
Abstract
Purpose
Aniridia (AN) is a rare congenital panocular disorder caused by the mutations of the paired box homeotic gene 6(PAX6) gene. The PAX6gene is also involved in other anterior segment malformations including Peters anomaly. We studied the PAX6gene mutations in a cohort of affected individuals with different clinical phenotype including AN, coloboma of iris and choroid, or anterior segment malformations.
Patients and methods
Six unrelated families and 10 sporadic patients were examined clinically. After informed consent was obtained, genomic DNA was extracted from the venous blood of all participants. Mutation screening of all exons of the PAX6gene was performed by direct sequencing of PCR-amplified DNA fragments. Multiplex ligation-dependent probe amplification (MLPA) was performed to detect large deletions.
Results
By clinical examination, the patients and the pedigrees were divided into the following three groups: AN, coloboma of iris and choroids, and the anterior segment malformations including peters anomaly. Sequencing of the PAX6gene, three intragenic mutations including a novel heterozygous splicing-site mutations c.357-3C>G (p.Ser119fsX) were identified in the patients of the AN group. A novel missense mutation c.643T>C (p.S216P) was detected in the anterior segment malformation group. The mutation p.S216P located in the homeodomain region of the PAX6 caused the phenotype of Peters anomaly in family A6 with different expressing. Through MLPA analysis, a large deletion including the whole PAX6gene and DKFZ p686k1684gene was detected in one sporadic patient from the AN group. Neither intragenic mutation nor large deletion was identified in the group with coloboma of iris and choroid.
Conclusion
Our findings further confirmed that different kind of mutations might cause different ocular phenotype, and clearly clinical phenotype classification might increase the mutation detection rate of the PAX6gene.
Similar content being viewed by others

Introduction
The paired box homeotic gene 6 (PAX6) gene is a key regulator in the development of the eye and central nervous tissues.1 It was identified as the candidate gene for aniridia (AN) by positional cloning in 1991.2 Aniridia (OMIM 106210) is a rare congenital disorder characterized by the complete or partial absence of the iris. It occurs in the general population with the frequency of about 1 in 64 000–96 000 live births.2 Aniridia may be isolated or accompanied by other ocular abnormalities (corneal opacification, cataract, glaucoma, fovea and optic nerve hypoplasia, and nystagmus).2, 3 About two-thirds of AN cases are families with an autosomal dominant mode of inheritance. In the remaining cases, no family history is found.2
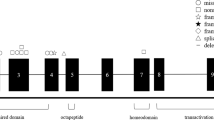
The PAX6 gene is located on chromosome 11p13 and contains 14 exons, including an alternatively sliced exon5a, in 22 kb genomic region. Therefore, there are two isoforms: PAX6 (−5a), comprising 422 amino acids, and PAX6 (+5a), comprising 436 amino acids.2, 3 The PAX6 gene is expressed early in the development of the eye, numerous regions of the brain, and the pancreas.2 The PAX6 contains an N terminal two DNA-binding domains: a bipartite paired domain (PRD), a paired-type homeodomain (HD) separated by a glycine-rich linker sequence, and a C-terminal praline, -serine, -threonine-rich transregulatory domain.2, 3
The PAX6 gene is the only gene identified to be responsible for AN. The mutations are either intragenic mutations or large deletions in the region of the PAX6 gene.1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15 To date, more than 300 intragenic mutations of the PAX6 have been described.1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15 Most of these mutations are nonsense mutations, frame-shifting insertions or deletion, and splicing mutations, which directly or indirectly introduce premature termination codons (PTCs), and are predominantly associated with AN.4, 5 In contrast, missense mutations usually generate distinctive non-AN phenotypes, including anterior segment anomalies, isolated forveal hypoplasia, and optic nerve malformations.4, 5, 10, 11, 12, 13, 14, 15
Peters anomaly is a rare disorder characterized by congenital central corneal opacity and variable degrees of iridolenticulocorneal adhesions. The corneal opacity is usually associated with defects in corneal endothelium, Descemet's member, and posterior stroma. The phenotype of Peters anomaly may vary greatly. Glaucoma may be present in over 50% of cases. Peters anomaly is most often sporadic but may be recessive or occasionally dominant in inheritance.16, 17, 18
In this study, we performed mutations screening of the PAX6 gene in a cohort of affected individuals with different clinical phenotype including AN, coloboma of iris and choroid, or anterior segment malformations including Peters anamoly. Four intragenic mutations and a large deletion were identified in this set of patients.
Patients and methods
Patients and DNA samples collection
This study was performed according to the tenets of the Declaration of Helsinki Principles for research involving human subjects. This study was approved by the Beijing Tongren Hospital Joint Committee on Clinical Investigation. After informed consents were obtained, participants underwent ophthalmologic examination including bilateral best corrected visual acuity using E decimal charts, slit-lamp biomicroscopy inspection of the anterior chamber, intraocular pressure (IOP) measurement by applanation tonometry (Goldmann), and fundus examination with a 66-diopter VOLK lens. Some patients underwent anterior chamber angle evaluation by gonioscopy (Goldmann) and A/B ultrasonic scan examination. On the basis of clinical phenotype, the patients were divided into the following three groups:
-
1)
Aniridia: completed absence of the iris with or without other eye abnormalities.
-
2)
Coloboma of the iris and choroid: part absence of iris (usually in the inferior part) and the corresponding choroid.
-
3)
Anterior chamber malformation: including Peter's anomaly.
Mutation screening of the PAX6 gene
Peripheral blood was obtained by venipuncture and genomic DNA was extracted according to standard protocols. Fourteen exons of the PAX6 gene were amplified by PCR from genomic DNA. Thirteen pairs of primers for the PAX6 gene were used, according to the article previously published.14 For direct sequencing, PCR products were purified (Shenneng Bocai PCR purification kit; Shenneng, Shanghai, China). An automatic fluorescence DNA sequencer (ABI, Prism 373A; Perkin–Elmer, Foster City, CA, USA), used according to the manufacturer's instructions, sequenced the purified PCR products in both forward and reverse directions. DNAssit Version 1.0 (http://ibioo.com) compared nucleotide sequences with the published DNA sequence of PAX6 (GenBank accession number NM_001604.3). For the PAX6 gene cDNA numbering, +1 corresponds to A in the ATG translation initiation codon of isoform PAX6 (−5a).
Multiplex ligation-dependent probe amplification
Multiplex ligation-dependent probe amplification (MLPA) was performed with SALSA MLPA Kits P219 (MRC-Holland b.v., Amsterdam, the Netherlands) according to the manufacturer's instructions. In brief, 100 ng DNA was denatured and hybridized with the SALSA probe mix overnight at 60 °C. The samples with ligase-65 were incubated for 15 min at 54 °C, after which PCR amplification was performed with the specific SALSA FAM PCR primers (MRC-Holland b.v.). The PCR products were separated by capillary electrophoresis on an automatic fluorescence DNA sequencer (ABI, Prism 373A). Data analysis was performed by exporting the peak areas to a Microsoft Excel file. Each peak was first normalized as described elsewhere,14 and the normalized peak was then divided by the mean of that peak in the control samples. The ratios outside the range of 0.7–1.3 times the control peak area were considered abnormal, with those below 0.7 represent deletions, and those above 1.3 represent duplications. For each MLPA analysis, several normal controls were included and the standard deviation for the normal samples was usually <10% of the mean. Each result was confirmed by two independent tests.
Restriction fragment length polymorphism analysis: variations found in the sequencing were confirmed with the restriction endonucleases HPY188I and BtscI (NewEngland Biolabs, Ipswich, MA, USA), which were used with all available family members and 100 normal controls. The reaction was performed in a 10 μl volume containing 9.5 μl PCR product and 0.5 μl enzyme (20 and 10 U/μl, respectively). The reaction was incubated overnight at 37 °C for HPY188I and 50 °C for BtscI. Later, the whole digest was run on a 1% agarose gel and visualized under ultraviolet light.
Bioinformatics analysis
The Garnier–Osguthorpe–Robson (GOR) software was used to predict the effect of the mutation on the secondary structure of PAX6 (http://www.brc.dcs.gla.ac.uk/~drg/courses/bioinformatics_mscIT/slides/slides7/sld017.htm).19 This method may infer the secondary structure of a sequence by calculating the probability for each of the four structure classes (helix, sheet, turn, and loop) based on the central residue and its neighbors from the calculated matrices.
Results
We have recruited six unrelated families (including 21 patients) and 10 sporadic patients. The inheritance pattern in the families was autosomal dominant (Figure 1).

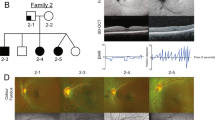
Family structure of six families in this study. Pedigree A1 with aniridia was subdivided into group 1. Pedigrees A2 and A3 with coloboma of iris and choroid were in group 2. Pedigrees A4–A6 with anterior chamber malformation including Peters anomaly were in group 3. Squares indicate males; circles indicate females; slashed symbols indicate deceased; solid symbols indicate affected; open symbols indicate unaffected; and symbols with dot in the center indicate carriers.
In group 1, which included one small pedigree (A1) and three sporadic patients, we identified three intragenic mutations and one large deletion. A splicing mutation c.357-3C>G (p.Ser119fsX32) detected in Family A1 was novel. This mutation co-segregated with the disease phenotype, but was not found in 100 normal controls (Figure 2c). Two remaining mutations c.11-2A>G (p.Ser4AsnfsX21) and 885G>A (p.W156X) identified in patients P3 and P4 have been described previously20, 21 (Figures 2a and b). By MLPA analysis, one large deletion including whole PAX6 gene and the DKFZ p686k1684 gene was found in patient P1 (Figure 3). All patients in this group presented bilateral completed AN, congenital cataract, and nystagmus (Figures 4a and b, Table 1).

DNA sequence chromatograms and co-segregation analysis of four intragenic mutations detected in this study. (a) Sequence presents the heterozygous splicing-site mutation c.11-2A>G (p.Ser4AsnfsX21) detected in patient P2. (b) Sequence shows the heterozygous nonsense mutation c.885G>A (p.W156X) identified in patient P3. (c) Upper, restriction fragment length analysis shows the c.357-3C>G (p.Ser119fsX) mutation abolishing a HPY188I site co-segregated with patients of family A1 (206, 123, and 83 bp), but not with unaffected individuals (123 and 83 bp); lower, sequence shows the heterozygous splicing-site mutation c.357-3C>G. (d) Upper, restriction fragment length analysis shows the mutation c.643T>C (p.S216P) abolishing a BtscI site co-segregated with patients and carrier of family A6 (214, 142, 72, and 30 bp), but not with unaffected individuals (142, 72, and 30 bp); lower, sequence shows the heterozygous missense mutation c.643T>C.

Normalized MLPA results of patient P1 of group 1. The height of the columns represents of the dosage of the respective segments in the genomic DNA with two alleles. The light blue columns represent the 11p13-specific probes. The orange columns represent the deleted probes. The dark blue columns represent the control probes. The allele dosage of the deleted probes was found in the range of about 0.5–0.7 of normal control, which corresponds to one allele.

Ophthalmological findings in patients from the different groups. (a) Photograph of anterior segment of patient P3 with complete absence of iris and congenital cataract. (b) His fundus image was normal. (c) Photograph of anterior segment of the right eye of patient P9 showed coloboma of iris in the inferior part and mild cataract. (d) Fundus image of the right eye of the proband of family A3, showing a large coloboma of choriod. (e) Photograph of anterior segment of the right eye of the proband of family A6, showing sclerocornea with diffuse corneal opacity and blood vessels, hypoplasia of iris. (f) Photographs of anterior segment of the left eye of the same patient of e, showing corneal opacity, iridocorneal adhesions, and anterior polar cataract. (g) Photograph of fundus of the right eye of the same patient of e, showing blurring appearance of the fundus. (h) Photograph of fundus of the right eye of the proband's mother, showing tessellated appearance and posterior staphyloma.
Neither mental retardation nor other general abnormalities was observed or documented in patient P1. In group 2, which contained two small pedigrees (A2 and A3) and six sporadic patients, neither intragenic mutation nor large deletion was identified. All patients in this group had bilateral coloboma of iris and choroid associated with other abnormalities (Figures 4c and d, Table 1).
In group 3, which included three small families (A4–A6) and one isolated patient, we identified one novel missense mutation c.643T>C (p.S216P) in family A6. This mutation also co-segregated with the disease phenotype, but was not detected in 100 normal controls (Figure 2d). Using GOR method, the result for secondary structure prediction suggested that the mutant PAX6 216P replace a β-sheet ‘E’ with a turn sheet ‘T’ at amino acid 210, and a turn sheet ‘T’ with a coil ‘C’ at amino acid 214 (Figures 5a and b). Family A6 had three affected members. The index patient, male aged 32 years, presented bilateral congenital nystagmus, sclerocornea with diffuse corneal opacity with blood vessels from the peripheral to the central, iridocorneal adhesions, broad-range peripheral synechiae of the anterior angle, and anterior polar cataract (Figures 4e and f). Owing to the corneal opacity, his fundus could not be observed clearly (Figure 4g). The result of his A/B ultrasonic scan examination indicated bilateral posterior staphyloma with the axial length of his right and left eye at a value of 25.03 and 24.85 mm, respectively. Interestingly, his mother aged 55 years, who was also carrying the mutation, did not have nystagmus, sclerocornea, iridoschisis, or congenital cataract. She only presented tessellated fundus and posterior staphyloma (Figure 4h). The patients in family A4 and A5 were diagnosed with Peters anomaly due to the corneal opacity, iridocorneal adhesions, and displacement of the pupil (corectopia). The only sporadic patient (P10) in group 3, who was a 21-year-old female, had progressive iris atrophy, pseudopolycoria, shallow of the anterior chamber, broad-based peripheral anterior synechiae, and high IOP. Their detailed clinical features and the PAX6 mutations screening results of this group were summarized in Table 2.

The effect of p.S216P on the secondary structure of the PAX6 using the GOR method and sequence alignment portion of the HD spanning the p.S216 of the PAX6 of human with other species. (a) The secondary structure of wild-type PAX6 around the site S216. (in blue). (b) The secondary structure of mutant P216 of the PAX6 of the corresponding region (in red). (c) Sequence alignment portion of the HD spanning the p.S216 of the PAX6 of human with other species. The mutation site is boxed in red letters. The color reproduction of this figure is available at the Eye journal online.
Discussion
In this study, we collected six families and 10 sporadic patients with different clinical phenotype and screened the PAX6 mutation. We identified four intragenic mutations and one large deletion.
The patients in group 1 presented similar phenotype, with the complete absence of iris, congenital nystagmus, and cataract. Both the novel splicing-site mutation c.357-3C>G (p.Ser119fsX32) detected in family A1 and the recurred mutation c.11-2A>G (p.Ser4AsnfsX21) identified in patient P2 cause frameshifts in the reading.21 Together with the recurred nonsense mutation p.W156X found in patient P3, the three intragenic mutations detected in group 1 result in premature termination of the PAX6 translation.22 As all premature stop codons are not located in the last exon of the PAX6 gene, there is a possibility that nonsense-mediated mRNA decay might be involved in the abnormal processing of RNAs. Therefore, the three intragenic mutations could have a similar effect on a deleted allele (whole PAX6 gene) detected in patient P1. The results are consistent with the hypothesis that haploinsufficiency of the gene causes the classical AN phenotype.4, 5
Only one missense mutation was identified in group 3. The novel missense mutation p.S216P detected in the family A6 was located in the highly conserved HD region (Figure 5c). The result of GOR suggested p.S216P leads to a secondary structure change between residues 210 and 216, which might interfere with the correct folding of the protein. It is known that PAX6 missense mutations may give rise to variant phenotypes and most of them are clustered in the highly conserved residues of the PRD.4, 5, 10, 11, 12, 13, 14, 15 In contrast, very few missense mutations have been found in the HD region.4, 5 Mutation p.S216P was the fourth mutation identified in the HD region. The previously reported three mutations p.R214G, p.R242T, and p.F258S in the HD region caused the phenotype of classical AN, unilateral partial AN, and large coloboma of the optic nerve, retina, and choroid, respectively.14, 22, 23 Mutation p.S216P was the first missense mutation identified in the HD region that gave rise to the phenotype of Peters anomaly. The patients in family A6 presented atypical phenotype of Peters anomaly. All patients had nystagmus, different extent of sclerocornea, iridocorneal adhesions, congenital cataract, tessellated fundus, and posterior staphyloma. However, the proband's mother, who also harbored this mutation, only presented tessellated fundus and posterior staphyloma. Such mild, incomplete penetrance, phenotypic changes had also been observed in one individual carrying mutation p.R242T, which is also located in the HD region.22 No PAX6 mutation was identified in families A3 and A4, which were also diagnosed with Peters anomaly. As Peters anomaly is genetically heterogeneous, the phenotypes of families A3 and A4 may be caused by mutations of other genes.17
In group 2, we detected neither intragenic mutation of the PAX6 gene nor large deletion in the PAX6 gene region. The patients in group 3 shared a common clinical feature, coloboma both of iris and choroid, which was similar with the optic fissure closure defect (OFCD) described by Morrison et al.21 They did not find any pathogenic mutations of the PAX6 gene in their cohort of 85 patients with OFCD.22
In conclusion, we identified four intragenic mutations and one large deletion in set of patients with different phenotype. In terms of genotype–phenotype correlation, the mutations that introduce PTCs or whole PAX6 gene deletion caused classical AN phenotype, whereas the missense mutation resulted in Peters anomaly. The PAX6 mutations are not responsible for coloboma of iris and choroid. Clearly clinical phenotype classification may increase the mutation detection rate of the PAX6 gene.

Accession codes
References
Heyningen V, Williamson KA . PAX6 in sensory development. Hum Mol Genet 2002; 11: 1161–1167.
Ton CC, Hirvonen H, Miwa H, Weil MM, Monaghan P, Jordan T et al. Positional cloning and characterization of a paired box- and homeobox-containing gene from the aniridia region. Cell 1991; 67: 1059–1074.
Glaser T, Walton DS, Maas RL . Genomic structure, evolutionary conservation and aniridia mutations in the human PAX6 gene. Nat Genet 1992; 2: 232–239.
Prosser J, van Heyningen V . PAX6 mutations reviewed. Hum Mutat 1998; 11: 93–108.
Tzoulaki L, White IM, Hanson IM . PAX6 mutations: genotype -phenotype correlations. BMC Genet 2005; 6: 27.
Fantes J, Redeker B, Breen M, Boyle S, Brown J, Fletcher J et al. Aniridia-associated cytogenetic rearrangements suggest that a position effect may cause the mutant phenotype. Hum Mol Genet 1995; 4: 415–422.
van Heyningen V, Boyd PA, Seawright A, Fletcher JM, Fantes JA, Buckton KE et al. Molecular analysis of chromosome 11 deletions in aniridia-Wilms tumor syndrome. Proc Natl Acad Sci USA 1985; 82: 8592–8596.
Drechsler M, Royer-Pokora BA . Line element is present at the site of a 300-kb deletion starting in intron 10 of the PAX6 gene in a case of familial aniridia. Hum Genet 1996; 98: 297–303.
Hanson IM, Seawright A, Hardman K, Hodgson S, Zaletayev D, Fekete G et al. PAX6 mutations in aniridia. Hum Mol Genet 1993; 2: 915–920.
Azuma N, Hotta Y, Tanaka H, Yamada M . Missense mutations in the PAX6 gene in aniridia. Invest Ophthalmol Vis Sci 1998; 39: 2524–2528.
Azuma N, Yamaguchi Y, Handa H, Hayakawa M, Kanai A, Yamada M et al. Missense mutation in the alternative splice region of the PAX6 gene in eye anomalies. Am J Hum Genet 1999; 65: 656–663.
Chao LY, Huff V, Strong LC, Saunders GF . Mutation in the PAX6 gene in twenty patients with aniridia. Hum Mutat 2000; 15: 332–339.
Dansault A, David G, Schwart C, Jaliffa C, Vieira V, de la Houssaye G et al. Three new PAX6 mutations including one causing an unusual ophthalmic phenotype associated with neurodevelopmental abnormalities. Mol Vis 2007; 13: 511–523.
Redeker EJW, de Visser ASH, Bergen AAB, Mannens MMAM . Multiplex ligation-dependent probe amplification (MLPA) enhances the molecular diagnosis of aniridia and related disorders. Mol Vis 2008; 14: 836–840.
Jia X, Guo X, Jia X, Xiao X, Li S, Zhang Q et al. A novel mutation of PAX6 in Chinese patients with new clinical features of Peters’ anomaly. Mol Vis 2010; 16: 676–681.
Traboulsi EI, Maumenee IH . Peters’ anomaly and associated congenital malformations. Arch Ophthalmol 1992; 110: 1739–1742.
Idrees F, Vaideanu D, Fraser SG, Sowden JC, Khaw PT . A review of anterior segment dysgeneses. Surv Ophthalmol 2006; 51: 213–231.
Harissi-Dagher M, Colby K . Anterior segment dysgenesis: Peters anomaly and sclerocornea. Int Ophthalmol Clin 2008; 48: 35–42.
Garnier J, Gibrat JF, Robson B . GOR method for predicting protein secondary structure from amino acid sequence. Methods Enzymol 1996; 266: 540–553.
Churchill AJ, Hanson IM, Markham AF . Prenatal diagnosis of aniridia. Ophthalmology 2000; 107: 1153–1156.
Chao LY, Mishra R, Strong LC, Saunders GF . Missense mutations in the DNA-binding region and termination codon in PAX6. Hum Mutat 2003; 21: 138–145.
Morrison D, FitzPatrick D, Hanson I, Williamson K, van Heyningen V, Fleck B et al. National study of microphthalmia, anophthalmia, and coloboma (MAC) in Scotland: investigation of genetic aetiology. J Med Genet 2002; 39: 16–22.
Azuma N, Yamaguchi Y, Handa H, Tadokoro K, Asaka A, Kawase E et al. Mutations of the PAX6 gene detected in patients with a variety of optic-nerve malformations. Am J Hum Genet 2003; 72: 1565–1570.
Acknowledgements
We thank the patients and their families for participating in this study. The study was supported by the Beijing National Science Foundation (No. 07G0069). The funding organization had no role in the design or conduct of this research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Zhang, X., Tong, Y., Xu, W. et al. Two novel mutations of the PAX6 gene causing different phenotype in a cohort of Chinese patients. Eye 25, 1581–1589 (2011). https://doi.org/10.1038/eye.2011.215
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/eye.2011.215
Keywords
This article is cited by
-
Diagnostik, Klinik und Genetik kongenitaler Hornhauttrübungen
Der Ophthalmologe (2022)
-
Novel mutations in PAX6, OTX2 and NDP in anophthalmia, microphthalmia and coloboma
European Journal of Human Genetics (2016)
-
Whole exome sequence analysis of Peters anomaly
Human Genetics (2014)
