
Abstract
Neuropathic pain-like joint symptoms (NP) are seen in a proportion of individuals diagnosed with osteoarthritis (OA) and post total joint replacement (TJR). In this study, we performed a genome-wide association study (GWAS) using NP as defined by the painDETECT questionnaire (score >12 indicating possible NP) in 613 post-TJR participants recruited from Nottinghamshire (UK). The prevalence of possible NP was 17.8%. The top four hits from the GWAS and two other biologically relevant single-nucleotide polymorphisms (SNPs) were replicated in individuals with OA and post TJR from an independent study in the same area (N=908) and in individuals from the Rotterdam Study (N=212). Three of these SNPs showed effect sizes in the same direction as in the GWAS results in both replication cohorts. The strongest association upon meta-analysis of a recessive model was for the variant allele in rs887797 mapping to the protein kinase C alpha (PRKCA) gene odds ratio (OR)possNP=2.41 (95% CI 1.74–3.34, P=1.29 × 10−7). This SNP has been found to be associated with multiple sclerosis and encodes a functional variant affecting splicing and expression of the PRKCA gene. The PRKCA gene has been associated with long-term potentiation, synaptic plasticity, chronic pain and memory in the literature, making this a biologically relevant finding.
Similar content being viewed by others

Introduction
Neuropathic pain-like joint symptoms (NP) have been reported in people with osteoarthritis (OA) of the knee or hip and in some people who have undergone total-joint replacement (TJR) for OA.1, 2 Estimates for NP post-TJR range from 1% to 63% in the literature depending on the methodology.2, 3, 4
Neuropathic pain is defined as ‘pain arising as a direct consequence of a lesion or disease affecting the somatosensory system’, adapted from the International Association for the Study of Pain (IASP) definition.5 Symptoms can include burning, hypersensitivity, prickling and numbness in both the affected areas and areas of the body distant from the site of damage.6 Treatments for NP have been reported to be of limited effectiveness for many individuals and the condition can have a large impact on quality of life.7, 8 There are numerous risk factors for NP identified in the literature such as nerve damage from surgery, chronic nociceptive input (as seen in chronic pain), complications from herpes zoster infection and diabetes.9, 10 There are some common risk factors for OA pain and NP such as age, past joint surgery and psychological factors.1, 7, 11
Heritability of NP has been estimated at 37% in the single published twin study on NP in humans.12 This is within the range of heritability estimates for other painful conditions such as back pain, migraine and sciatica that range from 21 to 58%.13, 14, 15, 16, 17
There have been numerous candidate gene studies on pain, including chronic pain post surgery.18 Genes reported in the literature on NP from candidate gene studies include the COMT gene, TRPV1 gene, P2X receptor genes and the CACNG2 gene.19, 20, 21, 22 The genetics of NP are still not fully understood.23 NP is thought to have distinct genetic mechanisms, and different types of hypersensitivity (eg, to heat or mechanical stimuli) and, according to mouse studies, different molecular mechanisms may be involved depending on the method for inducing NP.23
A genome-wide association scan (GWAS) can be used to study the genetic basis of complex traits so is an appropriate design to study NP which can have a complex aetiology. GWAS identifies the genetic locations (single-nucleotide polymorphisms (SNPs)) that differ significantly between cases and controls for a specific phenotype. The genes in which these loci are located offer clues about the mechanisms behind the phenotype.
To date only one GWAS has been published on NP, in individuals with diabetic neuropathy. Results from this GWAS identified SNPs in the GFRA2 and ZSCAN20 genes.24, 25 Zinc finger proteins are potentially relevant in the treatment of NP.26 Previous GWAS for migraine and chronic widespread pain (CWP) have identified susceptibility loci relating to genes involved in synaptic plasticity and some types of neuropathy, respectively.27, 28 A GWAS has also been published on acute post-surgical pain.29
The aim of this study was to identify genes associated with the risk of NP in individuals post TJR using a genome-wide approach. The replication analysis aimed to reproduce these findings in other groups containing individuals with knee and hip OA and knee pain.
Methods
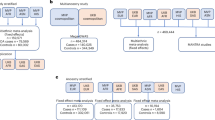
The study design is outlined in Figure 1.

Study design.
Participants
Nottingham discovery cohort
Participants were recruited post-total hip or knee replacement for OA (n=613) from secondary care in the Nottinghamshire area.
Nottingham replication cohort
Participants from an independent Nottingham-based study (n=908) including individuals with knee OA, hip OA, or both and individuals post-total hip or knee replacement were used as a replication cohort.
The North Nottinghamshire Research Ethics Committee gave approval for the ethics of both studies. All participants gave written, informed consent.
To improve statistical power, in each of the above two Nottingham groups, total hip replacement participants and total knee replacement participants were combined into one post-TJR group, as seen in previous GWAS analyses.
The Rotterdam study
The selected individuals were part of Rotterdam Study III (RS-III) that was started in 2006 and comprised of in total 3932 participants. A total of 212 women that reported knee pain had data on painDETECT and genetic data. This population-based cohort study has been previously described and is studied in the context of chronic disabling diseases in older adults.30 The Erasmus University Medical School medical ethics committee gave approval for this study. All participants gave written, informed consent.
Stage 1: GWAS
Blood samples from the participants in this study were processed to obtain genotype data. Genotype data were analysed using the Illumina 610 k array (https://www.ebi.ac.uk/ega/studies/EGAS00001001017). Only directly typed SNPs were used. Genotyping and QC were carried out as previously described.31 gPLINK software (version 1.07) was used to analyse GWAS data from this array.32 The results of this association are a list of genetic variants (SNPs) and information about their location in the genome, as well as an odds ratio (OR), chi-square value and P-value to indicate the level of association of the variants with the specified phenotype.
The results of this GWAS have been submitted to GWAS Central and can be accessed at: http://www.gwascentral.org/study/HGVST1846.
The statistics program R (version 3.0.2) was used to create Manhattan and QQ plots using the ‘ggplot2’ library and ‘qqplot’ script.
Post-genomic analysis was undertaken using the Database for Annotation, Visualisation and Integrated Discovery (DAVID).33 This is an online tool to which a list of genes can be submitted and subsequently results are generated regarding the genes’ involvement in biological processes.33 The gene list was comprised of genes corresponding to all SNPs with a P-value of P<0.0001 in the GWAS analysis. The BioCarta and Kegg pathways maps were used for functional annotation.
Stage 2: replication cohorts
The top four SNPs with a nominal P-value of P≤5 × 10−6 after the stage 1 GWAS analysis and two additional lower ranking but potentially biologically relevant SNPs were selected for replication (see Results). Genotype information for these SNPs from in silico and de novo genotype data were used for further analysis. In total, six SNPs were selected for replication analysis.
Stage 3: meta-analysis
The ‘meta’ library in the statistics program R (version 3.0.2) was used to run the meta-analysis using the three cohorts described above. Meta-analysis takes the effect size, standard error and sample size into account to give an overall effect from the different groups studied. If heterogeneity was significant between the cohorts in the meta-analysis, a Han Eskin random effects model was used as an alternative meta-analysis method as, compared with traditional models, it allows for more heterogeneity in the data.34
Phenotype
Individuals were assigned a phenotype by classifying them according to their scores on the painDETECT questionnaire. This is a seven-item questionnaire scored from 0 to 39 that uses a Likert scale for participants to describe the nature of their pain, in order to distinguish it from nociceptive pain. Questions are included on qualities such as burning pain, tingling, sudden pain and sensitivity to heat and cold. In all cohorts, scores of >12 were classified as ‘possible neuropathic pain’ according to the validated cut-offs for diagnosis by Freynhagen et al.35
This cut-off and not the stricter one of ≥19 for ‘likely NP’ was used to increase statistical power given the larger number of cases available and the similar clinical characteristics, particularly the use of anti-neuropathic medication in our data (possible NP 38.5% opioids, 11% antineuropathics, likely NP 37% opioids, 11% antineuropathics). Using this definition, we therefore had n=109 possible NP cases and n=504 controls. This gave a statistical power of 42% to detect an association with an OR of 2.1, and 97% power to detect an association with an OR of 2.9 with P<5 × 10−7 for a minor allele frequency (MAF) of 35%.
Results
Stage 1: GWAS
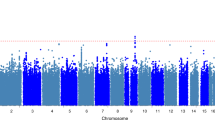
The results of the unadjusted GWAS on NP can be seen in Table 1, and Figures 2 and 3. A total of 548 381 SNPs were tested for association with NP. The genomic control inflation factor for the P-values was low (λ=0.99) and the quantile–quantile (QQ) plot indicated no substantial population stratification due to cryptic relatedness, population substructure or other biases (Figure 2).

QQ plot for the results of the GWAS (λ=0.99).

Manhattan plot showing the P-value of association tests for SNPs with possible NP in the Illumina array GWAS. P-values represent the association of the SNPs with possible NP.
The results of the GWAS are summarised in Manhattan plots of the P-values (Figure 3). Table 1 shows the OR and significance of the results from the Illumina array NP GWAS for four of the top-scoring SNPs and two SNPs of biological relevance. The top four SNPs with P≤5 × 10−6 were selected for further replication, as were two SNPs with higher P-values but in potential candidate genes, rs4866176 mapping to the brain-specific cadherin CDH18 gene and rs1133076 mapping to the thyroglobulin TG gene.
Pathway analysis
Pathway analysis was carried out on the GWAS results using a list of genes corresponding to SNPs with P<0.0001 in the GWAS (n=62; see Supplementary Table S1, Supplements). If the SNP mapped to an area within a gene, this gene was used. For intergenic SNPs, the two closest flanking genes on each side were used. The results of this analysis report no significant findings after adjusting for multiple testing with a Bonferroni correction (see Supplementary Table S2, Supplements).
Stage 2: replication cohorts
We sought to replicate the six selected SNPs for their association with NP in two independent replication cohorts. The results are shown in Table 1. As shown in Table 1, two of the SNPs selected from the GWAS in stage 1 for replication analysis show nominally significant P-values and effects in the same direction in one of the replication cohorts.
Stage 3: meta-analysis
We then combined discovery and replication results in a joint meta-analysis. The results can be seen in Table 1. Heterogeneity of the loci was tested using the Cochran Q test.
Due to the significant heterogeneity introduced to the model by the replication data in the rs887797, rs4866176, rs7734804, rs298235 and rs12596162 meta-analyses, a Han Eskin random effects model was used to account for this (Table 1). The additive model for the rs887797 SNP after this analysis gave a result of: OR=1.48 (95% CI 1.23–1.75), P=1.65 × 10−5. A recessive model for the rs887797 SNP was also used in a meta-analysis. A recessive model was used to test the nature of the effect of the risk allele, that is, to test if two copies of the risk allele were needed to increase the risk of possible NP. After Han Eskin analysis, the recessive model for rs887797 gave a result of OR=2.41 (95% CI 1.74–3.34, P=1.29 × 10−7; Figure 4).

Forest plot showing the results of an unadjusted Han Eskin analysis of the rs887797 SNP using a recessive model.
After adjusting for age, sex and BMI, Han Eskin analysis of the rs887797 SNP gave values of: ORpossNP=1.44 (95% CI 1.21–1.73, P=7.13 × 10−5) and ORpossNP=2.33 (95% CI 1.67–3.27, P=8.67 × 10−7) for the additive and recessive models, respectively. Upon combining the data from the two replication cohorts used, it was found that overall this SNP was nominally significant. The additive model for the rs887797 SNP in the combined Nottingham replication cohort and Rotterdam Study cohort gave OR=1.25 (95% CI 1.01–1.55), P=0.040 (not significant after adjusting for seven tests) and the recessive model gave OR=1.75 (95% CI 1.15–2.64), P=0.0076 (Bonferroni P-value P<0.053).
Finally, we attempted to replicate two of the top hits from the only published GWAS on NP. These SNPs were reported to be suggestively associated with diabetic neuropathy: rs17428041 (GFRA2, OR=0.67, P=1.77 × 10−7)24 and rs71647933 (ZSCAN20, OR=2.31, P=4.88 × 10−7).25 The effect of rs17428041 was not replicated in the results of our GWAS: OR=1.47, P=0.016. Similarly, after using a proxy for rs71647933 (rs12565140, r2=0.947) we found no association with NP in the results of our GWAS: OR=0.71 (95% CI 0.46–1.09, P=0.12).
Discussion
We report a suggestive association between a variant in the PRKCA gene and NP in people with knee pain, knee or hip OA and post TJR. The findings are biologically plausible and supported by previously published work in the literature. We were unable to confirm the recently published association between SNPs in the GFRA2 and ZSCAN20 genes and diabetic neuropathy.24, 25 However, it should be noted that diabetic neuropathy is not necessarily the same phenotype as neuropathic pain-like joint symptoms. The definition of NP used in these studies was partly based on use of prescription analgesic medication and partly on the results of sensory testing. However, this type of medication is commonly used even by people with no NP, including people post TJR with no NP. In our study, a validated screening questionnaire (painDETECT) was used, the location of pain is exclusively that of the OA-affected joint and further clinical history and demographics have been collected for all participants.
The top hit from our analysis maps to the PRKCA gene. This gene codes for protein kinase C alpha, a protein that has been linked with the nervous system and may contribute to central sensitisation in dorsal horn neurons.36 The PRKCA gene has also been found in the literature to be involved in long-term potentiation (LTP), a process involved in both memory and chronic pain.37 As well as this, the PRKCA gene has been implicated in related processes such as memory capacity and post-traumatic stress disorder (PTSD)38 and genetic variation in this gene has been linked to the neural basis of episodic memory.39 Although we do not reach the P<5 × 10−8 threshold for GWS, we show a plausible effect on NP post TJR.40 A role for the PRKCA gene in pain has been previously reported.41 The rs887797 variant identified in this paper is a variant already associated with multiple sclerosis.42 Therefore, although this association may not reach GWS it remains biologically plausible, although further work is needed to confirm its role on NP.
In the present GWAS, the intergenic rs12596162 SNP near the FOXL1 gene was associated with NP: OR=2.05 (95% CI 1.51–2.79), P=3.53 × 10−6. This gene codes for a forkhead/winged helix-box transcription factor.43 This gene and the rest of the FOX gene family are involved in many cellular processes.43 FOXL1 in particular was found in one study to be involved in the Wnt/β-catenin pathway44 that is important in the nervous system and has been implicated in NP and hip OA.45, 46
Thyroglobulin, encoded by the TG gene, is a protein necessary for normal thyroid function that has previously been related to NP and central sensitisation in the literature.47 The rs1133076 SNP mapping to this gene was suggested in this analysis to be associated with possible NP at the discovery stage with P=3.41 × 10−4. However this variant did not replicate in the additional cohorts and the evidence for association with NP for this gene is very weak.
The effect sizes we report here are larger than those reported in previous GWAS on pain traits such as migraine and CWP (OR=1.18 and OR=1.23, respectively).27, 28 The effect size for the GWAS on NP in diabetes was 2.31 for the SNP with the lowest P-value, which is consistent with our finding for the rs887797 SNP in the GWAS analysis (OR=2.00, see Table 1).
There are a number of limitations to this study. None of the variants identified by this study reaches GWS. This is not surprising given the small discovery and replication sample sizes available for this kind of study. A major issue with the use of GWAS is the potential for inflated associations.48 The statistical power for the rs887797 recessive model with the observed OR=2.41 was 56% for GWS. For the observed P-value, the statistical power was 66% given the observed minor allele frequency and the rare homozygote frequency (which is in HWE). Although the study was underpowered for GWS, the effect size is relatively large. To achieve 80% power with this effect size and the same proportion of cases to controls we would have needed 417 cases and 1 767 controls, a 25% larger sample size, assuming that in the additional sample the effect was the same.48 Due to the ‘winner’s curse’49 the effect size reported here is likely to be an overestimate given the small sample size used for the discovery phase, and sample sizes of at least twice those that were used are likely to be needed. Furthermore, heterogeneity between the groups used in the meta-analysis can limit the effects seen in the results though we attempted to address this by the use of a Han Eskin Random Effects analysis.34
The absence of a clinical NP diagnosis in these participants is another limitation of this study. However, the results of this questionnaire have been shown to correlate with brain activity in areas associated with NP in people with NP and OA.50
In summary, we report a biologically plausible genetic effects on possible NP in individuals with knee pain, OA and post TJR. Replication in further cohorts could improve sample size and P-values and it is hoped that this GWAS of neuropathic pain-like symptoms of the joint may encourage the collection of DNA and of painDETECT and similar instruments in other cohorts.
References
Hochman JR, Gagliese L, Davis AM, Hawker GA : Neuropathic pain symptoms in a community knee OA cohort. Osteoarthritis Cartilage 2011; 19: 647–654.
Wylde V, Hewlett S, Learmonth ID, Dieppe P : Persistent pain after joint replacement: prevalence, sensory qualities, and postoperative determinants. Pain 2011; 152: 566–572.
Buvanendran A, Kroin JS, Della Valle CJ et al: Perioperative oral pregabalin reduces chronic pain after total knee arthroplasty: a prospective, randomized, controlled trial. Anesth Analg 2010; 110: 199–207.
Haroutiunian S, Nikolajsen L, Finnerup NB, Jensen TS : The neuropathic component in persistent postsurgical pain: a systematic literature review. Pain 2013; 154: 95–102.
Treede R-D, Jensen TS, Campbell J et al: Neuropathic pain redefinition and a grading system for clinical and research purposes. Neurology 2008; 70: 1630–1635.
Kehlet H, Jensen TS, Woolf CJ : Persistent postsurgical pain: risk factors and prevention. Lancet 2006; 367: 1618–1625.
Valdes AM, Suokas AK, Doherty SA, Jenkins W, Doherty M : History of knee surgery is associated with higher prevalence of neuropathic pain-like symptoms in patients with severe osteoarthritis of the knee. Semin Arthritis Rheum 2013; 43: 588–592.
van Hecke O, Austin SK, Khan RA, Smith BH, Torrance N : Neuropathic pain in the general population: a systematic review of epidemiological studies. Pain 2014; 155: 654–662.
Dualé C, Ouchchane L, Schoeffler P et al: Neuropathic aspects of persistent postsurgical pain: a French multicenter survey with a 6-month prospective follow-up. J Pain 2014; 15: p 24.e1–24.e20.
Graven-Nielsen T, Wodehouse T, Langford RM, Arendt-Nielsen L, Kidd BL : Normalization of widespread hyperesthesia and facilitated spatial summation of deep-tissue pain in knee osteoarthritis patients after knee replacement. Arthritis Rheum 2012; 64: 2907–2916.
Novak JC, Lovell JA, Stuesse SL, Cruce WLR, McBurney DL, Crisp T : Aging and neuropathic pain. Brain Res 1999; 833: 308–310.
Momi SK, Fabiane SM, Lachance G, Livshits G, Williams FM : Neuropathic pain as part of chronic widespread pain: environmental and genetic influences. Pain 2015; 156: 2100–2106.
Honkasalo ML, Kaprio J, Winter T et al: Migraine and concomitant symptoms among 8167 adult twin pairs. Headache 1995; 35: 70–78.
Ziegler DK, Hur YM, Bouchard TJ, Hassanein RS, Barter R : Migraine in twins raised together and apart. Headache 1998; 38: 417–422.
Heikkila JK, Koskenvuo M, Heliovaara M et al: Genetic and environmental factors in sciatica. Evidence from a nationwide panel of 9365 adult twin pairs. Ann Med 1989; 21: 393–398.
Larsson B, Bille B, Pedersen NL : Genetic influence in headaches: a Swedish twin study. Headache 1995; 35: 513–519.
Bengtsson B, Thorson J : Back pain: a study of twins. Acta Genet Med Gemellol 1991; 40: 83–90.
Clarke H, Katz J, Flor H et al: Genetics of chronic post-surgical pain: a crucial step toward personal pain medicine. Can J Anesth 2015; 62: 294–303.
Belfer I, Shnol H, Finelli P: Molecular genetics of variability in human pain; in eLS. John Wiley & Sons, Ltd., 2013.
Nissenbaum J, Devor M, Seltzer Z et al: Susceptibility to chronic pain following nerve injury is genetically affected by CACNG2. Genome Res 2010; 20: 1180–1190.
Tsuda M, Kuboyama K, Inoue T, Nagata K, Tozaki-Saitoh H, Inoue K : Behavioral phenotypes of mice lacking purinergic P2X4 receptors in acute and chronic pain assays. Mol Pain 2009; 5: 28.
Valdes AM, De Wilde G, Doherty SA et al: The Ile585Val TRPV1 variant is involved in risk of painful knee osteoarthritis. Ann Rheum Dis 2011; 70: 1556–1561.
Young EE, Costigan M, Herbert TA, Lariviere WR : Heritability of nociception IV: neuropathic pain assays are genetically distinct across methods of peripheral nerve injury. Pain 2014; 155: 868–880.
Meng W, Deshmukh HA, van Zuydam NR et al: A genome-wide association study suggests an association of Chr8p21.3 (GFRA2) with diabetic neuropathic pain. Eur J Pain 2015; 19: 392–399.
Meng W, Deshmukh HA, Donnelly LA et al: A genome-wide association study provides evidence of sex-specific involvement of Chr1p35.1 (ZSCAN20-TLR12P) and Chr8p23.1 (HMGB1P46) with diabetic neuropathic pain. EBioMedicine 2015; 2: 1386–1393.
Krishna SS, Majumdar I, Grishin NV : Structural classification of zinc fingers: survey and summary. Nucleic Acids Res 2003; 31: 532–550.
Anttila V, Stefansson H, Kallela K, Unda Todt U, Terwindt GM, Calafato MS et al: Genome-wide association study of migraine implicates a common susceptibility variant on 8q22.1. Nat Genet 2010; 42: 869–873.
Peters MJ, Broer L, Willemen HLDM et al: Genome-wide association study meta-analysis of chronic widespread pain: evidence for involvement of the 5p15.2 region. Ann Rheum Dis 2013; 72: 427–436.
Kim H, Ramsay E, Lee H, Wahl S, Dionne RA : Genome-wide association study of acute post-surgical pain in humans. Pharmacogenomics 2009; 10: 171–179.
Hofman A, Breteler MB, van Duijn C et al: The Rotterdam Study: objectives and design update. Eur J Epidemiol 2007; 22: 819–829.
Zeggini E, Panoutsopoulou K, Southam L et al: Identification of new susceptibility loci for osteoarthritis (arcOGEN): a genome-wide association study. Lancet 2012; 380: 815–823.
Purcell S, Neale B, Todd-Brown K et al: PLINK: a toolset for whole-genome association and population-based linkage analysis. Am J Hum Genet 2007; 81: 559–575.
Huang, da W, Sherman BT, Lempicki RA : Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4: 44–57.
Han B, Eskin E : Random-effects model aimed at discovering associations in meta-analysis of genome-wide association studies. Am J Hum Genet 2011; 88: 586–598.
Freynhagen R, Baron R, Gockel U, Tolle TR : painDETECT: a new screening questionnaire to identify neuropathic components in patients with back pain. Curr Med Res Opin 2006; 22: 1911–1920.
Kawasaki Y, Kohno T, Zhuang ZY, Brenner GJ, Wang H, Van Der Meer C et al: Ionotropic and metabotropic receptors, protein kinase A, protein kinase C, and Src contribute to C-fiber-induced ERK activation and cAMP response element-binding protein phosphorylation in dorsal horn neurons, leading to central sensitization. J Neurosci 2004; 24: 8310–8321.
Price TJ, Inyang KE : Commonalities between pain and memory mechanisms and their meaning for understanding chronic pain. Prog Mol Biol Transl Sci 2015; 131: 409–434.
de Quervain DJF, Kolassa I-T, Ackermann S et al: PKCα is genetically linked to memory capacity in healthy subjects and to risk for posttraumatic stress disorder in genocide survivors. Proc Natl Acad Sci USA 2012; 109: 8746–8751.
MacLeod CA, Donaldson DI : PRKCA polymorphism changes the neural basis of episodic remembering in healthy individuals. PLoS One 2014; 9: e98018.
Welter D, MacArthur J, Morales J et al: The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Research 2014; 42 (Database issue): D1001–D1006.
Olah Z, Karai L, Iadarola MJ : Protein kinase Cα is required for vanilloid receptor 1 activation: evidence for multiple signaling pathways. J Biol Chem 2002; 277: 35752–35759.
Paraboschi EM, Rimoldi V, Soldà G et al: Functional variations modulating PRKCA expression and alternative splicing predispose to multiple sclerosis. Hum Mol Genet 2014; 23: 6746–6761.
NCBI. FOXL1 forkhead box L1 [Homo sapiens (human)]. Gene 2014. Available at http://www.ncbi.nlm.nih.gov/gene/2300.
Jiang D, Hwang KS, Bordelon Y, Apostolova LG : Plenary paper - cortical atrophy and gene expression in Parkinson's disease with mild cognitive impairment. J Am Geriatr Soc 2013; 61: S3.
Zhang Y-K, Huang Z-J, Liu S et al: WNT signaling underlies the pathogenesis of neuropathic pain in rodents. J Clin Invest 2013; 123: 2268–2286.
Castaño Betancourt MC, Cailotto F, Kerkhof HJ et al: Genome-wide association and functional studies identify the DOT1L gene to be involved in cartilage thickness and hip osteoarthritis. Proc Natl Acad Sci 2012; 109: 8218–8223.
Penza P, Lombardi R, Camozzi F, Ciano C, Lauria G : Painful neuropathy in subclinical hypothyroidism: clinical and neuropathological recovery after hormone replacement therapy. Neurol Sci 2009; 30: 149–151.
Ioannidis JP : Why most discovered true associations are inflated. Epidemiology 2008; 19: 640–648.
Garner C : Upward bias in odds ratio estimates from genome-wide association studies. Genet Epidemiol 2007; 31: 288–295.
Gwilym SE, Keltner JR, Warnaby CE, Carr AJ, Chizh B, Chessell I, Tracey I : Psychophysical and functional imaging evidence supporting the presence of central sensitization in a cohort of osteoarthritis patients. Arthritis Care Res 2009; 61: 1226–1234.
Acknowledgements
SCW was funded by a PhD studentship awarded by the University of Nottingham. This work was supported by a EULAR project grant to AMV (grant no. 108239) and by the Arthritis Research UK Pain Centre (grant no. 18769). We gratefully acknowledge the contributions of Sally Doherty and Maggie Wheeler to patient assessments at baseline, data collection and entry. JM was funded by The Netherlands Society for Scientific Research (NWO) VIDI Grant 917103521. The generation and management of GWAS genotype data for the Rotterdam Study (RS I, RS II, RS III) was executed by the Human Genotyping Facility of the Genetic Laboratory of the Department of Internal Medicine, Erasmus MC, Rotterdam, The Netherlands. The GWAS data sets are supported by the Netherlands Organisation of Scientific Research NWO Investments (no. 175.010.2005.011, 911-03-012), the Genetic Laboratory of the Department of Internal Medicine, Erasmus MC, the Research Institute for Diseases in the Elderly (014-93-015; RIDE2), the Netherlands Genomics Initiative (NGI)/Netherlands Organisation for Scientific Research (NWO) Netherlands Consortium for Healthy Aging (NCHA), project nr. 050-060-810. We thank Pascal Arp, Mila Jhamai, Marijn Verkerk, Lizbeth Herrera and Marjolein Peters, MSc, and Carolina Medina-Gomez, MSc, for their help in creating the GWAS database, and Karol Estrada, PhD, Yurii Aulchenko, PhD, and Carolina Medina-Gomez, MSc, for the creation and analysis of imputed data. The Rotterdam Study is funded by Erasmus Medical Center and Erasmus University, Rotterdam, Netherlands Organization for the Health Research and Development (ZonMw), the Research Institute for Diseases in the Elderly (RIDE), the Ministry of Education, Culture and Science, the Ministry for Health, Welfare and Sports, the European Commission (DG XII), and the Municipality of Rotterdam. We are grateful to the study participants, the staff from the Rotterdam Study and the participating general practitioners and pharmacists.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Warner, S., van Meurs, J., Schiphof, D. et al. Genome-wide association scan of neuropathic pain symptoms post total joint replacement highlights a variant in the protein-kinase C gene. Eur J Hum Genet 25, 446–451 (2017). https://doi.org/10.1038/ejhg.2016.196
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2016.196
This article is cited by
-
Die EAN-NeuPSIG-Leitlinie zur Diagnostik bei neuropathischen Schmerzen – eine Kurzfassung
Der Schmerz (2024)
-
Genome-wide enriched pathway analysis of acute post-radiotherapy pain in breast cancer patients: a prospective cohort study
Human Genomics (2019)
-
The Rotterdam Study: 2018 update on objectives, design and main results
European Journal of Epidemiology (2017)
