
Abstract
The active inflammatory phase of thyroid eye disease (TED) is mediated by the innate immune system, and management is aimed at aborting this self-limited period of autoimmune activity. In most patients with TED, ocular and adnexal changes are mild and management involves controlling thyroid dysfunction, cessation of smoking, and addressing ocular surface inflammation and exposure. In patients with acute moderate disease, this being sufficient to impair orbital functions, immunosuppression reduces the long-term sequelae of acute inflammation, and adjunctive fractionated low-dose orbital radiotherapy is used as a steroid-sparing measure. Elective surgery is often required following moderate TED, be it for proptosis, diplopia, lid retraction, or to debulk the eyelid, and this should be delayed until the disease is quiescent, with the patient stable and weaned off all immunosuppression. Thus, surgical intervention during the active phase of moderate disease is rarely indicated, although clinical experience suggests that, where there is significant orbital congestion, early orbital decompression can limit progression to more severe disease. Acute severe TED poses a major risk of irreversible loss of vision due to marked exposure keratopathy, ‘hydraulic’ orbital congestion, or compressive optic neuropathy. If performed promptly, retractor recession with or without a suture tarsorrhaphy protects the ocular surface from severe exposure and, in patients not responding to high-dose corticosteroid treatment, decompression of the deep medial orbital wall and floor can rapidly relieve compressive optic neuropathy, as well as alleviate the inflammatory and congestive features of raised orbital pressure.
Similar content being viewed by others

Introduction
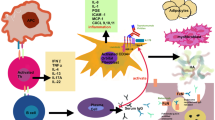
Thyroid eye disease (TED), also known as thyroid-associated ophthalmopathy, or Graves’ disease, is a form of ‘idiopathic’ lymphocytic orbital inflammation. Most commonly encountered in patients with autoimmune thyrotoxicosis, the other recognised risk factors for disease severity are gender (Although TED is more frequent in women, for severe disease, the female-to-male ratio is reversed (at 1 : 4).) and smoking.1 Although the pathogenesis remains incompletely understood, autoimmune activation of orbital fibroblasts, which in TED express the TSH receptor and produce both proinflammatory molecules and extracellular matrix components, is thought to play a central role. This effect is further compounded by the infiltration of immunocompetent Th1 and B lymphocytes, mast cells, and macrophages, with a release of the Th1-type pro-inflammatory cytokines IL-2, IF-γ, and TNF.2, 3 It is of interest that, in vitro, a subpopulation of orbital fibroblasts termed ‘pre-adipocytes’ has the potential to differentiate into mature adipocytes, and these could contribute to increased adipose tissue in vivo.4
Hypertrophy of extraocular muscles and enhanced adipogenesis, together with deposition of nonsulfated glycosaminoglycans and hyaluronate, compress the soft tissues within the confined bony orbit, with orbital functions impeded due to compressive congestion and the complications of acute and chronic inflammation.5 Unique among human ‘autoimmune’ diseases, active TED is self-limiting, manifesting with an active (dynamic) phase lasting up to 24 months, followed by an inactive (static) disease phase. Furthermore, most of the orbit lacks lymphoid tissue, unlike the target organs for other human autoimmune diseases, such as synovium in rheumatoid arthritis. It is possible that, for sustained activity to occur in any autoimmune disease, lymphoid neogenesis within the tissues may be required; this cannot occur in the orbit, and therefore might account for the typically self-limiting nature of TED.
In about 40% of patients, there is simultaneous onset of ocular and systemic symptoms.6 Orbital inflammatory disease occurs in up to 60% of individuals with hyperthyroidism and, of those patients presenting with clinical evidence for TED, ∼85% are biochemically hyperthyroid and 10% are hypothyroid.7 However, a small minority can be biochemically euthyroid at presentation, with the only peripheral abnormality being the presence of serum antithyroid microsomal antibodies, antithyroglobulin antibodies or antithyroid stimulating hormone receptor antibodies. Indeed, not infrequently, these raised antibodies can be the only finding in patients with a history of subtle lid fullness or retraction (Figure 1).

Upper lid retraction, with positive antithyroid antibodies being the only abnormality on serological testing.
The structures chiefly affected during the acute inflammatory phase of TED are the orbital extraocular muscles and orbital adipose tissue. As orbital congestion (and therefore venous permeability) increase, intercellular fluid, pro-inflammatory cytokines and lymphocytes accrue within the tissues. This cycle of increased retrobulbar hydrostatic pressure and secondary inflammation leads to exophthalmos, bulging of the eyelid sulci, and orbital congestion, manifest as conjunctival chemosis, raised intraocular pressure, impaired motility, and optic nerve compromise (Table 1). Where a healthy orbital septum is effective in retaining the orbital contents, progressive exophthalmos cannot occur and a rapid escalation in retrobulbar orbital pressure can lead to optic neuropathy with few if any ‘classic’ TED signs (Figure 12). This ‘crowding’ effect is most marked at the orbital apex, where efferent venous drainage, arterial perfusion pressure, and axonal flow within the optic nerve can be rapidly compromised: with a relative arterial perfusion watershed at the posterior optic nerve, a rise in orbital pressure can lead to posterior ischaemic optic neuropathy and irreversible blindness. Although severe thyroid eye disease is rare (affecting only one in twenty of all TED patients),6 it carries a major risk of sight loss due to corneal exposure, orbital congestion and optic neuropathy, and such patients should therefore be treated urgently to avoid irreversible loss of vision.
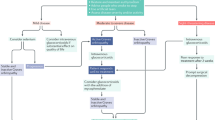
Medical treatment of acute thyroid eye disease
Mild TED
Of those patients who suffer acute TED, symptoms and signs tend to be mild, often resolving as thyroid function improves, with a small minority requiring subsequent surgery once the self-limiting inflammatory phase has passed. The mainstay of treatment is to control thyroid function, to cease smoking (this habit reducing the response of TED to treatment),8 and to provide supportive ocular lubrication. Among patients with mild TED, progression to more severe disease, as reflected in a change in the clinical activity score (CAS),9 occurs in about 15–25%.
Several in vitro studies implicate oxygen-free radicals in the pathogenesis of active TED.1, 10, 11 Increased oxidative stress is reported in hyperthyroidism, and oxygen-free radicals are considered to play a role in the pathogenesis of TED,12 with serum selenoprotein P, an index of the oxidative state, being reduced in TED patients.13 Furthermore, clinical studies indicate that oral Selenium, an essential component of anti-oxidant enzymes,14 can slow progression of the disease in patients with mild Graves' orbitopathy.15, 16 In a randomised placebo-controlled trial in patients with mild active TED, Marcocci et al15 compared the effect of a six month treatment regimen with oral sodium selenite (100 μg twice daily), or oral pentoxifylline (600 mg twice daily), against a placebo.15 Compared with placebo, at the end of 6 months treatment, and 6 months after cessation of treatment, oral selenite (but not pentoxifylline) was associated with a significant improvement in the quality of life, reduced ocular disease, and less risk of disease progression.
Moderate to severe TED
The definition recommended by EUGOGO for moderate to severe TED is that sight-threatening disease is absent but that symptoms are sufficiently disabling to warrant immunosuppression during active disease, or surgery if inactive.17, 18 Patients typically have at least one of the following features: (i) >2 mm of upper eyelid retraction, (ii) moderate to severe soft tissue involvement, (iii) >3 mm of exophthalmos than would be expected for racial and gender variations, and (iv) diplopia. This definition is not intended to reflect disease activity, as all but the second of these criteria can persist indefinitely after all inflammation has waned. In those patients with moderately severe inflammatory TED, increasing orbital congestion can lead to a wide spectrum of symptoms and signs alluded to in (ii) above (Table 1; Figures 2, 3, 4, 5, 6, 7, 8, 9, 10, 11); these are related to disruption of ocular surface protection, increased muscle volume with reduced compliance, and impeded venous drainage of the orbit. Treatment of moderately active TED is rarely surgical, and medical measures, which aim to diminish and shorten the acute inflammatory phase, are generally highly effective if given sufficiently early (Table 2). Finally, and in active disease only, adjunctive low-dose external beam orbital radiotherapy is frequently used as a steroid-sparing measure.

Primary upper eyelid retraction in acute thyroid ophthalmopathy in a patient with bilateral compressive optic neuropathy (top). CT imaging identified enlarged recti with crowding of the orbital apices (bottom).

Active TED with upper lid retraction and lower lid displacement secondary to proptosis.

Superior limbic keratitis.

Acute thyroid orbitopathy presenting with orbital congestion, gross proptosis and ocular surface exposure, right microbial keratitis, and left corneal perforation.

Acute presentation of active TED with upper eyelid retraction, oedematous upper and lower eyelids, proptosis, and compressive optic neuropathy.

Atypical presentation with congested orbit, chemosis (top), and enlarged superior rectus muscle (bottom).

‘Hydrostatic’ orbital signs with orbital congestion, dilated episcleral vessels and chemosis.

Active TED with impaired ocular ductions and diplopia.

CT imaging identifying enlarged extraocular muscles, proptosis and a ‘taut’ optic nerve.

CT imaging showing enlarged and anteriorly displaced lacrimal glands.
Immunosuppression, biological agents, and immune mediators in TED
Corticosteroids have been used in the treatment of TED for over 50 years.19 Although their exact mechanism of action remains elusive, in TED they are thought to reduce transcription of intra- and extra-cellular pro-inflammatory proteins in orbital fibroblasts and Th1 lymphocytes. Numerous different oral and parenteral regimes are reported (Table 3), with no clear consensus on the optimum dosage, dosing intervals, and duration of treatment.20 Treatment is most effective when given early in the course of active disease, with the literature now favouring high-dose intravenous glucocorticoid pulses for moderate to severe TED, with response rates of about 80% for parenteral treatment, as compared with about 60% for the oral route.21 Although the parenteral route has fewer side effects than oral administration (56% and 85%, respectively),22, 23 it is nevertheless associated with significant morbidity in up to 6% of cases, and should be avoided in patients with a history of hepatic, cardiovascular or renal morbidity, uncontrolled hypertension, and diabetes.24, 25, 26, 27, 28 The risk of serious systemic side effects appears to be dose related, being reported mainly when consecutive or alternating doses greater than 500mg have been given.29 In a comparison of oral corticosteroids and intravenous administration, Kahaly et al30 reported a cohort of 70 consecutive patients with severe active TED randomised to receive either intravenous methyl prednisolone (500 mg methylprednisolone once weekly for 6 weeks, then 250 mg once weekly for 6 weeks) or oral prednisolone (100 mg daily for a week with a gradual taper over 3 months).30 A positive clinical response was observed in 77% for the intravenous group compared with 51% of those receiving oral treatment, with a similar outcome reported in a second cohort of patients with moderately active TED treated with an identical protocol (72% vs 49% respectively).31 Other observed benefits included increased quality of life scores and decreased need for subsequent ophthalmic surgery in the intravenous treatment group.
Where steroids are not tolerated, or are ineffective, other steroid-sparing agents and so-called ‘biologics’ have been used with varying effect. Azathioprine (AZT) has not been shown to be of benefit as a single agent32 but, in patients treated with glucocorticoids, AZT with or without low-dose orbital radiotherapy may yet have a role in active TED—this being the subject of the current multicentre CIRTED study.33 Although oral cyclosporine has been reported to be less effective than glucocorticoid treatment, combined therapy is considered to be more effective in patients refractory to treatment with either as monotherapy.34
The use of biological agents in TED is limited to case reports and uncontrolled studies. Nevertheless, the experience with Rituximab (RTX), a chimeric mouse monoclonal anti-human CD20 antibody that blocks B-cell proliferation and maturation is encouraging.35, 36, 37, 38 Salvi et al38, using parenteral RTX in a non-randomised cohort of nine patients with active TED, reported a greater improvement in clinical activity after RTX (1000 mg intravenous infusion, twice at two-week interval), and with fewer side effects, than those treated with a standard intravenous glucocorticoid regimen (500 mg i.v. weekly for 16 weeks). Significantly, even after two years of follow-up, there were no relapses of inflammatory orbital disease in the RTX group, this thought to be due to persistent peripheral blood B-cell depletion in the RTX group.38
Finally, monoclonal antibodies against TNF-α (including infliximab, adalimumab, and etanercept) have been used successfully in the treatment of rheumatoid arthritis and Crohn's disease, and also show promise in the treatment of TED.39, 40, 41 Serum TNF-α levels have been associated with more severe forms of TED and early reports indicate that infliximab can rapidly reduce orbital inflammation and improve visual function, with relatively few side effects. Etanercept is similarly effective in about 60% of patients, although almost 30% experience a flare-up of disease after cessation of the drug. It is likely that patients who are genetically predisposed to be high producers of TNF-α will benefit the most, as genetic TNF polymorphisms have previously been implicated in other forms of idiopathic orbital and ocular inflammation.42
In rare cases, TED can progress to severe orbital disease despite medical treatment, and surgical decompression (v.i.), which disrupts the cycle of increased orbital congestion, vascular engorgement and further inflammation, should be considered during the acute inflammatory phase.
Orbital radiotherapy
Orbital lymphocytes are sensitive to radiotherapy,43 and low-dose orbital radiotherapy, while not used as monotherapy for acute TED, is a well-established adjunctive treatment,45 and is particularly effective in improving muscle motility during active disease.46 Although a typical radiotherapy dose is 20 Gy, given in 10 fractions of 2 Gy, lower doses may be equally as effective.44
In moderately severe non-sight-threatening disease, radiotherapy combined with intravenous methylprednisolone is effective in 88% of patients, this being more effective than oral prednisolone and radiotherapy. Radiotherapy should be used with caution in diabetic patients (particularly in those who are also hypertensive, due to the risk of new or deteriorating diabetic retinopathy),47 and preferably restricted to patients older than 35 years of age due to the long latency of radiotherapy-induced tumours.
Exposure keratopathy
The urgency and extent of treatment for keratopathy depends on the degree of ocular surface exposure. Since corneal ulceration, perforation and secondary endophthalmitis can occur (Figure 5), urgent action should be taken to prevent a catastrophic loss of vision. Intensive topical lubricants and antibiotics should be started, and the use of moist chambers considered, if necessary using cellophane food wrapping affixed to the periocular skin with ointment or vaseline. To maintain corneal protection, such patients are likely to require urgent surgery (v.i.).
Optic neuropathy
As the orbital apex pressure increases, blurred vision, impaired colour appreciation, and/or new visual field defects indicate early compressive optic neuropathy (or dysthyroid optic neuropathy, DON). Although typically occurring in the setting of an inflamed and congested orbit, DON can also occur with few overt inflammatory signs. Furthermore, stable exophthalmometry should not be taken to indicate normal orbital pressure, but may instead be due to containment of the orbital tissues by a healthy orbital septum. In some such patients, the hydrostatic pressure of the orbit can escalate quickly, and severe optic neuropathy ensues with startling rapidity. Thus, signs of ocular surface inflammation can be absent, with the only clinical features being a firm orbit on retropulsion, optic neuropathy with or without optic disc swelling, and severe crowding of the orbital apex seen on imaging (Figure 12).

Bilateral optic neuropathy in a patient with healthy orbital septae and without a change in exophthalmometry.
The optimum treatment for optic neuropathy is high-dose systemic immunosuppression, with prompt surgical decompression performed when the response to medical treatment is inadequate. Various regimes have been described (Table 3),24 and although none have been universally adopted, a commonly used protocol for intravenous methyl prednisolone is 500 mg/g daily for 3 days with a view to urgent surgical decompression for those patients who fail to respond. Low-dose orbital radiotherapy is often used as an adjunctive treatment, be cannot be used alone in view of the delay in treatment response.
Surgical treatment of acute thyroid eye disease
Exposure keratopathy
Surgical options for patients with severe exposure keratopathy (Figure 5) depend on the degree of proptosis, and whether the thyroid activity is adequately controlled. In those who remain markedly hyperthyroid, a prompt endocrinological assessment should be obtained, and the patient considered for urgent upper lid lowering with, or without, temporary suture tarsorrhaphy. In the interim, corneal protection can be achieved with a moist chamber (as discussed above), in addition to frequent topical lubrication. Where the thyroid activity is controlled, high-dose corticosteroids should be started, and, in the presence of moderate or marked exophthalmos, orbital decompression should be considered with simultaneous lid lowering. The latter can be achieved through the posterior (conjunctival) approach, or via an anterior (skin crease) approach; in either case, the levator complex should be recessed adequately. Patients with severe exposure typically have up to 7–10 mm of retraction of the upper lid from the superior limbus, and will therefore require extensive release of the central aponeurosis, lateral horn and Muller’s muscle from the tarsus and conjunctiva to achieve sufficient corneal protection. Release of the most medial retractors should be graded, or ‘softened’, so as to avoid a flat post-operative lid contour. Such patients should however be warned of the inherent unpredictability of lid lowering in the acute phase of TED, and the likelihood of requiring later corrective surgery for lid height, contour, or both.
Although the use of Botulinum toxin to lower the upper eyelid has been suggested, this approach is inadvisable because its effect is limited, unpredictable, and delayed by up to 48 h or longer. Corneal protection is further jeopardised if the neighbouring superior rectus muscle is affected (even if transiently), as this reduces Bell’s phenomenon and ocular elevation, with the risk of exposure being even higher if the inferior rectus muscle has been involved.
Optic neuropathy
In some patients, high-dose systemic steroids alone (either intravenous or oral) are sufficient to relieve tissue pressure at the orbital apex, and a gradual taper, without recurrence of the optic neuropathy, can be achieved. However, where a prompt and adequate response is not seen with medical treatment, surgical decompression of the posteromedial orbit should be considered, with numerous studies reporting a rapid and beneficial effect on vision;48, 49, 50, 51, 52 the objective is to relieve the hydrostatic pressure at the orbital apex and, by doing so, reduce orbital congestion and improve vascular perfusion and axonal flow within the optic nerve. Surgically, this is achieved by removing the bony medial wall as far posteriorly as the annulus of Zinn, this including the posterior and mid-ethmoid air cells, the posteromedial orbital floor, and the posterior half of the maxillo-ethmoidal strut (Figure 13). Although surgery can be performed endoscopically, these authors prefer the retrocaruncular transconjunctival approach for a number of reasons. Firstly, the approach affords a wide ‘open-sky’ visualisation of the entire medial orbital wall and maxillo-ethmoidal ‘strut’, allowing effective decompression of the orbital apex with a wide excision of the medial periosteum—this being essential to allow maximum soft tissue decompression. Secondly, a retrocaruncular incision leaves no visible scar and involves minimal tissue injury, with a rapid post-operative recovery. Finally, where a greater reduction of proptosis is desired, the conjunctival incision can be extended into the inferomedial conjunctival fornix to allow adequate exposure to decompress the anterior orbital floor medial to the infra-orbital nerve. In general, the rate of recovery tends to reflect the duration of pre-existing optic neuropathy, and restoration of full visual functions can occur almost immediately in those patients with an acute history. In a number of patients, a marked and rapid improvement has been observed despite 6 months or longer of pre-existing compressive optic neuropathy.

Diagram to show extent of medial orbital orbital decompression. Note, to achieve an effective decompression, the posterior half of the bony bar (‘strut’) between the maxillary antrum and the ethmoid air cells should be removed.
Medial wall orbital decompression for optic neuropathy—technique
Briefly, decompression of the medial wall is performed as follows. Single 2/0 silk traction sutures are placed through the medial aspects of the upper and lower lids, and a retrocaruncular incision is made in the conjunctiva, this extended towards the supero- and inferomedial conjunctival fornices with tissue scissors. An assistant protects the lacrimal sac with a small malleable retractor and, with a second 16 mm wide retractor, the surgeon gently displaces the orbital soft tissues laterally (Figure 14). With blunt dissection, a small periosteal elevator is used to expose the medial orbital wall behind the posterior lacrimal crest, the maxillary-ethmoidal interface (‘strut’) and the medial orbital floor, taking care not to in-fracture prematurely the thin lamina papyracea. The periosteum is opened along an anatomically vertical incision (with care not to disrupt the ethmoid air cells), and peeled intact off the full extent of the medial wall and the medial floor of the orbit; an intact periosteum helps to prevent the prolapse of orbital fat into the surgical field (Figure 15). Noting the tighter periosteal attachment along the ethmoid-maxillary suture, the posteromedial orbital floor is infractured first and removed to the most posterior aspect of the maxillary antrum; the medial wall is similarly infractured and decompressed using Tilley’s ethmoidectomy forceps. Decompression of the medial wall should extend superiorly to expose the floor of the anterior cranial fossa (this being at the level of the anterior ethmoidal vessels) and posteriorly to a depth ∼1 cm deeper than the back wall of the maxilla, the latter acting as an important landmark in medial decompression. The fine bone and mucosal linings of the posterior and mid-ethmoid air cells are also removed, without which subsequent soft tissue prolapse is limited. The posterior half of the intervening bone (or ‘strut’) between the maxillary and ethmoid sinuses should also be removed before extensive excision of the periosteum to permit widespread prolapse of orbital soft tissues.

Surgical approach to the medial orbital wall.

Schematic diagram to illustrate the approach to the extraperiosteal space of the medial orbital wall.
Depending on the degree of proptosis, with adequate exposure, the orbital floor can also be decompressed laterally as far as the infra-orbital nerve, with care taken to diathermy the major vessels passing from the orbit to the nerve. No surgical drain is required as, in the unlikely event of a post-operative haemorrhage, blood will drain freely into the sinuses. Finally, the conjunctiva is closed with two 7/0 absorbable sutures to prevent local prolapse of adipose tissue. The patient is double padded, nursed semirecumbent, advised against blowing the nose for 2 weeks, and discharged the following day on oral antibiosis and a reducing course of oral prednisolone.
In those patients with a severely ‘hydraulic’ picture and in whom there is a moderate degree of proptosis, the lateral wall can also be decompressed; if this is performed first, access to the medial wall is improved. In patients with gross proptosis (>28 mm), a full three-wall decompression can be achieved through a lower lid swinging flap alone (Figure 16) but, if available surgical time is limited, it is acceptable to perform a retrocaruncular medial wall and medial floor decompression with a view to further floor and lateral wall decompression after the acute inflammatory phase. Finally, in rare cases where compressive optic neuropathy persists despite adequate anatomical decompression of the medial wall, a deep lateral wall decompression can further relieve compression of the posterior orbit.

Axial and coronal CT images before (top) and after (bottom) bilateral bony decompression of the medial wall and medial floor of the orbit for optic neuropathy.
Summary
Acute thyroid eye disease can be severe and sight-threatening, and can occur at any time after the onset of TED. Corticosteroids remain the mainstay of treatment, with pulsed intravenous methyl prednisolone being more effective, and with fewer overall side effects, than the oral route. Neither route is without risk, although those associated with intravenous methyprednisolone tend almost exclusively occur with consecutive or alternate daily doses >500 mg. Newer biologics offer hope of more targeted treatments, these blocking the production of Th1 cytokines and their target receptors, with future treatments likely to be focused on orbital fibroblast activation and differentiation. The role of surgery in acute thyroid eye disease is to prevent the complications of severe keratopathy, orbital congestion and apical compression, and is necessary early in the active phase of severe disease where medical treatment is inadequate to restore orbital functions.
References
Bartalena L, Marcocci C, Tanda ML, Manetti L, Dell’Unto E, Bartolomei MP et al. Cigarette smoking and treatment outcomes in Graves ophthalmopathy. Ann Intern Med 1998; 129: 632–635.
Kumar S, Bahn RS . Relative overexpression of macrophage-derived cytokines in orbital adipose tissue from patients with graves’ ophthalmopathy. J Clin Endocrinol Metab 2003; 88: 4246–4250.
Laban-Guceva N, Bogoev M, Antova M . Serum concentrations of interleukin (IL-)1alpha, 1beta, 6 and tumor necrosis factor (TNF-) alpha in patients with thyroid eye disease (TED). Med Arh 2007; 61: 203–206.
Feldon SE, O’loughlin CW, Ray DM, Landskroner-Eiger S, Seweryniak KE, Phipps RP . Activated human T lymphocytes express cyclooxygenase-2 and produce proadipogenic prostaglandins that drive human orbital fibroblast differentiation to adipocytes. Am J Pathol 2006; 169: 1183–1193.
Naik VM, Naik MN, Goldberg RA, Smith TJ, Douglas RS . Immunopathogenesis of thyroid eye disease: emerging paradigms. Surv Ophthalmol. 2010; 55: 215–226.
Bahn RS, Heufelder AE . Pathogenesis of Graves' ophthalmopathy. N Engl J Med 1993; 329: 1468–1475.
Soeters MR, van Zeijl CJ, Boelen A, Kloos R, Saeed P, Vriesendorp TM et al. Optimal management of Graves orbitopathy: a multidisciplinary approach. Neth J Med 2011; 69: 302–308.
Cawood TJ, Moriarty P, O'Farrelly C, O'Shea D . Smoking and Thyroid-associated ophthalmopathy: A novel explanation of the biological link. J Clin Endocrinol Metab 2007; 92: 59–64.
Terwee CB, Prummel MF, Gerding MN, Kahaly GJ, Dekker FW, Wiersinga WM . Measuring disease activity to predict therapeutic outcome in Graves’ ophthalmopathy. Clin Endocrinol (Oxf) 2005; 62: 145–155.
Heufelder AE, Wenzel BE, Bahn RS . Methimazole and propylthiouracil inhibit the oxygen free radical-induced expression of a 72 kilodalton heat shock protein in Graves’ retroocular fibroblasts. J Clin Endocrinol Metab 1992; 74: 737–742.
Burch HB, Lahiri S, Bahn RS, Barnes S . Superoxide radical production stimulates retroocular fibroblast proliferation in Graves’ ophthalmopathy. Exp Eye Res 1997; 65: 311–316.
Lu R, Wang P, Wartofsky L, Sutton BD, Zweier JL, Bahn RS et al. Oxygen free radicals in interleukin-1betainduced glycosaminoglycan production by retro-ocular fibroblasts from normal subjects and Graves’ ophthalmopathy patients. Thyroid 1999; 9: 297–303.
Dehina NE, Minich W, Behrends T, Morgenthaler NG, Kohrle J, Schomburg L . Circulating selenoprotein P concentrations are decreased in patients with Graves’ disease and correlate inversely to severity of orbitopathy. Acta Med Port 2009; 22: 1.
Hoffmann PR, Berry MJ . The influence of selenium on immune responses. Mol Nutr Food Res 2008; 52: 1273–1280.
Marcocci C, Kahaly GJ, Krassas GE, Bartalena L, Prummel M, Stahl M et alEuropean Group on Graves' Orbitopathy. Selenium and the course of mild Graves' orbitopathy. N Engl J Med 2011; 364: 1920–1931.
Duntas LH . The evolving role of selenium in the treatment of Graves' disease and ophthalmopathy. J Thyroid Res 2012; 2012: 736161.
Bartalena L, Baldeschi L, Dickinson A, Eckstein A, Kendall-Taylor P, Marcocci C et alEuropean Group on Graves' Orbitopathy (EUGOGO). Consensus statement of the European Group on Graves' orbitopathy (EUGOGO) on management of GO. Eur J Endocrinol 2008; 158: 273–285.
Bartalena L, Baldeschi L, Dickinson AJ, Eckstein A, Kendall-Taylor P, Marcocci C et al. Consensus statement of the European group on Graves' orbitopathy (EUGOGO) on management of Graves' orbitopathy. Thyroid 2008; 18: 333–346.
Hales IB, Thomas ID . Treatment of thyroid ophthalmopathy with corticoid analogues. Australas Ann Med 1962; 11: 113–117.
Wiersinga WM . Autoimmunity in Graves' ophthalmopathy: the result of an unfortunate marriage between TSH receptors and IGF-1 receptors? Clin Endocrinol Metab 2011; 96: 2386–2394.
Zang S, Ponto KA, Kahaly GJ . Clinical review: Intravenous glucocorticoids for Graves' orbitopathy: efficacy and morbidity. J Clin Endocrinol Metab 2011; 96: 320–332.
Marcocci C, Bartalena L, Tanda ML, Manetti L, Dell’Unto E, Rocchi R et al. Comparison of the effectiveness and tolerability of intravenous or oral glucocorticoids associated with orbital radiotherapy in the management of severe Graves’ ophthalmopathy: results of a prospective, single-blind, randomized study. J Clin Endocrinol Metab 2001; 86: 3562–3567.
Bartalena L, Baldeschi L, Dickinson AJ, Eckstein A, Kendall-Taylor P, Marcocci C et al. Consensus statement of the European Group on Graves’orbitopathy (EUGOGO) on management of GO. Eur J Endocrinol 2008; 158: 273–285.
Marinó M, Morabito E, Brunetto MR, Bartalena L, Pinchera A, Marocci C . Acute and severe liver damage associated with intravenous glucocorticoid pulse therapy in patients with Graves' ophthalmopathy. Thyroid 2004; 14: 403–406.
Weissel M, Hauff W . Fatal liver failure after high-dose glucocorticoid pulse therapy in a patient with severe thyroid eye disease. Thyroid 2000; 10: 521.
Gursoy A, Cesur M, Erdogan MF, Corapcioglu D, Kamel N . New-onset acute heart failure after intravenous glucocorticoid pulse therapy in a patient with Graves' ophthalmopathy. Endocrine 2006; 29: 513–516.
Tigas S, Papachilleos P, Ligkros N, Andrikoula M, Tsatsoulis A . Hypokalemic paralysis following administration of intravenous methylprednisolone in a patient with Graves' thyrotoxicosis and ophthalmopathy. Hormones (Athens) 2011; 10: 313–316.
Wichary H, Gasińska T . Methylprednisolone and hepatotoxicity in graves' ophthalmopathy. Thyroid 2012; 22: 64–69.
Kahaly GJ . Management of moderately severe Graves’ orbitopathy. In Wiersinga WM, Kahaly GJ (eds) Graves' Orbitopathy. A multidisciplinary Approach–Questions and Answers 2nd Edn, Karger Basel, 2010 pp 120–155.
Kahaly GJ, Pitz S, Hommel G, Dittmar M . Randomized, single blind trial of intravenous versus oral glucocorticoid monotherapy in Graves’ orbitopathy. J Clin Endocrinol Metab 2005; 90: 5234–5240.
Aktaran S, Akarsu E, Erbağci I, Araz M, Okumuş S, Kartal M . Comparison of intravenous methylprednisolone therapy vs oral methylprednisolone therapy in patients with Graves' ophthalmopathy. Int J Clin Pract 2007; 61: 45–51.
Perros P, Weightman DR, Crombie AL, Kendall-Taylor P . Azathioprine in the treatment of thyroid-associated ophthalmopathy. Acta Endocrinologica 1990; 122: 8–12.
Rajendram R, Lee RW, Potts MJ, Rose GE, Jain R, Olver JM et al. Protocol for the combined immunosuppression and radiotherapy in thyroid eye disease (CIRTED) trial: a multi-centre, double-masked, factorial randomised controlled trial. Trials 2008; 9: 6.
Prummel MF, Mourits MP, Berghout A, Krenning EP, van der Gaag R, Koornneef L et al. Prednisone and cyclosporine in the treatment of severe Graves' ophthalmopathy. New England Journal of Medicine 1989; 321: 1353–1359.
Douglas RS, Gupta S . The pathophysiology of thyroid eye disease: implications for immunotherapy. Curr Opin Ophthalmol 2011; 22: 385–390.
Bonara P, Vannucchi G, Campi I, Rossi S, Cantoni F, Frugoni C et al. Rituximab induces distinct intraorbital and intrathyroidal effects in one patient satisfactorily treated for Graves' ophthalmopathy. Clin Rev Allergy Immunol 2008; 34: 118–123.
Nielsen CH, El Fassi D, Hasselbalch HC, Bendtzen K, Hegedüs L . B-cell depletion with rituximab in the treatment of autoimmune diseases. Graves' ophthalmopathy the latest addition to an expanding family. Expert Opin Biol Ther 2007; 7: 1061–1078.
Salvi M, Vannucchi G, Campi I, Currò N, Beck-Peccoz P . New immunomodulators in the treatment of Graves' ophthalmopathy. Ann Endocrinol (Paris) 2008; 69: 153–156.
Komorowski J, Jankiewicz-Wika J, Siejka A, Lawnicka H, Klysik A, Gos R et al. Monoclonal anti-TNFalpha antibody (infliximab) in the treatment of patient with thyroid associated ophthalmopathy. Klin Oczna 2007; 109: 457–460.
Durrani OM, Reuser TQ, Murray PI . Infliximab: a novel treatment for sight-threatening thyroid associated ophthalmopathy. Orbit 2005; 24: 117–119.
Paridaens D, van den Bosch WA, van der Loos TL, Krenning EP, van Hagen PM . The effect of Etanercept on Graves' ophthalmopathy: a pilot study. Eye 2005; 19: 1286–1289.
Verity DH, Wallace GR, Vaughan RW, Ohno S, Kondeatis E, Madanat W et al. HLA and TNF polymorphisms in ocular Behçet’s disease. Tissue Antigens 1999; 54: 264–272.
Kahaly GJ, Roesler HP, Kutzner J, Pitz S, Muller-Forell W, Beyer J et al. Radiotherapy for thyroid-associated orbitopathy. Exp Clin Endocrinol Diabetes 1999; 107 (Suppl 5): S201–S207.
Kahaly GJ, Rosler HP, Pitz S, Hommel G . Low- versus high-dose radiotherapy for Graves’ ophthalmopathy: a randomized, single blind trial. J Clin Endocrinol Metab 2000; 85: 102–108.
Bartalena L, Marcocci C, Tanda ML, Rocchi R, Mazzi B, Barbesino G et al. Orbital radiotherapy for Graves' ophthalmopathy. Thyroid 2002; 12: 245–250.
Prummel MF, Terwee CB, Gerding MN, Baldeschi L, Mourits MP, Blank L et al. A randomized controlled trial of orbital radiotherapy versus sham irradiation in patients with mild Graves’ ophthalmopathy. J Clin Endocrinol Metab 2004; 89: 15–20.
Marcocci C, Bartalena L, Rocchi R, Marinò M, Menconi F, Morabito E et al. Long-term safety of orbital radiotherapy for Graves' ophthalmopathy. J Clin Endocrinol Metab 2003; 88: 3561–3566.
Liao SL, Chang TC, Lin LL . Transcaruncular orbital decompression: an alternate procedure for Graves ophthalmopathy with compressive optic neuropathy. Am J Ophthalmol 2006; 141: 810–818.
McCann JD, Goldberg RA, Anderson RL, Burroughs JR, Ben Simon GJ . Medial wall decompression for optic neuropathy but lateral wall decompression with fat removal for non vision-threatening indications. Am J Ophthalmol 2006; 141: 916–917.
Metson R, Pletcher SD . Endoscopic orbital and optic nerve decompression. Otolaryngol Clin North Am 2006; 39: 551–561.
Siracuse-Lee DE, Kazim M . Orbital decompression: current concepts. Curr Opin Ophthalmol. 2002; 13: 310–316.
Eloy P, Trussart C, Jouzdani E, Collet S, Rombaux P, Bertrand B . Transnasal endoscopic orbital decompression and Graves' ophtalmopathy. Acta Otorhinolaryngol Belg 2000; 54: 165–174.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Verity, D., Rose, G. Acute thyroid eye disease (TED): Principles of medical and surgical management. Eye 27, 308–319 (2013). https://doi.org/10.1038/eye.2012.284
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/eye.2012.284
Keywords
This article is cited by
-
Machine learning-based prediction of diagnostic markers for Graves’ orbitopathy
Endocrine (2023)
-
Impact of sphenoid trigone size and extraocular muscle thickness on the outcome of lateral wall orbital decompression for thyroid eye disease
Oral and Maxillofacial Surgery (2023)
-
Intraocular pressure improvement in patients receiving teprotumumab for the treatment of thyroid eye disease: a case series
Journal of Medical Case Reports (2022)
-
Evaluating the interreader agreement and intrareader reproducibility of Visual Field Defects in Thyroid Eye Disease– Compressive Optic Neuropathy
Eye (2022)
-
A data-driven approach for the discovery of biomarkers associated with thyroid eye disease
BMC Ophthalmology (2021)
