
Abstract
Vascular Ehlers–Danlos Syndrome (vEDS), also known as EDS type IV, is considered to be an autosomal dominant disorder caused by sequence variants in COL3A1, which encodes the chains of type III procollagen. We identified a family in which there was marked clinical variation with the earliest death due to extensive aortic dissection at age 15 years and other family members in their eighties with no complications. The proband was born with right-sided clubfoot but was otherwise healthy until he died unexpectedly at 15 years. His sister, in addition to signs consistent with vascular EDS, had bilateral frontal and parietal polymicrogyria. The proband and his sister each had two COL3A1 sequence variants, c.1786C>T, p.(Arg596*) in exon 26 and c.3851G>A, p.(Gly1284Glu) in exon 50 on different alleles. Cells from the compound heterozygote produced a reduced amount of type III procollagen, all the chains of which had abnormal electrophoretic mobility. Biallelic sequence variants have a significantly worse outcome than heterozygous variants for either null mutations or missense mutations, and frontoparietal polymicrogyria may be an added phenotype feature. This genetic constellation provides a very rare explanation for marked intrafamilial clinical variation due to sequence variants in COL3A1.
Similar content being viewed by others

INTRODUCTION
Vascular Ehlers–Danlos Syndrome (vEDS; OMIM# 130050), also known as EDS type IV, is considered to be an autosomal dominant disorder that results from sequence variants in COL3A1 (2q32.2) (OMIM#120180). Vascular EDS is associated with a life-long high risk for arterial rupture or dissection and gastro-intestinal rupture.1 As a consequence, lifespan is reduced for most affected individuals. Heterozygous missense or splice-site mutations account for about 95% of identified variants (Ehlers–Danlos Syndrome variant databases: https://eds.gene.le.ac.uk/home.php?select_db=COL3A1; unpublished data from the Collagen Diagnostic Laboratory, Department of Pathology, University of Washington). In about 5% of families, a variant that results in a premature-termination codon and mRNA instability, or in the inability of proα1(III) chains to be incorporated into the type III procollagen homotrimer, account for the disorder.2 These variants lead to haploinsufficiency via nonsense-mediated messenger RNA decay or failure of protein–protein interaction, and result in both reduced penetrance and delayed onset of complications with clinical manifestations apparently limited to vascular events.
Although vEDS is almost always inherited in an autosomal dominant fashion, the possibility of recessive inheritance was raised by Sulh et al3 but no variants were identified in COL3A1. Confirmation that autosomal recessive inheritance could occur was established by the identification of an 11-year-old girl who was homozygous for a null mutation carried by each of the clinically unaffected parents.4 The girl died at the age of 11 years due to tissue fragility and gastro-intestinal rupture. She was also found to have cerebrocortical dysplasia with predominantly frontal location.
We report siblings who are compound heterozygous for COL3A1 sequence variants. This family calls attention to two considerations: first, that marked intrafamilial variation in the phenotype could be explained by biallelic variants and second, that individuals with rare autosomal recessive forms of vascular-type EDS may have structural alterations in the brain, with or without cognitive alterations.
Materials and methods
DNA isolation and sequence analysis
Nine family members were available for this study (Table 1). There was no blood available for mutation testing in the deceased maternal grandfather (I-3). Genomic DNA was isolated from peripheral blood by standard procedures. The COL3A1 coding sequence, the intron–exon borders and parts of the 5′ and 3′ regions were PCR amplified and sequenced using automated dideoxy-chain termination sequence analysis in DNA from proband (III-2), and compared with the reference sequence NM_000090.3 with Mutation Surveyor. Exons were numbered as outlined in the Ehlers–Danlos Syndrome variant database.5 The nomenclature used to identify exon number in COL3A1 reflects the historical attempt to have the same exons of the triple helical domains in all fibrillar collagens that have the same number. In COL3A1 the fourth exon, which encodes residues 112–149 of the protein, appears to have fused the sequences equivalent to exons 4 and 5 in other fibrillar collagens. As a consequence, it is named exon 4/5 and the subsequent exon is designated exon 6 (but is exon 5 in the ENSEMBL assembly (ensemble.org)) and this difference extends throughout the gene. In other family members, only the specific sequences that surrounded the sequence variants identified were analyzed. In DNA from the proband sequences were also determined for the coding parts and exon–intron borders of FBN1 (65 exons, reference sequence NM_000138.4, OMIM 134797), TGFBR1 (exons 1–9, reference sequence NM_004612.2, OMIM 190181) and TGFBR2 (exons 1–7, reference sequence NM_003242.5, OMIM 190182), and supplemental MLPA analyses of the FBN1, TGFBR1, and TGFBR2 genes were performed. Sequence primer and details of sequence and MLPA (Multiplex Ligation-dependent Probe Amplification) analyses are available upon request.
Analysis of collagens produced by cultured cells
Skin biopsies were done with appropriate consent and the dermal fibroblasts were isolated using standard practices. Collagenous proteins were biosynthetically labeled and then type I and III collagen and procollagen were examined by sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE) using previously described methods.6 The mobilities of the chains and the relative amounts of proα chains of type I and III procollagens, and the α-chains of collagens were assessed.
Results
Clinical descriptions
The proband was born with right-sided clubfoot and developed unilateral hydrocele of the testis (III-2, see Table 1 for summary including evaluation of the clinical major and minor features listed according to the current Villefranche diagnostic criteria for vEDS7). He had crowded teeth, retrognathia, and was treated with braces from 12 years of age. He was otherwise healthy until he died unexpectedly at 15 years of age. The family reported he had backaches for a week prior to his death. Autopsy revealed three dissections: a transverse dissection including nearly the whole circumference of the thoracic aorta at the level of ligamentum arteriosus, and separate dissections of the right renal artery and the left femoral artery. There had been extensive bleeding into the thoracic and abdominal cavities. Histological examination revealed ischemia of the myocardium, acute renal tubular necrosis (probably due to reduced blood flow) and cystic medial necrosis in the aortic wall. The brain had normal macroscopic appearance, and cerebral dissection at autopsy revealed no obvious macroscopic anomalies. Neither brain imaging nor histological cerebral examinations were performed. Owing to the extensive vascular involvement in combination with long and slender fingers in the boy, the pathologists suspected Marfan syndrome on autopsy.
The parents (II-5 and II-6), an older brother (III-1), and a younger sister (III-3) were referred to the clinical geneticist. The sister, 6 years old at the time, had long and slender fingers with small joint hypermobility in hands and positive thumb, and wrist signs. The mother (II-6) had positive thumb and wrist signs, and a ‘marfanoid’ facial appearance. At that time, aortic root measurements were normal in both the girl and her mother; the mother had minimal mitral insufficiency. Examinations of the father (II-5) and brother (III-1) of the proband were normal. Analysis of FBN1 was not pursued.
At age 14, the girl (III-3) was referred for new evaluation. By then she had crowded teeth in a small lower jaw and translucent skin with visible subcutaneous vessels on her legs and thorax. The parents reported an episode with spontaneous small superficial vessel rupture on her lower left leg. Additional evaluation that included echocardiography, ultrasonography of the abdominal aorta, CT of the thorax, MRI of lumbosacral regions, and ocular examinations were all normal.
At 19 years of age, III-3 was further evaluated. Family history with early death due to cerebral bleeding in the maternal grandfather (I-3), previous findings in the deceased proband and photographs, and further history about the proband, together with new clinical evaluation of the young woman, led to consideration of the vascular type of EDS as an alternative diagnosis. The siblings III-2 and III-3 both manifested pale, translucent skin with visible subcutaneous vessels and facial features characteristic of EDS type IV with a small lower jaw, prominent eyes, pinched nose, and thin upper lip. The sister had no congenital anomalies and her motor and language development were normal. At this evaluation, she reported several episodes of spontaneous small superficial vessel ruptures on her hands and lower extremities. She had surgery at 16 and 19 years because of bilateral rupture of medial meniscus and anterior cruciate ligament, respectively, due to skiing and tennis trauma. For years, she had intermittent stomach aches, periodic diarrhea, and occasionally she observed red blood in her stool. She had regular migraines with aura since early teenage years and tinnitus for the last few months. On examination she had thinned skin on her knees. She did not have varicose veins. She had a normal armspan to height ratio, but a reduced upper to lower segment ratio. Her Beighton score for joint mobility was zero, but she had small joint hypermobility in her hands with positive thumb and wrist signs.
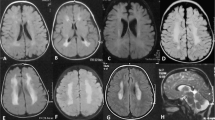
Sequence analysis of COL3A1 identified biallelic sequence variants (see below) consistent with the diagnosis of vascular EDS in III-3. Because of her symptoms, supplemental clinical examinations were done. Aortic dimensions were normal throughout the length of the vessel. Ultrasound revealed a stenotic process in the right external iliac artery (data not shown). Echocardiography revealed a structurally normal heart with minimal late-systolic mitral leakage. The stiffness index of aortic wall elasticity was at the upper limits of normal.8,9 An abdominal CT scan showed increased wall thickness of the transverse colon; however, stool calprotectin was normal, consistent with a low risk of inflammatory bowel disease. Owing to the risk of perforation, colonoscopy was not done. MRI scan of her small bowel was performed with normal results. Capsule endoscopy was planned, but not conducted due to patient distress. MR arteriography (Figure 1a) and CT angiography revealed a stenotic process in her right internal carotid (ICA) and ultrasonography of neck vessels proved to represent a dissection (data not shown). The left ICA was ectatic. Follow-up MR imaging 1 year later showed a newly established network of collateral vessels along the previously dissected right ICA, and also verified a dissection flap in the ectatic left ICA (Figure 1b). She had cerebral cortical dysplasia with thickened frontoparietal cortices bilaterally, small gyri and findings consistent with pachy micropolygyria (Figures 1c and d). She had normal cognitive functioning, a recent neurological examination was normal and she had not experienced epileptic seizures.

(a) MR angiography (MRA) of patient III-3, coronal view; Cr: cranial, Ca: caudal, R: right, L: left, thick arrows: internal carotid arteries; thin arrows: vertebral arteries; arrowhead: basilar artery. There is occlusion/subocclusion along the dissected right ICA. The left ICA is ectatic with irregular caliber in the precranial segment. (b) 3T MRA 10 months later reveals an established collateral network along the dissected segment of the right ICA indicating recanalized arterial segment, and also verifies a dissection flap along the ectatic left ICA. The caliber of the right ICA is still smaller than that of the left. (c) MRI of the head, T2-weighted axial view; A: anterior, P: posterior, R: right, L: left, and (d) T1-weighted saggital view from left side. Both views showing thickened cortices bilaterally, predominantly frontal but extending over parietal lobes limited by the central sulci, small gyri and findings consistent with pachy micropolygyria.
The mother of the siblings (II-6) was diagnosed with multiple sclerosis in her thirties and presented with mild spastic paraparesis and disturbed ambulation. She had rectal incontinence. All three childbirths had been without complications. She did not have facial features characteristic of EDS type IV. She had thinning of skin on her legs and knees, and visible subcutaneous vessels on her chest and back. She did not have varicose veins. Scars were normal. Her joint mobility was normal, but she had positive wrist and thumb signs. She had no arterial aneurysms but echocardiography identified reduced elasticity of the ascending aorta. A CT scan of her lungs revealed bilateral emphysema. As a part of the evaluation of the family, the 57-year-old maternal aunt of the proband (II-7) and her two children were referred for counseling and clinical examination. None of them exhibited clinical features of EDS, and they all had echocardiography done with normal results. The maternal grandfather (I-3) died from cerebral hemorrhage at age 43. His wife (I-4), mother of II-6, died in her sixties due to stomach cancer.
The father of the proband (II-5) was healthy at age 63. On clinical examination, he had neither facial nor skin features suggestive of EDS type IV. Echocardiography and aortic elasticity index were normal. Two of his healthy siblings sought counseling. A 47-year-old paternal aunt of the proband (II-3) had no abnormal echocardiographic findings. She had onset of varicose veins in her twenties, and had repeated surgery to remove them without complications. A healthy 61-year-old paternal uncle (II-4) of the proband had umbilical and inguinal hernias. He had normal results on cardiologic examination. The 85-year-old paternal grandmother (I-2) was healthy except for angina pectoris.
DNA studies
The proband (III-2) and his sister (III-3) were each heterozygous for two COL3A1 sequence variants: c.1786C>T, p.(Arg596*) in exon 26, which encodes part of the triple helical domain of proα1(III), and c.3851G>A, p.(Gly1284Glu) in exon 50, which encodes a portion of the carboxyl-terminal propeptide (submitted to https://eds.gene.le.ac.uk/home.php?select_db=COL3A1 as AN_003368 (DB-ID COL3A1_00451 and DB-ID COL3A1_005855,10; Figure 2a). In DNA from the proband, there were no sequence variants in FBN1, TGFBR1, and TGFBR2. The mother (II-6) was heterozygous for the COL3A1 c.3851G>A, p.(Gly1284Glu) sequence variant, and the father (II-5) was heterozygous for the COL3A1 c.1786C>T, p.(Arg596*) sequence variant, which confirmed that the variants were in trans. The unaffected brother (III-1) of the proband had neither variant. Mutation status in the extended family is displayed in Table 1. None of the individuals with the variant in the father’s family had complications or clinical features consistent with the diagnosis of vEDS.

Analysis of COL3A1 genomic sequence and of collagenous proteins synthesized in dermal fibroblasts. (a) Single amino acid codons of gDNA sequencing results for family members as indicated. (b) Protein electrophoresis studies of cultured dermal fibroblasts: Procollagens were separated under reducing conditions on SDS-PAGE. In proteins secreted into the medium from the individual with two mutant alleles (III-3), the amount of type III procollagen and collagen, α1(III)3, are reduced and the electrophoretic mobility of the proα1(III) chains (b) and of the trimer of type III collagen (c) are slowed. The amount of type III procollagen secreted by the cells from II-5 was reduced. There was subtle shift in electrophoretic mobility of some proα1(III) chains made by the cells from II-6.
Protein studies
Cultured dermal fibroblasts from III-3 produced a reduced amount of type III procollagen, the chains of which all had an abnormal electrophoretic mobility (Figure 2b). In the collagens (produced by removal of the propeptides with pepsin), there is a small amount of α1(III) chains that migrates just above the normal α1(I) chains that is consistent with failure of formation of intermolecular disulfide bond formation by the two cysteine residues at the end of the triple helical domain. We have noted this effect with sequence variants that alter glycine residues in the last 30 residues of the triple helical domain (unpublished observations). Dermal fibroblasts from II-6, heterozygous for the missense mutation, produced mostly normal and a small amount of abnormal type III procollagen in which there was subtle overmodification of the chains. Dermal fibroblasts from II-5 heterozygous for the nonsense mutation produced a reduced amount of apparently normal type III procollagen.
Discussion
COL3A1 encodes the proα1(III) chains of type III procollagen, a homotrimer formed by the association of three proα1(III) chains into a procollagen molecule that is proteolytically cleaved to collagen by removal of peptides from both ends. The protein is an integral component of the walls of hollow organs. About 2/3rds of the described sequence variants result in substitutions for the glycine residues in the 342 uninterrupted Gly-X-Y triplets of the triple helical domain and most of the rest alter splice sites (unpublished data from the Collagen Diagnostic Laboratory, Department of Pathology, University of Washington; Ehlers–Danlos Syndrome variant databases https://eds.gene.le.ac.uk/home.php?select_db=COL3A1 ). Both types of variants generally result in in-frame alterations of sequence. A dominant negative effect leads to seven out of eight molecules that have one or more abnormal chains with associated defects in secretion, processing, and incorporation into extracellular fibrils.11 Deletions or duplications of single exons or the whole COL3A1 gene constitutes a small proportion of the cases (~2%). Fewer than 5% of all families identified with vEDS have variants that result in mRNA instability or proα1(III) chain instability such that only half the normal amount of type III procollagen is produced.2
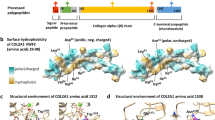
The carboxyl-terminal propeptide must fold into an appropriate structure stabilized by disulfide bonds, after which trimer assembly is mediated by a unique sequence in the propeptide that determines the specificity of chain–chain interaction. Sequence variants that alter sequences in the propeptide can result in failure of chain association, in which case the proα1(III) chain is usually very unstable. Alternatively, a change in the rate of association can result in increased modification of the triple helical domain of the chain. The affected siblings in our study were compound heterozygotes for a missense mutation that altered the sequence of the carboxyl-terminal propeptide and a nonsense mutation that presumably led to instability of the mRNA from that allele. In the presence of both variants, cultured dermal fibroblasts produced a reduced amount of type III procollagen, and the chains of which all had abnormal electrophoretic mobility were consistent with overmodification. Overmodification results from slow folding of the triple helical domain and reflects increased hydroxylation glycosylation of lysyl residues.12 Substitution of glycine by glutamic acid at position 1284 of the proα1(III) chain (p.Gly1284Glu) predicts the change of a highly conserved amino acid that is adjacent to the one of two cysteine residues involved in interchain, intramolecular disulfide bond formation that initially stabilizes the trimer, and is also embedded in a six-residue domain that coordinates a calcium ion and appears to facilitate the interaction of three chains13 (Figure 3). The large charged side chain should distort the calcium-binding region, and on this basis the effect observed on post-translational modification must slow the assembly or stabilization of the proα1(III) chain trimer, and may distort the region near the carboxyl-terminal end of the triple helix responsible for initiation of triple helix propagation.

Sequence of the C-terminal propeptide of the proα1(III) chain. The sequence begins with 1 (residue 1222 of the chain) that is the site of the enzymatic cleavage from the remainder of the chain. The last residue, 245 in this figure, is position 1466 of the proα1(III) chain, the last encoded residue of the chain. There are eight cysteine residues in the chain (numbered 1–8). There are three sets of intrachain disulfide bonds (1–4, 5–8 and 6–7). Cysteine residues 2 and 3 are involved in interchain disulfide bonds that stabilize the trimer. The glycine (G) that precedes cysteine 3 is substituted by glutamic acid (E) in the products of one of the mutant alleles. ▴, regions of α-helix; ▪, regions of β-sheet; •, residues involved in coordinating a Ca++ ion. The standard single letter code is used to designate amino acids. The chain-association region is underlined and the regions of the structure as determined by Bourhis et al13 are described (stalk, base, and petal). This figure is adapted from Figure 1 along with information from publication by Bourhis et al, and the spacing of residues is retained to account for regions that are missing in the C-propeptides of other fibrillar collagen genes.
To our knowledge there has been one previous publication on a patient with biallelic COL3A1 variants.4 The individual described was an 11-year-old girl homozygous for a single basepair duplication that led to a downstream premature termination codon c.479dupT, p.(Lys161Glnfs*45) that was shown to cause mRNA decay and production of no type III procollagen. She developed acute abdominal pain, and during surgery when attempting to repair a necrotic and perforated jejunum, extreme intestinal arterial and tissue fragility were encountered. The girl died 6 days after intervention from multisystem failure. In addition to clinical features, characteristic of EDS type IV, she had delayed motor development, pulmonary valve stenosis, and absence epilepsy. Her brain MRI revealed diffuse cortical dysplasia with a predominantly frontal location and ventricular dilatation.
The girl reported by Plancke et al4 and our proband’s sister both had brain alterations that, to our knowledge, have not been reported in individuals with vEDS due to heterozygosity for dominant variants. We cannot be sure if the vEDS phenotype with biallelic COL3A1 sequence variants and cerebrocortical findings in two separate patients are coincidental findings, and we recognize that the two phenotypes could reflect the presence of variants in a second gene. Type III collagen appears to function in cerebral cortical development through its role in pial basal membrane organization.14 The protein serves as the ligand for GPR56, a member of the G-protein-coupled receptor family.15 Sequence variants in GPR56 cause recessively inherited bilateral frontoparietal polymicrogyria, a severe developmental anomaly associated with mental retardation and seizures in humans.16,17 Our clinical findings and those described by Plancke et al4 suggest that the interaction of GPR56 and type III (pro)collagen identifies two parts of a pathway involved in frontotemporal brain formation. This complex developmental process depends on gene network regulation,18 and we cannot exclude the possibility that mutations in other genes involved in brain development could explain the cortical dysplasia in our patient.
Although each parent carried one of the two sequence variants that we identified, the father (c.1786C>T, p.(Arg596*)) did not have abnormal clinical findings at age 63 and the mother (c.3851G>A, p.(Gly1284Glu)) did not express a complete picture of vascular-type EDS by age 60. We do not know if the maternal grandfather, who died from cerebral hemorrhage in his early forties, was heterozygous for the missense mutation. If so, this would be consistent with a pattern of variable expression in families with this type of sequence variant.
The father of the siblings harbored the nonsense mutation (c.1786C>T, p.(Arg596*)) that was predicted to lead to mRNA instability, and was shown to give no protein expression from that allele (Figure 2b). Several family members on the paternal side who were heterozygous for the variant had no major cardiovascular findings. In other contexts, COL3A1 null mutations occur with clinical manifestations that, when present, primarily involve the vascular system but with later age at onset, compared with individuals with missense mutations, or no manifestations at all2 consistent with findings in this arm of the family.
Our study mirrors findings in Marfan syndrome, in which almost all individuals are heterozygous for sequence variants in FBN1. Three studies have now identified families in which there are probands with biallelic FBN1 variants in families with affected individuals with Marfan features among maternal and paternal relatives.19, 20, 21 In these families there are both healthy heterozygotes and heterozygotes with a partial or complete Marfan syndrome phenotype and in one family the proband had a lethal newborn phenotype that resembled neonatal MFS.20
Sequence variants in COL3A1 that lead to premature termination codons and mRNA instability, and variants that alter sequences in the carboxyl-terminal propeptide are both less frequent than would be expected based on experience of variants in another fibrillar collagen gene, COL1A1. In COL3A1, fewer than 5% of the identified sequence variants result in premature termination codons,2 and only a handful alter sequences in the carboxyl-terminal propeptide. Close to half the variants identified in COL1A1 result in the creation of premature termination codons and a mild phenotype of osteogenesis imperfecta (OI), OI type I (see Database of Collagen Mutations https://oi.gene.le.ac.uk/home.php?select_db=COL1A1, and unpublished data from the Collagen Diagnostic Laboratory, Department of Pathology, University of Washington). There is nothing in the structure of the two genes that would appear to lead to the difference in frequency with which such variants are observed in the clinical setting. Thus, it is likely that the phenotype that results from nonsense mutations in COL3A1 often does not bring people to clinical attention, or the manifestations occur at an age when variants in this gene would not be considered likely. In addition, variation in other genes may influence the penetrance of the genotype, and there do seem to be important modifying factors that remain to be discovered to further explain the marked variation in expression in patients with null variants.
There are two mouse models for COL3A1 ‘null’ alleles. One was engineered to remove the promotor and the first two exons of the gene, whereas the second was a spontaneous 185 kb deletion that removes exons 1–39 of COL3A1 and about 145 kb of upstream DNA that does not contain protein coding genes.22,23 Consistent with the idea that the genetic background can influence the phenotype, heterozygous mice from the first group do not develop aneurysms or dissection and most of the homozygotes die in the peripartum period. In contrast, the heterozygous mice with the large deletion often develop aortic dissection or rupture and die. The first mice are on a BALB/c background, while the second have the mutation on a mixed background that includes C57BL/6 and 129Ola. No cerebral imaging was reported on homozygous mice from the first group. This would have been of interest in regard to the possibility of cerebral anomalies as an autosomal recessive phenotype. In the Smith et al23 study, no mice homozygous for the deletion were ever identified. Histological examination of brain tissue in any surviving knockouts from this line would also be of interest as mentioned above in regard to possible cerebral anomalies.
Treatment of mice in the first group with low-dose doxycycline, an inhibitor of metalloproteinases, appeared to change aortic morphology.24,25 No similar trials have been reported with the second group, which could provide a better model because of the chance to affect the rate of aortic rupture and dissection in the heterozygotes and offer the potential to expand the limited therapy available.26
In conclusion, our report provides a rare explanation for variable expression of the vascular EDS phenotype in families with COL3A1 sequence variants and, further, suggests that a novel syndromic form of recessively inherited vascular Ehlers–Danlos includes both tissue fragility and cerebrocortical anomalies.
References
Pepin M, Schwarze U, Superti-Furga A, Byers PH : Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N Engl J Med 2000; 342: 673–680.
Leistritz DF, Pepin MG, Schwarze U, Byers PH : COL3A1 haploinsufficiency results in a variety of Ehlers-Danlos syndrome type IV with delayed onset of complications and longer life expectancy. Genet Med 2011; 13: 717–722.
Sulh HM, Steinmann B, Rao VH et alEhlers-Danlos syndrome type IV D: an autosomal recessive disorder. Clin Genet 1984; 25: 278–287.
Plancke A, Holder-Espinasse M, Rigau V, Manouvrier S, Claustres M, Khau Van KP : Homozygosity for a null allele of COL3A1 results in recessive Ehlers-Danlos syndrome. Eur J Hum Genet 2009; 17: 1411–1416.
Dalgleish R : The human type I collagen mutation database. Nucleic Acids Res 1997; 25: 181–187.
Bonadio J, Holbrook KA, Gelinas RE, Jacob J, Byers PH : Altered triple helical structure of type I procollagen in lethal perinatal osteogenesis imperfecta. J Biol Chem 1985; 260: 1734–1742.
Beighton P, De PA, Steinmann B, Tsipouras P, Wenstrup RJ : Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos national foundation (USA) and Ehlers-Danlos support group (UK). Am J Med Genet 1998; 77: 31–37.
O'Rourke MF, Staessen JA, Vlachopoulos C, Duprez D, Plante GE : Clinical applications of arterial stiffness; definitions and reference values. Am J Hypertens 2002; 15: 426–444.
Rosca M, Magne J, Calin A, Popescu BA, Pierard LA, Lancellotti P : Impact of aortic stiffness on left ventricular function and B-type natriuretic peptide release in severe aortic stenosis. Eur J Echocardiogr 2011; 12: 850–856.
Dalgleish R : The Human Collagen Mutation Database 1998. Nucleic Acids Res 1998; 26: 253–255.
Myllyharju J, Kivirikko KI : Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet 2004; 20: 33–43.
Kirsch E, Krieg T, Remberger K, Fendel H, Bruckner P, Muller PK : Disorder of collagen metabolism in a patient with osteogenesis imperfecta (lethal type): increased degree of hydroxylation of lysine in collagen types I and III. Eur J Clin Invest 1981; 11: 39–47.
Bourhis JM, Mariano N, Zhao Y et alStructural basis of fibrillar collagen trimerization and related genetic disorders. Nat Struct Mol Biol 2012; 19: 1031–1036.
Jeong SJ, Li S, Luo R, Strokes N, Piao X : Loss of Col3a1, the gene for Ehlers-Danlos syndrome type IV, results in neocortical dyslamination. PLoS One 2012; 7: e29767.
Luo R, Jeong SJ, Jin Z, Strokes N, Li S, Piao X : G protein-coupled receptor 56 and collagen III, a receptor-ligand pair, regulates cortical development and lamination. Proc Natl Acad Sci USA 2011; 108: 12925–12930.
Chang BS, Piao X, Bodell A et alBilateral frontoparietal polymicrogyria: clinical and radiological features in 10 families with linkage to chromosome 16. Ann Neurol 2003; 53: 596–606.
Piao X, Hill RS, Bodell A et alG protein-coupled receptor-dependent development of human frontal cortex. Science 2004; 303: 2033–2036.
Radmanesh F, Caglayan AO, Silhavy JL et alMutations in LAMB1 cause cobblestone brain malformation without muscular or ocular abnormalities. Am J Hum Genet 2013; 92: 468–474.
de Vries BB, Pals G, Odink R, Hamel BC : Homozygosity for a FBN1 missense mutation: clinical and molecular evidence for recessive Marfan syndrome. Eur J Hum Genet 2007; 15: 930–935.
Karttunen L, Raghunath M, Lonnqvist L, Peltonen L : A compound-heterozygous Marfan patient: two defective fibrillin alleles result in a lethal phenotype. Am J Hum Genet 1994; 55: 1083–1091.
van Dijk FS, Hamel BC, Hilhorst-Hofstee Y et alCompound-heterozygous Marfan syndrome. Eur J Med Genet 2009; 52: 1–5.
Liu X, Wu H, Byrne M, Krane S, Jaenisch R : Type III collagen is crucial for collagen I fibrillogenesis and for normal cardiovascular development. Proc Natl Acad Sci USA 1997; 94: 1852–1856.
Smith LB, Hadoke PW, Dyer E et alHaploinsufficiency of the murine Col3a1 locus causes aortic dissection: a novel model of the vascular type of Ehlers-Danlos syndrome. Cardiovasc Res 2011; 90: 182–190.
Briest W, Cooper TK, Tae HJ, Krawczyk M, McDonnell NB, Talan MI : Doxycycline ameliorates the susceptibility to aortic lesions in a mouse model for the vascular type of Ehlers-Danlos syndrome. J Pharmacol Exp Ther 2011; 337: 621–627.
Tae HJ, Marshall S, Zhang J, Wang M, Briest W, Talan MI : Chronic treatment with a broad-spectrum metalloproteinase inhibitor, doxycycline, prevents the development of spontaneous aortic lesions in a mouse model of vascular Ehlers-Danlos syndrome. J Pharmacol Exp Ther 2012; 343: 246–251.
Ong KT, Perdu J, De BJ et alEffect of celiprolol on prevention of cardiovascular events in vascular Ehlers-Danlos syndrome: a prospective randomised, open, blinded-endpoints trial. Lancet 2010; 376: 1476–1484.
Acknowledgements
We gratefully acknowledge the family members described herein and their agreement on publication of these cases. We thank the University Hospital of North Norway for creating space for work like this, and the Medical Genetics Department at Oslo University Hospital for Marfan syndrome-related DNA analysis in the proband. Funds from the Freudmann Fund for Research in Ehlers–Danlos Syndrome at the University of Washington and from the Department of Pathology provided partial support for these studies.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Jørgensen, A., Fagerheim, T., Rand-Hendriksen, S. et al. Vascular Ehlers–Danlos Syndrome in siblings with biallelic COL3A1 sequence variants and marked clinical variability in the extended family. Eur J Hum Genet 23, 796–802 (2015). https://doi.org/10.1038/ejhg.2014.181
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2014.181
This article is cited by
-
The Ehlers–Danlos syndromes
Nature Reviews Disease Primers (2020)
-
Periodontal Ehlers–Danlos syndrome is associated with leukoencephalopathy
neurogenetics (2019)
-
Vascular Ehlers-Danlos Syndrome Presenting as a Pulsatile Neck Mass: a Case Report and Review of Literature
Journal of General Internal Medicine (2018)
