
Abstract
Progressive pseudorheumatoid dysplasia (PPD) is a rare autosomal recessive disease that causes progressive joint stiffness and pain. It is associated with loss-of-function mutations in the WISP3 gene. We describe two sisters suffering from PPD in whom molecular genetic analysis revealed a homozygous deletion of exon 1 and of the 5′UTR of the WISP3 gene. This is the first time that a gross deletion has been described as the causal mutation in PPD.
Similar content being viewed by others

PPD is a rare autosomal recessive disease characterised by progressive joint stiffness, motor weakness, gait disturbances, articular pain and contractures.1,2 This non-inflammatory arthropathy was first described by Wynne-Davies et al.4 and independently by Spranger et al.3 Classically, the disease presents with decreased joint mobility, in particular reduced hip movements, and a progressive involvement of metacarpophalangeal joints, proximal interphalangeal joints, distal interphalangeal joints, wrists, elbows, knees, shoulders and ankles.1,3,4 In most cases, principal disability arises from hip involvement resulting in pain, limitation of movements and contractures.4 In addition to the hip, hand involvement is one of the most frequent and typical features of PPD.2 The initial symptoms of the disease manifest primarily in childhood, between 3 and 8 years of age.2,4 All patients previously described were normal at birth2–4 and did not have any other dysmorphic characteristics.4 There are no extra-skeletal manifestations of the disease. The stature of patients with PPD is normal in infancy, but deviates generally to lower than the third percentile in adulthood.2 PPD is a progressive disease, although the rate of progression varies between patients.1 The population incidence of PPD is estimated to be ~1 in a million in the United Kingdom,4 but it appears to be higher in the Middle-East and Gulf States as well as in Turkey, where a founder effect has been suggested.2,5
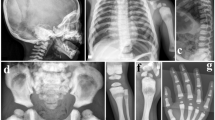
Radiographs from patients with PPD have some typical features. Radiographs from the hip show a large capital femoral epiphysis, a short femoral neck and a decreased intra-articular space (Figure 1).1,2,4 The hands show enlarged epiphyses and metaphyses of the metacarpals and proximal and middle phalanges, often mistaken as ‘joint swelling’, and periarticular osteoporosis (Figure 1).1,2,4 Periarticular osteoporosis can also be present at other joints. Another typical feature is vertebral flattening or platyspondyly (Figure 1).1,4,6 PPD never presents with erosions, periostitis or joint destructions.1,4

(a) Radiographs of the hips of patient 1 (1) and patient 2 (2). Both radiographs show large capital femoral epiphysis, a short femoral neck and a decreased intra-articular space. Radiographs of the lumbar spine of patient 1 show platyspondyly (3–4). Radiographs of the hand of patient 2 (5) showing widened epiphyses and metaphyses of the metacarpals and proximal and middle phalanges. (b) qPCR data using primer sets 4 (blue) and 5 (red) on genomic DNA from all family members. The values for the fold differences compared with controls are shown on the y-axis. C1 control 1; C2 control 2; F father; M mother; P1 patient 1; P2 patient 2. (c) Gel image of the PCR analysis with primer sets 1, 2, 3 and 6 on control and patient DNA showing that no product could be obtained for primer sets 3 and 6 that map within the homozygous deletion. The first lane is the 100 bp DNA ladder. PCR products are all ~500 bp in size. (d) The WISP3 gene, located on chromosome 6q21, consists of five coding exons (boxes, ATG start codon in exon 1). Molecular analysis of WISP3 by quantitative real-time PCR revealed a homozygous deletion of the 5′UTR and the first exon of the WISP3 gene (qPCR primer sets are depicted as arrows) with a maximal deleted region of 9 kb (box).
Most patients are initially misdiagnosed because of the rarity of the disease and the lack of awareness of most clinicians. The presentation of PPD can mimic juvenile idiopathic arthritis1,3,4 as osseous joint swelling can be mistaken for synovitis; although there is no evidence of inflammation in PPD as inflammatory markers (such as ESR and CRP) are always within the normal range. Radiographs can also help to distinguish between these two diseases as destructive or erosive bone changes are never seen on radiographs from PPD patients.1,3,4
As there is no active inflammation, the response to the conventional anti-rheumatic treatment with disease-modifying antirheumatic drugs and nonsteroidal anti-inflammatory drugs is disappointing.1,4 The exact pathomechanism is still unclear; consequently, no specific treatment is currently available.2 Hip replacement may be useful in some cases. There are no longitudinal studies, but patients who underwent hip replacement seem to suffer from less pain and regain walking ability.2
In 1999, loss-of-function mutations in the WISP3 gene were identified as being responsible for PPD.6 Here, we describe two sisters suffering from PPD in whom molecular genetic analysis showed a homozygous deletion of exon 1 and of the 5′UTR of the WISP3 gene. To the best of our knowledge, this is the first time that a gross deletion has been detected as the causal mutation in PPD.
The first patient is a currently 27-year-old woman of Moroccan origin who presented for the first time in our hospital at the age of 7 because of articular pain and short stature. A normal pregnancy was reported, and no abnormalities were noted as a baby. At birth, her length was at the 50th percentile. From the age of 18 months, her length curve dropped to the third percentile and was even lower from the age of 3 years on. At the age of 4, she underwent a bilateral valgisation osteotomy of the tibia because of Blount’s syndrome. Physical examination at that time showed short stature, just below the third percentile, and bilateral prominent genu valgum with an impaired gait and thickening of the small finger joints with restricted flexion and extension. Laboratory tests were normal without signs of inflammation. Radiographs showed global osteopenia, ovoid vertebrae and widened distal epiphyses from the metacarpal bones, the proximal phalanges and the distal phalanges (Figure 1a). The diagnosis of PPD was made, and the patient began intensive physiotherapy. She underwent a total hip replacement surgery of the left side at the age of 18 and of the right side at the age of 19, with a satisfactory outcome. Physical examination currently shows a body height of 132 cm, an impaired gait, fixed flexion of hips and knees, impossible dorsiflexion of the wrists and bony thickening of the interphalangeal joints with functional limitations. Despite these physical limitations, she functions quite well in daily life and has almost no need for painkillers.
The second patient is the sister of the first patient who is 3 years younger. She first presented in our centre at the age of 7 because of articular pain in the legs. Physical examination showed a normal length and weight, a slightly impaired gait, bony thickening of the interphalangeal joints, incomplete extension of the knees, impaired flexion, abduction and internal rotation of the hips and limited flexion of the cervical spine. Radiographs show widened epiphyses of the phalanges (Figure 1a). Because her sister was diagnosed a couple of years before with PPD, the diagnosis was obvious, although she exhibited a milder clinical presentation. At present, physical examination shows finger deformations; slightly limited motion of the shoulders, elbows, wrists and hips; and cervical impairment. She does not take any drugs and is well adapted to daily life.
Their parents, as well as their two older brothers, are healthy and do not suffer from the disease. There is no known consanguinity between the parents, but they both originated from the same village. Both sisters asked for genetic counselling and all individuals involved gave written informed consent according to the Helsinki declaration.
DNA extracted from peripheral white blood cells was obtained from both affected siblings and parents according to standard procedures. Molecular mutation analysis was performed and resulted in a repeated failure to amplify the first exon using the traditional PCR method. Using quantitative real-time PCR, as previously described,6 a homozygous deletion of the 5′UTR and the first exon of the WISP3 gene was detected (Figure 1b). We designed additional primers (Figures 1c and d; sequences available on request) and were able to delineate the deletion to a maximum of 9 kb extending into the 5′UTR. Parents are both heterozygous carriers of the deletion (Figure 1b). No mutation was detected in the amplified and sequenced exons 2–5 of the WISP3 gene.
In 1999, Hurvitz et al.7 showed for the first time an association between PPD and mutations in the CCN (for CTGF, cyr61/cef10, nov) family member WISP3 by using a positional-candidate approach. The CCN family encodes cysteine-rich secreted growth factors that regulate cell proliferation, differentiation, migration and adhesion. The high conservation of cysteines in the CNN proteins suggests that these residues are essential for their function.2,7 To date, more than 20 mutations in the WISP3 gene have been published. All of these mutations are loss-of-function mutations, including deletions, frame shifts and missense mutations.2 There is a large molecular spectrum of mutations, as mutations have been identified in all protein domains of WISP3, and no clear genotype–phenotype correlation has been established.2
Currently, little is known about the biological activity of WISP3. The level of expression of WISP3 is rather low.7 The transcript is detected mainly in mesenchymal cells and tissues such as chondrocytes, synoviocytes and bone marrow progenitor cells induced to undergo in vitro chondrogenesis.2,7 WISP3 appears to play a role in the expression of collagen type II, aggrecan and SOX9, which explains why PPD is characterised by an arthropathy primarily affecting the articular cartilage.8 It appears that WISP3 is a potential stimulator of anabolic pathways in cartilage. Because WISP3 depletion or overexpression fails to show a pathological phenotype in mice,9 zebrafish were used for further in vivo research. Overexpression of WISP3 in zebrafish led to the inhibition of BMP and WNT signalling, whereas depletion of WISP3 led to no or decreased inhibition of these pathways.10 Therefore, the dysregulation of BMP and/or WNT signalling may contribute to cartilage failure in PPD patients.
The mutation in the WISP3 gene was confirmed in our two patients. A homozygous deletion of the 5′UTR and the first coding exon of the WISP3 gene was detected. To the best of our knowledge, no large deletions in the WISP3 gene have been described previously. The deletion of the 5′ region will most likely prevent mRNA transcription and will therefore result in WISP3 insufficiency. The detection of the mutation in both sisters confirms the diagnosis of PPD and shows that both parents are most likely distant relatives.
References
References
el-Shanti HE, Omari HZ, Qubain HI . Progressive pseudorheumatoid dysplasia: report of a family and review. J Med Genet 1997; 34: 559–563.
Garcia Segarra N, Mittaz L, Campos-Xavier AB, Bartels CF, Tuysuz B, Alanay Y et al. The diagnostic challenge of progressive pseudorheumatoid dysplasia (PPRD): a review of clinical features, radiographic features, and WISP3 mutations in 63 affected individuals. Am J Med Genet C Semin Med Genet 2012; 160C: 217–229.
Spranger J, Albert C, Schilling F, Bartsocas C, Stoss H . Progressive pseudorheumatoid arthritis of childhood (PPAC). A hereditary disorder simulating rheumatoid arthritis. Eur J Pediatr 1983; 140: 34–40.
Wynne-Davies R, Hall C, Ansell BM . Spondylo-epiphysial dysplasia tarda with progressive arthropathy. A ‘new’ disorder of autosomal recessive inheritance. J Bone Joint Surg 1982; 64: 442–445.
Delague V, Chouery E, Corbani S, Ghanem I, Aamar S, Fischer J et al. Molecular study of WISP3 in nine families originating from the Middle-East and presenting with progressive pseudorheumatoid dysplasia: identification of two novel mutations, and description of a founder effect. Am J Med Genet 2005; 138A: 118–126.
Van Esch H, Bauters M, Ignatius J, Jansen M, Raynaud M, Hollanders K et al. Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am J Hum Genet 2005; 77: 442–453.
Hurvitz JR, Suwairi WM, Van Hul W, El-Shanti H, Duperti-Fuga A, Roudier J et al. Mutations in the CCN gene family member WISP3 cause progressive pseudorheumatoid dysplasia. Nat Genet 1999; 23: 94–98.
Sen M, Cheng YH, Goldring MB, Lotz MK, Carson DA . WISP3-dependent regulation of type II collagen and aggrecan production in chondrocytes. Arthritis Rheum 2004; 50: 488–497.
Kutz WE, Gong Y, Warman ML . WISP3, the gene responsible for the human skeletal disease progressive pseudorheumatoid dysplasia, is not essential for skeletal function in mice. Mol Cell Biol 2005; 25: 414–421.
Nakamura Y, Weidinger G, Liang JO, Aquilina-Beck A, Tamai K, Moon RT et al. The CCN family member Wisp3, mutant in progressive pseudorheumatoid dysplasia, modulates BMP and Wnt signaling. J Clin Invest 2007; 117: 3075–3086.
Data Citations
Van Esch, H HGV Database (2015) http://dx.doi.org/10.6084/m9.figshare.hgv.726
Acknowledgements
We would like to thank the family for their cooperation. BN received a PhD fellowship of the FWO Flanders. HVE is a senior clinical investigator of the FWO Flanders.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Neerinckx, B., Thues, C., Wouters, C. et al. A homozygous deletion of exon 1 in WISP3 causes progressive pseudorheumatoid dysplasia in two siblings. Hum Genome Var 2, 15049 (2015). https://doi.org/10.1038/hgv.2015.49
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2015.49
This article is cited by
-
A retrospective study of nine patients with progressive pseudorheumatoid dysplasia: to explore early diagnosis and further treatment
Clinical Rheumatology (2022)
-
Progressive Pseudorheumatoid Dysplasia resolved by whole exome sequencing: a novel mutation in WISP3 and review of the literature
BMC Medical Genetics (2019)
-
Progressive pseudorheumatoid dysplasia: a rare childhood disease
Rheumatology International (2019)
