
Abstract
Genetic diagnostics of phenylketonuria (PKU) and tetrahydrobiopterin (BH4) deficient hyperphenylalaninemia (BH4DH) rely on methods that scan for known mutations or on laborious molecular tools that use Sanger sequencing. We have implemented a novel and much more efficient strategy based on high-throughput multiplex-targeted resequencing of four genes (PAH, GCH1, PTS, and QDPR) that, when affected by loss-of-function mutations, cause PKU and BH4DH. We have validated this approach in a cohort of 95 samples with the previously known PAH, GCH1, PTS, and QDPR mutations and one control sample. Pooled barcoded DNA libraries were enriched using a custom NimbleGen SeqCap EZ Choice array and sequenced using a HiSeq2000 sequencer. The combination of several robust bioinformatics tools allowed us to detect all known pathogenic mutations (point mutations, short insertions/deletions, and large genomic rearrangements) in the 95 samples, without detecting spurious calls in these genes in the control sample. We then used the same capture assay in a discovery cohort of 11 uncharacterized HPA patients using a MiSeq sequencer. In addition, we report the precise characterization of the breakpoints of four genomic rearrangements in PAH, including a novel deletion of 899 bp in intron 3. Our study is a proof-of-principle that high-throughput-targeted resequencing is ready to substitute classical molecular methods to perform differential genetic diagnosis of hyperphenylalaninemias, allowing the establishment of specifically tailored treatments a few days after birth.
Similar content being viewed by others

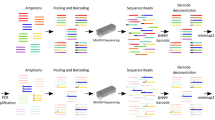
Introduction
Hyperphenylalaninemia (HPA) is a medical condition characterized by excessive blood levels (>120 μmol/l) of phenylalanine.1 HPA is caused in 98% of the cases by inherited loss-of-function mutations in the phenylalanine-4-hydroxylase (PAH, EC 1.14.16.1) gene, which cause phenylketonuria (PKU, MIM#261600), and has a population prevalence of 1 in 10 000. In the remaining 2% of cases, HPA is caused by genetic defects in enzymes involved in the synthesis and regeneration of the PAH active cofactor tetrahydrobiopterin (BH4).2, 3, 4 PAH is the rate-limiting enzyme in phenylalanine catabolism, mediating its hydroxylation to tyrosine. This reaction is BH4-dependent, which is de novo synthesized from guanosine triphosphate (GTP) via a sequence of three enzymatic steps carried out by GTP cyclohydrolase I (EC 3.5.4.16), 6-pyruvoyl-tetrahydropterin synthase (PTPS, EC 4.2.3.12), and sepiapterin reductase (SR, EC1.1.1.153) encoded by GCH1, PTS, and SPR, respectively. Regeneration of the BH4 cofactor requires pterin-4a-carbinolamine dehydratase (PCD, EC 4.2.2.96), encoded by PCBD1, and dihydropteridine reductase (EC 1.6.99.7), encoded by QDPR. BH4 deficiency due to autosomal recessive mutations in any of these enzymes, except for SR, causes severe forms of HPA; although PCD deficiency is considered a transient form of HPA, most PCD-deficient patients usually do not show significant clinical abnormalities.
BH4-deficient HPA (BH4DH) is in general more severe than PKU, and with regard to patients’ response to therapy and treatment it is substantially different.2, 3 Furthermore, knowing whether a patient has residual PAH enzymatic activity can be relevant for the therapeutic approach to PKU because some mutations are responsive to Kuvan. For all these reasons, it is essential to perform a differential HPA diagnosis using enzymatic, biochemical, and genetic approaches in order to implement specific treatments for PKU and BH4DH. However, the extensive allelic and genetic heterogeneity of HPA, combined with the low throughput and poor scalability of conventional gene-by-gene screening methods, hinders a quick, comprehensive, and cost-effective molecular diagnostic for PKU and BH4DH. For instance, only for PAH, more than 800 mutations have been described in the Human Gene Mutation Database (HGMD; www.hgmd.cf.ac.uk), including single-nucleotide variants (SNVs), short insertions and deletions (InDels), and large structural variants (SVs).
In this study, we sought to assess the amenability of high-throughput sequencing for the genetic diagnosis of PKU and BH4DH as an accurate, comprehensive, and cost-effective alternative to conventional genetic testing in the medical diagnostics context. We performed an in-solution hybrid capture to enrich the complete genomic sequence of the PAH, GCH1, PTS, and QDPR genes in a heterogeneous panel of PKU and BH4DH patients. We assessed the performance of this assay in a validation cohort of 95 patients, in whom we detected all the previously known mutations, and then used it in a discovery cohort of 11 samples with unknown mutations.
Materials and methods
Subjects
High-quality genomic DNA from 107 unrelated samples (106 PKU and BH4DH patients and one control sample) was obtained from peripheral blood lymphocytes, using standard protocols. The validation cohort included 95 patients and one control sample that had previously undergone conventional biochemical and genetic diagnosis at the Centro de Diagnóstico de Enfermedades Moleculares (CEDEM) in Madrid, Spain,5 and all disease-causing mutations in PAH, GCH1, PTS, and QDPR were confirmed using Sanger sequencing or multiplex ligation-dependent probe amplification (MLPA), except in the case of patient H8, for whom the second PAH allele was unknown. The discovery cohort consisted of 11 HPA consecutive samples, received at the CEDEM from Chile for genetic diagnosis for which no mutations were known. All samples of the discovery cohort were anonymized in order to ensure the protection of their identity, and the list of confirmed mutations was not provided to the investigators performing the bioinformatics mutation analysis until the end of the variant prioritization process. Informed consent was provided by the parents of patients in the discovery cohort.
Capture and multiplexed resequencing of the PAH and GCH1, genes
To carry out DNA capture, we designed a custom NimbleGen SeqCap EZ Choice Library (Roche, Inc., Madison, WI, USA) to target the complete genomic sequence of the PAH, GCH1, PTS, and QDPR genes and 10 kb of genomic sequence flanking at the 5′ and 3′ ends of each gene, accounting for a total of 226 543 bp. DNA baits were selected using the most stringent settings for probe design (uniqueness tested using the Sequence Search and Alignment using Hashing Algorithm (SSAHA)).6 No probe redundancy was allowed in the final capture design. The BED file of the probe sequences is available on request from the authors.
Genomic capture was carried out following the instructions of the NimbleGen SeqCap EZ Library SR User’s Guide v3.0 (Roche, Inc.) that are available for free download, to perform sequence capture from pooled libraries prepared using the TruSeq DNA Sample Preparation Kits (Illumina, Inc., San Diego, CA, USA). Briefly, genomic DNA (1 μg) from blood was sonicated using a Covaris S2 instrument (Covaris, Inc., Woburn, MA, USA) to obtain fragments of approximately 200–300 bp with 3′ and 5′ overhangs. Then, DNAs were subjected to three enzymatic steps: end repair, A-tailing, and ligation to Illumina paired-end indexed adapters, as outlined in the DNA Truseq protocol (Illumina, Inc.). Once the DNA libraries were indexed, they were PCR-amplified (seven cycles) and pooled before in-solution hybridization to a custom NimbleGen SeqCap EZ Choice Library (Roche, Inc.) of complementary oligonucleotide DNA baits (pools of 8, 12, 16, and 24 samples in the validation cohort and a pool of 11 samples for the discovery cohort). After stringent washing, the captured libraries were PCR-amplified (17 cycles) and sent for sequencing to generate 2 × 100 bp paired-end reads using a HiSeq 2000 instrument (Illumina, Inc.), in the case of the validation cohort and the control sample. The samples of the discovery cohort were sequenced using a MiSeq (Illumina, Inc.) to generate 2 × 250 bp paired-end reads. The resulting fastQ files were analyzed using an in-house-developed pipeline described below.
Bioinformatics analysis of DNA variants
The authors involved in the analysis of the mutations performed this study blindly; that is, they had no information about the pathogenic variants known to be present in the samples of the validation cohort and, of course, in the discovery cohort. All the bioinformatics tools in this study were run using standard parameters unless stated otherwise. Image analyses, base calling, and de-multiplexing on each lane of data were performed using Illumina’s Sequencing Analysis Pipeline version 1.7.0 (Illumina, Inc.). Reads were aligned to the human reference genome hg19 using the Burrows–Wheeler Aligner (bwa aln) version 0.5.9,7 allowing for maximally six mismatches and one gap of up to 20 bp. We also performed local re-alignment around potential insertions/deletions and SNP clusters, base-quality recalibration, and duplication marking using the GATK pipeline8 and picard-tools (http://picard.sourceforge.net). The resulting alignments were used as input for three different variant prediction tools, namely GATK Unified Genotyper,9 samtools mpileup10 and SHORE (http://1001genomes.org). Only SNVs and InDels of up to 30 bp and found within 150 bp of the ends of the enriched targets were considered for subsequent analysis. Functional annotation of high-quality variants was performed using Annovar,11 providing a comparison of the predicted variants to the National Center for Biotechnology Information (NCBI) SNP Database build 132 (dbSNP132), the March 2010 pilot release of the 1000 Genomes project (1000G; www.1000genomes.org), conservation around variants based on phastCons,12 segmental duplication filter, gene annotation (exon/intron/UTR), amino-acid substitutions and splice variants based on UCSC Genome Browser13 tracks, as well as multiple estimates of the impact of amino-acid substitution on the structure and function of proteins (tools: Sift,14 Polyphen2,15 PhyloP,16 and MutationTaster17). The reference sequences used for the four genes targeted in this study were NM_000277 (PAH), NM_000320 (QDPR), NM_000161 (GCH1), and NM_000317 (PTS). To identify large InDels and SVs we used Pindel,18 Conifer,19 and PeSV-Fisher (http://gd.crg.eu/tools). An in silico analysis of the effect on splicing of the novel intronic SNVs of sample H8 of the validation cohort was performed using different tools: Berkeley Drosophila Genome Project Splice Site Prediction (www.fruitfly.org/seq_tools/splice.html), Analyzer Splice Tool (http://ibis.tau.ac.il/ssat/SpliceSiteFrame.htm), and Human Splicing Finder (www.umd.be/HSF/).
Identification of PKU and BH4DH mutations
In order to identify the pathogenic mutations that could cause PKU and BH4DH, we applied the following cascade of filtering steps:20
-
1
We required all candidate variants on both sequenced DNA strands and to account for ≥15% of total reads at that site.
-
2
Common polymorphisms (≥5% in the general population) were discarded using comparison with dbSNP 132, the 1000G, the Exome Variant Server (http://evs.gs.washington.edu), and an in-house exome variant database to filter out both common benign variants and recurrent artifact variant calls. However, as these databases contain known disease-associated mutations, all detected variants were compared with gene-specific mutation databases (www.hgmd.cf.ac.uk and www.pahdb.mcgill.ca).
-
3
Then, we screened for mutations that could give rise to premature protein truncating mutations, that is, stop mutations, damaging missense variants, splice sites, exonic deletions/insertions, and large genomic rearrangements.
-
4
Variants were ranked based on evolutionary conservation and potential deleteriousness of the affected nucleotide using Sift,14 Polyphen2,15 PhyloP,16 and MutationTaster.17
All newly identified variants identified in this study have been submitted to PAHdb (http://www.pahdb.mcgill.ca/), which is the reference database for hyperphenylalaninemia mutations.
Validation of newly identified SVs and SNVs
Validation of variants of the discovery cohort, the previously unknown novel intronic SNVs in patient H8 and parents, and the chromosomal breakpoints for the identified deletions in samples of the validation cohort was performed using standard Sanger sequencing protocols. The sequences of the primers used for the validation of the breakpoints of the deletions can be found in Supplementary Table 1.
Results
PAH, PTS, QDPR, and GCH1 enrichment
We designed oligonucleotides to target all exons, introns, and 10 kb of the 5′ and 3′ flanking genomic regions of the PAH (NM_000277), PTS (NM_000317), QDPR (NM_000320), and GCH1 (NM_000161) genes that are responsible for PKU and BH4DH. After the removal of repetitive sequences, only 75.42% of the targeted bases could be covered using capture baits for a final targeted region of 170 865 bp divided into 195 individual regions, with lengths ranging from 67 to 7815 bp (average of 876 bp) (Table 1). As expected, the target regions that precluded bait tilling correspond mainly to intronic and intergenic sequences, and only a small proportion corresponds to protein coding regions. The reasons for including the untranslated fraction of the four genes were (a) to have a complete definition of the non-coding variability and (b) to favor the detection and sizing of large SVs within the four genes.
Sequencing statistics
In the validation cohort, an evenly distributed mean depth of coverage of 279X on average across samples for the target genes was achieved, with a coefficient of variation of 42.17%. In fact, 99.89% of all targeted bases were covered using at least five reads (the minimum that we require for variant calling) and 83.63% using at least 100 reads (Table 2). To determine if the coverage was substantially lower for any region, we calculated the proportion of base pairs for each locus that was captured using <50 reads. The proportion of these poorly covered regions accounted for 0.047 for PAH, 0.049 for PTS, 0.067 for GCH1, and 0.044 for QDPR. Across targets, only 0.012% of the targeted bases were not covered using any read (Supplementary Table 2). Owing to the lower throughput of the MiSeq sequencer, the average coverage achieved in the discovery cohort was 34.46X, with a coefficient of variation of 19.52% across the 11 samples. In all, 98.77% of all targeted bases were covered using at least 5 reads and 79.81% using at least 20 reads. Across targets, only 0.05% of the targeted bases were not covered using any read (Table 2).
As expected, these low-covered genomic regions in both cohorts of patients are characterized using low complexity and a high content of GC base pairs. Sequence targets with these two characteristics usually preclude enrichment, resulting in reduced coverage for these sites. However, as shown above, this was the case for only a very small proportion of all the bases intended to be captured in this study. For a comprehensive summary of the obtained sequencing results, see Supplementary Table 3. From these data we can conclude that all the samples, regardless of the pool sizes in the pre-capture step and the sequencer used, are uniformly covered at depths that in all the cases exceed by far the minimum coverage required for a reliable variant calling.
Identification of PAH, PTS, QDPR, and GCH1 mutations in the validation cohort
The selection of the samples for the validation cohort was done with the idea of including as many different types of PKU and BH4DH mutations as possible, including SNVs, InDels, and large SVs, to simulate a real-world diagnostic scenario, so that we could test the effectiveness of our approach for all these types of genetic variation. To assess the sensitivity of our assay to the detection of pathogenic mutations, we blindly inspected all the mapped sequence reads from the 95 HPA patients with previously defined mutations in PAH, GCH1, PTS, or QDPR, and one control sample.
On average, for every sample, we were able to detect 121 SNVs (4 novel) and 22 InDels (14 novel) in PAH, 16 SNVs (1 novel) and 3 InDels (2 novel) in PTS, 45 SNVs (1 novel) and 13 InDels (8 novel) in GCH1, and 63 SNVs (1 novel) and 8 InDels (4 novel) in QDPR (Supplementary Table 2). Then, we applied our variant prioritization strategy to identify pathogenic mutations present in the validation cohort. Using this strategy we detected 76 different pathogenic mutations on the four targeted genes (63 in PAH, 4 in GCH1, 6 in PTS, and 3 in QDPR) in their correct heterozygous/homozygous state across the 95 HPA patients included in the validation cohort (some variants were detected in more than one patient). More elaborately, we identified 48 missense, 7 nonsense (stop-gain), and 12 splice site (mRNA splicing) SNVs, and 4 frameshift deletions, 1 frameshift insertion, and 1 non-frameshift deletion, known to cause PKU and BH4DH (Supplementary Table 4). In addition, we also were able to detect four large deletions involving various PAH exons and introns. No spurious calls were detected in the control sample.
Identification of PAH, PTS, QDPR, and GCH1 mutations in the discovery cohort
The discovery cohort consisted of 11 consecutive samples received for HPA diagnosis, for which no mutations were previously known. On average, for every sample, we were able to detect 108 SNVs (4 novel) and 10 InDels (5 novel) in PAH, 12 SNVs (1 novel) and 1 InDels (1 novel) in PTS, 33 SNVs (3 novel) and 7 InDels (4 novel) in GCH1, and 50 SNVs (1 novel) and 4 InDels (1 novel) in QDPR (Table 2). After applying the filters for causative mutations, we detected 12 different pathogenic mutations, all of them in PAH, across the 11 patients of the discovery cohort (some variants were detected in more than one patient). Specifically, we found 6 missense, 1 nonsense (stop-gain), and 5 splice-site (mRNA splicing defect) SNVs known to cause PKU (Table 3). We confirmed all the variants using Sanger sequencing in samples from the parents. We identified the two causative alleles in all of the 11 patients.
Sensitivity and specificity of the assay
This study led to the identification of all the previously known mutations, including SNVs, InDels, and large SVs, in their correct heterozygous/homozygous state of the validation cohort. Also, we reached a 100% diagnostic rate in the discovery cohort. No false positives or spurious calls were detected either in the control sample or in any of the patients. In the case of the validation cohort, we would have achieved a diagnostic rate of 94/95 (98.94%), as for one of the PKU patients we could not identify his second mutant PAH allele (which was also previously unknown). Patient H8 carries the missense mutation c.143T>C (p.(L48S)) in heterozygosity, and no second mutation or deletion was detected using standard techniques. It was hypothesized that the second mutation could lie deep in an intron producing an aberrant splicing process. Examination of the targeted resequencing results of this sample confirmed the presence of c.143T>C (p.(L48S)) and a number of SNVs, most of which were common polymorphisms present in dbSNP. Three intronic SNVs were novel and we validated them using Sanger sequencing in the index case and in parental samples. One of them, g.103284549A>G, was present on the same allele as c.143T>C (p.(L48S)) and was also detected in two other samples of this study, suggesting that it is a relatively common SNV in the Spanish population. The other two, g.103277024A>G and g.103290290A>G, were present on the allele with a yet unidentified mutation. Bioinformatics analysis predicts the activation of a 5′ splice site for the first change and no clear effect for the second. Further studies are necessary to investigate the potential pathogenicity of these variants.
Characterization of large SVs
Conventional mutation detection methods tend to overlook large SVs. A major step forward for high-throughput sequencing technologies with respect to classical molecular approaches is the possibility of detecting large genomic rearrangements at the same time as SNVs and InDels, without the need for additional assays specific for large SVs, such as array-comparative genomic hybridization (aCGH), qPCR-based methods, MLPA, or quantitative multiplex PCR of short fluorescent fragments. In our study, the combination of paired-end mapping, split-read analysis, and normalized depth of coverage strategies allowed the blind identification of all 3 known large PAH deletions in three patients of the validation cohort: two different deletions involving exon 5 (1670 bp in patient A1 and 5351 bp in patient C9) and a deletion covering exons 6, 7 and 8 (4799 bp in patient G3), which were previously identified using MLPA analysis.21 In addition, we identified a previously unknown (missed during conventional PAH screening and not present in the public databases) 899-bp deletion in intron 3 (Figure 1) in patient C9, of unknown clinical significance, which was further confirmed using PCR and Sanger sequencing.

Detection of large structural variants. Representation of the normalized read counts in the y-axis. X-axis represents the position on chromosome 12 in windows of 100 bp. Colored peaks indicate the four large PAH deletions identified in the three samples of the validation cohort.
We have also been able to identify using paired-end mapping and split-read analysis the breakpoints of three of them (Table 4), with a complete concordance between the predictions of our algorithms and the Sanger sequencing validations (Supplementary Figure 1). Noteworthily, for sample A1, by carefully analyzing the depth of coverage data we were able to estimate the coordinates of the breakpoints on the targeted sequence within 200 bp of those of the previous results obtained using long-PCR and Sanger sequencing analysis.21 Actually, it was reported that this deletion starts in the middle of a simple TA repeat sequence in intron 4 and that the 3′ deletion breakpoint lies near a TG repeat in intron 5. Thus, we suspect that we were unable to detect in patient A1, using paired-end mapping or split-read analysis, the breakpoints of this deletion (1670 bp) because they might have evaded hybrid capture (no oligonucleotides were designed to target repetitive sequences) and no sequencing data were available.
Reproducibility
Thirty-one out of the 76 pathogenic mutations detected using our analysis in the validation cohort were present in two or more patients. This represents a reproducibility of 100% for pathogenic variant calls between two or more samples (based on the results for more than 40% of the mutations included in the validation cohort). As most of the samples bearing these mutations were multiplexed in independent pre-capture pools of different sample sizes, and were also run in different sequencer lanes, we can conclude that our approach offers great robustness and reproducibility to the detection of pathogenic HPA variants. Although the coverage for a given mutation can vary significantly between samples, the proportion of reads supporting the non-reference allele was always maintained (Supplementary Table 4).
Discussion
We are witnessing the early stages of the transition of high-throughput sequencing from basic research to clinical diagnostics.22 It is expected that during the following years high-throughput sequencing technologies will transform molecular diagnostics in the same way that they have transformed genomic research.23 To date, Sanger sequencing of individual DNA fragments has been the gold-standard molecular approach in the genetic diagnostics of inherited disorders, including PKU and BH4DH. However, this rather costly, stepwise, and time-consuming technology will be gradually replaced by high-throughput sequencing technologies, which offer higher throughput and scalability and, as a corollary, reduced costs per sequenced nucleotide and a shorter turnaround time.
Here we present a complete workflow for the molecular diagnosis of PKU and BH4DH based on the pooled target enrichment and multiplexed high-throughput sequencing of the PAH, PTS, QDPR, and GCH1 genes. We have validated this new approach in a cohort of 95 HPA patients with previously known pathogenic mutations in PAH, QDPR, GCH1, and PTS genes, and one control sample. After mapping the sequencing reads to the reference genome and performing variant calling and filtering, our bioinformatics pipeline successfully retrieved all the known pathogenic mutations (including SNVs, InDels, and large deletions) in their correct heterozygous/homozygous state, without detecting spurious calls in any of the patients or in the control sample. Then we used the same assay in a discovery cohort of 11 HPA patients without previously characterized pathogenic mutations, reaching a diagnostic rate of 100%.
The main consequence of including the non-coding regions of the four genes is that more sequencing is needed to warrant a minimum average depth of coverage. However, this approach allows exploring intronic mutations affecting potential splicing sites and facilitates the detection of large SVs and their breakpoints within the captured genes. Also, given that the total length of the captured genomic regions is relatively short (≈170 kb), the inclusion of the intronic regions has no significant economic consequences on the total cost of the assay, thanks to the high throughput of the current NGS sequencers.
To date, the standard genetic screening for PKU and BH4DH has consisted of Sanger sequencing of all exonic regions and intronic boundaries, plus MLPAs, SNP or CGH arrays to test for deletions and duplications in the PAH, PTS, QDPR, and GCH1 genes,21 although, most commonly, previous biochemical and enzymatic testing has been employed prior to the sequencing analysis to allow the differentiation of PKU from BH4DH. We estimate that our high-throughput sequencing-based assay has an overall cost of less than ɛ200 in consumables per sample, which represents 60–80% of cost savings per sample and makes the whole diagnostic process at least eight times faster when compared with the techniques currently used for the molecular diagnosis of PKU and BH4DH. In addition, our strategy offers a complete definition of the captured genes, without the need, anymore, for stepwise testing and choosing which gene to sequence first. We anticipate that these differences will become even more significant because of the constantly dropping sequencing costs24 and optimized library preparation and sequencing protocols. For the discovery cohort, in which the MiSeq was used, the complete process of library preparation, sequence enrichment, high-throughput sequencing, and bioinformatics analysis was completed in five days after the reception of the DNA samples. The challenge for shortening the time to give results will allow the implementation of high-throughput sequencing in a new era for newborn screening avoiding the time-consuming biochemical, enzymatic approaches to distinguish between PKU and the different BH4 deficiencies and making possible the early implementation of accurate treatments a few days after birth.
We think that, as is currently the norm with Sanger sequencing, clinical high-throughput testing would likely be externalized to certified laboratories. Ideally, these high-throughput specialized laboratories would receive and centralize the samples from multiple hospitals, rather than setting up high-throughput facilities in each hospital. This approach should reduce costs to the health system and will allow the specialized laboratories to work with batches of samples large enough to exploit the full potential of the high-throughput instruments.
In conclusion, this represents, to the best of our knowledge, the first study to successfully use targeted high-throughput sequencing to detect PKU and BH4DH pathogenic mutations in a clinic-like scenario, allowing the establishment of specifically tailored treatments sooner than would be possible using conventional molecular methods. Our approach has shown great accuracy and efficiency and meets the sensitivity and specificity required for genetic diagnostics, being ready to substitute classic molecular tools in the medical diagnostics of PKU and BH4DH.
References
Scriver CR, Kaufman S, Eisensmith R, Woo SLC : The hyperphenylalaninemias; in Scriver CR, Beaudet AL, Sly WS, Valle D (eds): The Metabolic and Molecular Bases of Inherited Disease 8th edn. New York: McGraw-Hill, 2001, pp 1667–1724.
The Metabolic and Molecular Bases of Inherited Disease 8th edn. New York, NY: McGraw-Hill, 2001; 1667–1724.
Werner ER, Blau N, Thony B : Tetrahydrobiopterin: biochemistry and pathophysiology. Biochem J 2011; 438: 397–414.
Opladen T, Hoffmann GF, Blau N : An international survey of patients with tetrahydrobiopterin deficiencies presenting with hyperphenylalaninaemia. J Inherit Metab Dis 2012; 35: 963–973.
Asociación Española para el Estudio de los Errores Congénitos del Metabolismo: Guía clínica para el diagnóstico, tratamiento y registro de pacientes con hiperfenilalaninemia en España, 2011, . ISBN 846947314X, 9788469473146.
Ning Z, Cox AJ, Mullikin JC : SSAHA: a fast search method for large DNA databases. Genome Res 2001; 11: 1725–1729.
Li H, Durbin R : Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009; 25: 1754–1760.
McKenna A, Hanna M, Banks E et al: The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010; 20: 1297–1303.
DePristo MA, Banks E, Poplin R et al: A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011; 43: 491–498.
Li H, Handsaker B, Wysoker A et al: The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009; 25: 2078–2079.
Amstutz U, Andrey-Zurcher G, Suciu D, Jaggi R, Haberle J, Largiader CR : Sequence capture and next-generation resequencing of multiple tagged nucleic acid samples for mutation screening of urea cycle disorders. Clin Chem 2011; 57: 102–111.
Siepel A, Bejerano G, Pedersen JS et al: Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 2005; 15: 1034–1050.
Kent WJ, Sugnet CW, Furey TS et al: The human genome browser at UCSC. Genome Res 2002; 12: 996–1006.
Kumar P, Henikoff S, Ng PC : Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009; 4: 1073–1081.
Adzhubei IA, Schmidt S, Peshkin L et al: A method and server for predicting damaging missense mutations. Nat Methods 2010; 7: 248–249.
Cooper GM, Stone EA, Asimenos G, Green ED, Batzoglou S, Sidow A : Distribution and intensity of constraint in mammalian genomic sequence. Genome Res 2005; 15: 901–913.
Schwarz JM, Rodelsperger C, Schuelke M, Seelow D : MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 2010; 7: 575–576.
Ye K, Schulz MH, Long Q, Apweiler R, Ning Z : Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 2009; 25: 2865–2871.
Krumm N, Sudmant PH, Ko A et al: Copy number variation detection and genotyping from exome sequence data. Genome Res 2012; 22: 1525–1532.
Walsh T, Lee MK, Casadei S et al: Detection of inherited mutations for breast and ovarian cancer using genomic capture and massively parallel sequencing. Proc Natl Acad Sci USA 2010; 107: 12629–12633.
Desviat LR, Perez B, Ugarte M : Identification of exonic deletions in the PAH gene causing phenylketonuria by MLPA analysis. Clin Chim Acta 2006; 373: 164–167.
Voelkerding KV, Dames SA, Durtschi JD : Next-generation sequencing: from basic research to diagnostics. Clin Chem 2009; 55: 641–658.
Desai AN, Jere A : Next-generation sequencing: ready for the clinics? Clin Genet 2012; 81: 503–510.
Wetterstrand KA. DNA sequencing costs: data from the NHGRI Large-Scale Genome Sequencing Program, 2012. Available at www.genome.gov/sequencingcosts.
Acknowledgements
We thank the subjects and referring physicians who participated in this study. This project was funded using the Spanish Plan Nacional SAF2008-00357 (NOVADIS); the Instituto de Salud Carlos III (FIS/FEDER PI11/00733); and the European Commission 7th Framework Program, Project N. 261123 (GEUVADIS), and Project N. 262055 (ESGI). D.T. is a PhD student supported by the Spanish Ministry of Economy and Competiveness.
AUTHOR CONTRIBUTIONS
This study was conceived and designed by DT, BP, LRD, LA, MU and XE. Selection of samples was performed by BP and VC. High-throughput sequencing libraries were prepared by DT and JG. The bioinformatics pipeline and the high-throughput sequencing analysis were performed by DT, CT, GE, and SO. Sanger confirmation of mutations was performed by BP, LRD, and RN. The manuscript was written by DT, BP, LRD, and XE.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Rights and permissions
About this article
Cite this article
Trujillano, D., Perez, B., González, J. et al. Accurate molecular diagnosis of phenylketonuria and tetrahydrobiopterin-deficient hyperphenylalaninemias using high-throughput targeted sequencing. Eur J Hum Genet 22, 528–534 (2014). https://doi.org/10.1038/ejhg.2013.175
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2013.175
Keywords
This article is cited by
-
Genetic etiology and clinical challenges of phenylketonuria
Human Genomics (2022)
-
The study of the full spectrum of variants leading to hyperphenylalaninemia have revealed 10 new variants in the PAH gene
Metabolic Brain Disease (2019)
-
Spectrum of PAH gene variants among a population of Han Chinese patients with phenylketonuria from northern China
BMC Medical Genetics (2017)
-
The complete European guidelines on phenylketonuria: diagnosis and treatment
Orphanet Journal of Rare Diseases (2017)
-
Correlation between genotype and the tetrahydrobiopterin-responsive phenotype in Chinese patients with phenylketonuria
Pediatric Research (2015)
