNPs for genotyping in 186 patients and 186 controls from Ireland (stage 2). Nineteen SNPs were selected from this and previous studies for follow-up in an extended Norwegian series (530 patients and 1142 controls), the Irish series and a US series (221 patients and 221 controls) (stage 3). After correction for multiple testing, markers within ubiquitin specific peptidase 24 (USP24) are significantly associated with PD within Norwegian, Irish, and US series combined (rs13312: odds ratio (OR) 0.78, P<0.001; rs487230: OR 0.80, P=0.001). Independently, the association for rs13312 is strongest in the extended Norwegian series (OR 0.76, P=0.005), although not significant after correction for multiple testing (P≤0.003 is considered significant). ORs in the Irish series are almost identical, and a similar but a weaker effect was observed for the US series. No marker showed consistent association with AAO. Our data indicate that genetic variability in USP24 is associated with PD. Although our work extends and confirms a previous report, the observed effect size does not explain the PARK10 linkage peak.
Similar content being viewed by others
Introduction
Parkinson's disease (PD; OMIM no. 168600) is a common neurodegenerative disease affecting ∼2% of people aged 65 years and older.1 Linkage studies have identified several genetic forms of PD over the past decade; however, the cause of disease in the majority of PD patients remains unknown. Several linked PARK loci still await the identification of the pathogenic gene.
Hicks et al2 linked a locus on chromosome 1p32 (PARK10, OMIM %606852) to late-onset PD in Iceland with a logarithm-of-odds (LOD) score of 4.9. Markers D1S2874 and D1S475 define an LOD minus one (LOD−1) interval of ∼8 Mb around the peak. Analyzed under an additive model, carriers of one or two ‘at-risk’ alleles have an estimated 30 times and 59 times the risk of developing PD, respectively, compared with noncarriers.2 Interestingly, Li et al3 linked a directly overlapping locus using age at onset (AAO) as a quantitative trait of PD in sib-pairs from the US Caucasian population (LOD score 3.4).
Several studies have investigated the PARK10 locus for single-nucleotide polymorphisms (SNPs) that are associated with susceptibility to PD or AAO, nominating several genes: human immunodeficiency virus type I enhancer binding protein 3 (HIVEP3), eukaryotic translation initiation factor 2B, subunit 3 gamma (EIF2B3), embryonic lethal, abnormal vision-like 4 (ELAVL4), and ubiquitin specific peptidase 24 (USP24).4, 5, 6, 7, 8 A recent genome-wide association study in PD identified two PARK10 SNPs in high linkage disequilibrium (LD) within the CUB domain containing protein 2 (CDCP2) gene.9 Despite comprehensive efforts by us and other groups, this finding could not be replicated.10, 11, 12, 13, 14, 15 Furthermore, a gene expression study recently highlighted ring finger protein 11 (RNF11) as a PARK10 candidate gene.16 It remains unresolved whether any of these candidate genes can account for the PARK10 linkage or are involved in the etiology of PD.
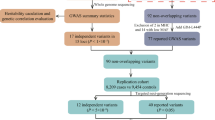
Population history and genetic data tell us that Iceland's heritage is primarily of Scandinavian descent, with a minor Celtic matrilineal component.17, 18 Assuming that the PARK10 mutation predates the settlement of Iceland from the ninth century and onward, we hypothesized that the 1p32 PD susceptibility gene may be identified in Scandinavian or Celtic PD patients. Consequently, we fine-mapped the PARK10 LOD−1 interval in Norwegian (stage 1) and Irish (stage 2) patient–control series. In stage 3, we used an extended series from Norway, the Irish series, and a Caucasian series from the United States to follow-up associated SNPs, and those SNPs in candidate genes reported to be associated with susceptibility to PD or AAO (Figure 1).

Study overview. Stage 1: fine-mapping of the PARK10 locus in patients and controls from central Norway. Stage 2: fine-mapping of ‘tagging’ SNPs in patients and controls from Ireland on the basis of the LD structure obtained from controls in the Norwegian exploratory series. Stage 3: Replication of 19 candidate SNPs from stages 1, 2 and candidate SNPs from previous studies in an extended series from central Norway, the Irish series and a Caucasian US series.
Patients and methods
Patients
All PD patients (n=924) were examined and observed longitudinally by an experienced movement disorder specialist (TL, JMG, RJU, ZKW, or JOA). Assessment of motor symptoms was carried out according to the Unified Parkinson's Disease Rating Scale and Hoehn and Yahr staging.19, 20 The diagnostic evaluation was based on clinical information, disease development, and response to levodopa, with PD diagnosed according to published criteria.21 Patients with atypical symptoms, LRRK2 mutations, or early-onset patients with parkin mutations were excluded. The controls reported no history of neurodegenerative disease; however, they were not all neurologically examined. The Norwegian exploratory series (stage 1) consisted of 180 patients and 180 age- (±4 years), gender-, and ethnicity-matched controls originating from central Norway. Mean age was 69±10 standard deviation (SD) years (range; 49–96) for both patients and controls. Mean AAO in patients was 57±11 SD years (range 25–88). The male-to-female ratio was 1.4:1 in both patients and controls. Three controls and six patients were eliminated from the Stage 1 Norwegian series due to diagnostic uncertainty during follow-up or inconsistent genotypes for markers. The addition of 356 patients and 965 unmatched controls resulted in 530 PD patients and 1142 controls from the same geographical region in central Norway being included in the extended Norwegian series (stage 3). Mean age in the extended series was 72±11 SD years (range 29–98) for patients and 73±11 SD (range 29–98) for controls. Mean AAO in patients was 59±11 SD years (range 25–88). The male-to-female ratio was 1.6:1 in patients and 0.9:1 in controls.
Irish samples (stage 2) included 186 patients and 186 controls matched for age (±4 years), gender, and ethnicity, with a mean age of 61±12 SD years (range 33–90) in both patients and controls. The male-to-female ratio was 0.6:1 in both patients and controls, and the mean AAO in PD cases was 50±11 SD years (range 18–77). Thirteen patients were eliminated from the Stage 2 Irish series due to diagnostic uncertainty during follow-up or inconsistent genotypes for markers, resulting in 173 PD patients and 173 age- and gender-matched controls in stage 3. Mean age in both patients and controls was 61±12 SD years (range 33–90), whereas mean AAO in patients was 49±11 SD years. Information regarding AAO was not available for 44 (25%) of the Stage 3 Irish patients.
The US samples (Stage 3) included 221 Caucasian patients and 221 age- (±4 years), gender-, and ethnicity-matched controls. Mean age was 70±10 SD years (range 36–89) in both patients and controls. The male-to-female ratio was 0.9:1 in both patients and controls, and the mean AAO in patients was 62±12 years (range 23–85). Differences between the Norwegian, Irish, and US samples were evident, with a younger AAO and a higher proportion of female patients in the Irish series. Participants provided informed consent and the ethical review boards at each Institution involved approved the study.
Marker selection and genotyping
We used a three-stage design to investigate the PARK10 locus (Figure 1). Stage 1: the original fine-mapping was performed by genotyping a set of SNPs spanning the PARK10 locus in a subset of 180 patients and 180 matched controls from central Norway. Additional SNPs were then selected on the basis of the empirical LD structure obtained from controls in the first set of markers. This procedure was repeated several times to ensure coverage of the locus (see Figure 2 and statistical analysis section below). Stage 2: 75 SNPs spanning the PARK10 locus were selected and genotyped in the Irish series on the basis of the LD structure obtained from Norwegian controls in stage 1. We have previously shown that the LD structure within the PARK10 locus is similar in Norwegian and Irish controls.13 Stage 3: SNPs were selected for genotyping in the follow-up either on the basis of evidence of association (P≤0.05) in the fine-mapping sets or because they were candidate genes based on previous reports. These SNPs were genotyped in an extended Norwegian series (530 PD patients and 1142 controls), including the samples genotyped in stage 1, the Irish samples (identical to stage 2), and the patient–control series from the United States. Only SNPs with minor allele frequency (MAF) over 1% among all series combined were included in the final association analysis (stage 3).

Metric LDU map of the top LOD−1 interval of the PARK10 locus between markers D1S2874 and D1S475. The LDU map provides information about LD patterns in the region. LDUs have an inverse relationship with LD with regions of extensive recombination having many LDUs. All SNPs genotyped during the fine-mapping part of the study are represented, including five SNPs in the ELAVL4 gene and 28 SNPs within the CDCP2 gene loci previously reported4, 13. The candidate genes HIVEP3 and EIF2B3 are located in the telomere of this interval, and thus not included in the figure.
Genotyping for the original fine-mapping in the Norwegian and Irish series (stages 1 and 2) was done by PCR and restriction enzyme digest or by using TaqMan chemistry on an ABI7900 genetic analyzer. Genotyping of new variants and SNPs in candidate genes (stage 3) was performed on the Sequenom MassArray iPlex platform (primer sequences available on request). Duplicate samples were included for genotype quality control.
Statistical analysis
Numerical variables were summarized with the sample mean, SD, and range. As a statistical control for systematic genotyping error, evidence for departure from Hardy–Weinberg equilibrium was assessed for each variant. For the Norwegian, Irish, and US matched series, individual associations between PD and each marker were measured by odds ratios (ORs), and corresponding 95% CIs were obtained from single-variable conditional logistic regression models. For the Norwegian extended series and combined series (extended Norwegian, Irish, and US), individual associations between PD and each marker were measured by ORs and 95% CIs obtained from logistic regression models adjusted for age, sex, and series (combined series only). In PD patients, linear regression models adjusted for sex were used to examine individual associations between AAO and each marker, separately for each series. Samples genotyped for markers in stages 1 and 2 were not included in the stage 3 follow-up genotyping for the same markers (Table 1). LD between markers in study controls was assessed with pairwise r2-values as implemented in the Haploview software.22 In addition, a metric LD unit (LDU) map was constructed.23 For each family of statistical tests, adjustment for multiple testing using the single-step minP procedure was performed,24 with 10 000 permutations of genotype labels to determine the level of significance that controls the family-wise error rate at 5%.24 P-values less than or equal to this level are considered significant.
Results
In our fine-mapping study, SNPs genotyped in the PARK10 LOD−1 interval are represented as an LDU map (Figure 2).23 The 188 SNPs included should provide reasonable coverage of the interval, as the mean LDU between markers was 0.40 (range, 0–3.0), with one marker for every 50 kb. Associations between individual SNPs and susceptibility to PD in the Norwegian series are shown in Supplementary Table 1 (stage 1; 180 PD patients and 180 controls). Associations between 75 individual SNPs and susceptibility to PD in the Irish series (stage 2) are shown in Supplementary Table 2.
We selected markers for further study on the basis of the associations between SNPs and susceptibility to PD in the Stage 1 Norwegian series (Supplementary Table 1): rs11579642 intragenic (OR: 1.49, P=0.042), rs2783175 in the thioredoxin domain containing 12 (TXNDC12) gene (OR: 2.79, P=0.002), and rs1970951 in the glutathione peroxidase 7 (GPX7) gene (OR: 0.51, P=0.001). There was also evidence for association between SNPs in an LD block between rs6662414 and rs1242317. One associated SNP (rs6588441) in the zyg-11 homolog B (ZYG11B) gene (OR: 1.84, P=0.001) was in high LD (r2=0.66–0.96) with all other SNPs in this LD-block and was selected for follow-up genotyping. The last SNP selected from the Stage 1 Norwegian series was rs8990 located in the 24-dehydrocholesterol reductase (DHCR24) gene (OR: 0.58, P=0.052). Two SNPs were also selected for genotyping on the basis of the results in the Irish series (Supplementary Table 2): rs734908 intragenic (OR: 0.64, P=0.015) and rs213498 in the single stranded DNA binding protein 3 (SSBP3) gene (OR: 0.71, P=0.028). In addition, SNPs in HIVEP3, EIF2B3, and USP24 were selected for follow-up genotyping as they have been nominated in previous studies.5, 6, 8 RNF11 was recently characterized as a PARK10 candidate gene. On the basis of HapMap data (http://www.hapmap.org), we selected two RNF11 SNPs with MAF≥0.1 for follow-up genotyping.16 Finally, a coding SNP (rs3820198) in the low-density lipoprotein receptor-related protein 8 apolipoprotein ɛ receptor (LRP8) gene was selected for follow-up genotyping as a biological candidate.25 Thus, a total of 19 SNPs were selected for follow-up genotyping in the combined Norwegian, Irish, and US series (stage 3). Associations between the 19 SNPs and susceptibility to PD or AAO are shown in Tables 1 and 2, respectively. Allele frequencies for the 19 SNPs genotyped in stage 3 are shown in Supplementary Table 3.
In the stage 3 Norwegian series, all four SNPs in USP24 were associated with susceptibility to PD with estimated ORs ranging from 0.76 to 0.81 (0.01>P>0.004) (Table 1). However, only P-values ≤0.003 are considered statistically significant after correction for multiple testing. ORs were almost identical in the Irish series, and there was a similar, although weaker, trend in the US series (Table 1).
Joint analysis of Norwegian, Irish, and US samples was significant after correction for multiple testing for rs13312 (OR: 0.78, 95% CI: 0.67–0.90, P=0.0007) and rs487230 (OR: 0.80, 95% CI: 0.70–0.91, P=0.0013), whereas rs287235 (OR: 0.81, 95% CI: 0.71 – 0.93, P=0.0035) and rs1165226 (OR: 0.86, 95% CI: 0.76–0.97, P=0.015) showed the same trend. LD structure for USP24 SNPs is shown in Figure 3. No other SNP was consistently associated with susceptibility to PD across series, and there were no SNPs that were consistently associated with AAO.

Haploview plot for SNPs in the USP24 gene. LD between markers is measured by pairwise r2 as calculated from 1142 Norwegian controls. Black cells=high LD, gray cells=intermediate LD, White cells=weak LD.
Discussion
Herein, we present the data from a fine-mapping and candidate gene investigation of the PARK10 locus in PD. SNPs were genotyped across the PARK10 locus in a Norwegian (Stage 1) and Irish (Stage 2) PD patient–control series (Figure 1). In stage 3, we investigated associated SNPs from stages 1 and 2 and SNPs previously reported to be associated with susceptibility to PD or AAO in an extended follow-up sample.
Three main conclusions can be derived from our study:
-
i)
None of the SNPs associated with susceptibility to PD in the fine-mapping study (stages 1 and 2) remained significant in the follow-up sample (stage 3; see Table 1). Thus, initial associations could be excluded when the SNPs were genotyped in a larger sample set.
-
ii)
Most previously reported associations between SNPs and susceptibility to PD or AAO within the PARK10 locus could not be replicated in our study. We selected a few of the most interesting SNPs from each candidate gene in an attempt to directly replicate previous associations (stage 3). This approach is in agreement with recent recommendations for replication studies.26, 27 From their fine-mapping study, Oliviera et al8 reported SNPs in HIVEP3 gene to be associated with susceptibility to PD. In a recent study by the same group, the association between HIVEP3 SNPs and PD was still present when genotyping an extended series that included the original samples.6 We found no association between HIVEP3 SNPs and PD (Table 1), including the most significant SNP from Li et al,6 rs1105414 (previously known as rs3856255). Hence, our data do not support a role for HIVEP3 in the etiology of PD. Furthermore, we could not replicate the reported association between SNPs in EIF2B3 or USP24 and AAO (Table 2). Our group has previously reported that SNPs in ELAVL4 are associated with PD in the Irish series, but not in the Norwegian or US series. This lack of consistency makes ELAVL4 less likely as a candidate gene for PARK10, although replication may be warranted for ELAVL4 in samples of Celtic descent.4
-
iii)
USP24 SNPs are associated with susceptibility to PD in the combined Norwegian, Irish, and US series after correction for multiple testing, and ORs observed are consistent with a previous report from the United States.5 Furthermore, Oliviera et al8 found these USP24 SNPs to be associated with AAO in PD. This finding must be interpreted with caution, as susceptibility to PD and AAO are different traits. Taken together, these results warrant follow-up of USP24 in independent series. On the basis of the level of LD between the USP24 SNPs included in this study, replicating the ‘top hit’ (rs13312) may suffice (Figure 3). We estimate that 1050 PD patients and 1050 matched controls are necessary in a follow-up study to detect ORs of 0.8 or stronger with 80% power at the 5% significance level, on the basis of the observed MAF in controls (>20%) using an additive model.
There are limitations to our study. Although we constructed an LDU map to ensure coverage of the region (Figure 2), not all genetic variability may be captured using our strategy, as our fine-mapping lacks power to detect subtle effects. There is an imbalance in gender between patients and controls in the Norwegian series. To deal with this, we have adjusted for gender in all logistic regression models involving the Norwegians. Power calculations show that 180 PD patients and 180 matched controls, as used in Stage 1, had 80% power at the 5% significance level to detect ORs of 1.9 or greater. This is with an additive model assuming an MAF in controls of 10% or greater. Our study is improved by the use of homogeneous patient–control series from Norway and Ireland to fine-map the PARK10 locus, as these populations represent the founders of the Icelandic population where PARK10 was originally linked.17, 18
Since the positional cloning of parkin in autosomal recessive young onset PD,28 the ubiquitin-proteasomal system has been implicated in the etiology of PD. This makes USP24 an interesting candidate beyond the fact that the gene is located within the PARK10 locus. However, the PARK10 linkage peak still remains unaccounted for, as we observed effect sizes (ORs 0.78–0.8 for the minor allele, corresponding to ORs 1.20–1.22 for the major allele) for USP24 SNPs that are far below what one would expect on the basis of the original linkage from Iceland. Hicks et al2 estimated a 30 times increased risk for carriers of one disease allele. A possible explanation for the observed association in this study is that these SNPs could be in weak LD with functional variants in USP24. The DeCODE group could resolve this issue by screening USP24 for mutations in PARK10-linked families. If no mutations are found, the possibility remains that the gene has yet to be found, or that the original PARK10 linkage is an artifact. Alternatively, as we used an association approach and not linkage, the USP24 gene may contain ‘risk-alleles’ that are independent of the PARK10 linkage.
References
de Rijk MC, Launer LJ, Berger K et al: Prevalence of Parkinson's disease in Europe: a collaborative study of population-based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology 2000; 54 (11 Suppl 5): S21–S23.
Hicks AA, Petursson H, Jonsson T et al: A susceptibility gene for late-onset idiopathic Parkinson's disease. Ann Neurol 2002; 52: 549–555.
Li YJ, Scott WK, Hedges DJ et al: Age at onset in two common neurodegenerative diseases is genetically controlled. Am J Hum Genet 2002; 70: 985–993.
Haugarvoll K, Toft M, Ross OA et al: ELAVL4, PARK10, and the celts. Mov Disord 2007; 22: 585–587.
Li Y, Schrodi S, Rowland C, Tacey K, Catanese J, Grupe A : Genetic evidence for ubiquitin-specific proteases USP24 and USP40 as candidate genes for late-onset Parkinson disease. Hum Mutat 2006; 27: 1017–1023.
Li YJ, Deng J, Mayhew GM, Grimsley JW, Huo X, Vance JM : Investigation of the PARK10 gene in Parkinson disease. Ann Hum Genet 2007; 71 (Part 5): 639–647.
Noureddine MA, Qin XJ, Oliveira SA et al: Association between the neuron-specific RNA-binding protein ELAVL4 and Parkinson disease. Hum Genet 2005; 117: 27–33.
Oliveira SA, Li YJ, Noureddine MA et al: Identification of risk and age-at-onset genes on chromosome 1p in Parkinson disease. Am J Hum Genet 2005; 77: 252–264.
Maraganore DM, de Andrade M, Lesnick TG et al: High-resolution whole-genome association study of Parkinson disease. Am J Hum Genet 2005; 77: 685–693.
Myers RH : Considerations for genomewide association studies in Parkinson disease. Am J Hum Genet 2006; 78: 1081–1082.
Clarimon J, Scholz S, Fung HC et al: Conflicting results regarding the semaphorin gene (SEMA5A) and the risk for Parkinson disease. Am J Hum Genet 2006; 78: 1082–1084; author reply 1092–1094.
Elbaz A, Nelson LM, Payami H et al: Lack of replication of thirteen single-nucleotide polymorphisms implicated in Parkinson's disease: a large-scale international study. Lancet Neurol 2006; 5: 917–923.
Farrer MJ, Haugarvoll K, Ross OA et al: Genomewide association, Parkinson disease, and PARK10. Am J Hum Genet 2006; 78: 1084–1088; author reply 92–94.
Goris A, Williams-Gray CH, Foltynie T, Compston DA, Barker RA, Sawcer SJ : No evidence for association with Parkinson disease for 13 single-nucleotide polymorphisms identified by whole-genome association screening. Am J Hum Genet 2006; 78: 1088–1090; author reply 1092–1094.
Li Y, Rowland C, Schrodi S et al: A case-control association study of the 12 single-nucleotide polymorphisms implicated in Parkinson disease by a recent genome scan. Am J Hum Genet 2006; 78: 1090–1092; author reply 1092–1094.
Anderson LR, Betarbet R, Gearing M et al: PARK10 candidate RNF11 is expressed by vulnerable neurons and localizes to Lewy bodies in Parkinson disease brain. J Neuropathol Exp Neurol 2007; 66: 955–964.
Goodacre S, Helgason A, Nicholson J et al: Genetic evidence for a family-based Scandinavian settlement of Shetland and Orkney during the Viking periods. Heredity 2005; 95: 129–135.
Helgason A, Sigurethardottir S, Nicholson J et al: Estimating scandinavian and gaelic ancestry in the male settlers of Iceland. Am J Hum Genet 2000; 67: 697–717.
Fahn S, Elton RL, Members of the UPDRS development committee: Unified Parkinson's disease rating scale; in Fahn S, Marsden CD, Calne D, Goldstein M (eds):: Recent Development in Parkinson's Disease. Florham Park: Macmillan Health Care Information, 1987, pp 153–163.
Hoehn MM, Yahr MD : Parkinsonism: onset, progression, and mortality. 1967. Neurology 2001; 50: S11–S26.
Gelb DJ, Oliver E, Gilman S : Diagnostic criteria for Parkinson disease. Arch Neurol 1999; 56: 33–39.
Barrett JC, Fry B, Maller J, Daly MJ : Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 2005; 21: 263–265.
Maniatis N, Collins A, Xu CF et al: The first linkage disequilibrium (LD) maps: delineation of hot and cold blocks by diplotype analysis. Proc Natl Acad Sci USA 2002; 99: 2228–2233.
Westfall PH, Young SS : Resampling-Based Multiple Testing: Examples and Methods for P-value Adjustment. New York: John Wiley and Sons, 1993, pp 66.
Li YJ, Hauser MA, Scott WK et al: Apolipoprotein E controls the risk and age at onset of Parkinson disease. Neurology 2004; 62: 2005–2009.
Chanock SJ, Manolio T, Boehnke M et al: Replicating genotype–phenotype associations. Nature 2007; 447: 655–660.
Clarke GM, Carter KW, Palmer LJ, Morris AP, Cardon LR : Fine mapping versus replication in whole-genome association studies. Am J Hum Genet 2007; 81: 995–1005.
Kitada T, Asakawa S, Hattori N et al: Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998; 392: 605–608.
Acknowledgements
We gratefully thank the patients, families and controls for their participation in this study. Mayo Clinic Jacksonville is a MK Udall Parkinson's Disease Research Center of Excellence (NINDS P01 NS40256) and we thank all collaborators of the Udall Center. This study was also supported by the National Institutes of Health (R01 AG022579), the Research Council of Norway (Grant no. 153487/V50) and Reberg's Legacy (JOA).
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Rights and permissions
About this article
Cite this article
Haugarvoll, K., Toft, M., Skipper, L. et al. Fine-mapping and candidate gene investigation within the PARK10 locus. Eur J Hum Genet 17, 336–343 (2009). https://doi.org/10.1038/ejhg.2008.187
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2008.187
Keywords
This article is cited by
-
GLIS1–3 transcription factors: critical roles in the regulation of multiple physiological processes and diseases
Cellular and Molecular Life Sciences (2018)
-
Neural ablation of the PARK10 candidate Plpp3 leads to dopaminergic transmission deficits without neurodegeneration
Scientific Reports (2016)
-
A customized high-resolution array-comparative genomic hybridization to explore copy number variations in Parkinson’s disease
neurogenetics (2016)
-
Parkinson’s disease mouse models in translational research
Mammalian Genome (2011)
