
Abstract
Von Hippel–Lindau (VHL) disease is an inherited tumor syndrome caused by germline mutations in the VHL tumor suppressor gene. It is characterized by hemangioblastoma in the central nervous system and retina, renal cell carcinoma, pancreatic tumor and cysts, and pheochromocytoma. In this study, we detected 26 germline mutations in the VHL gene of Korean patients, of which 1 was a novel mutation, c.417_418insT. We also integrated our data from this study with the published literature to identify 55 VHL germline mutations in Koreans, and identified a unique hotspot at codon 70. Nine unrelated patients (9/55, 16.4%) had the same amino-acid substitution at codon 70 (Glu70Lys) and showed VHL type 1 phenotypes. Although this mutation was shown to have a mild effect on VHL function, four of the nine patients (44.4%) subsequently developed multiple central nervous system hemangioblastomas or retinal hemangioblastoma. However, this hotspot has not been identified in Chinese or Japanese patients. This study provides information on the spectrum of VHL mutations in Korean VHL disease and contributes to a better understanding of VHL disease in terms of improvements in the clinical management of VHL families.
Similar content being viewed by others
Introduction
Von Hippel–Lindau (VHL) disease is an inherited tumor syndrome predisposing to a variety of benign and malignant tumors and cysts. Typical VHL tumors are hemangioblastoma (HAB) in the central nervous system (CNS) and retina, renal cell carcinoma (RCC), pheochromocytoma, endolymphatic sac tumors and islet cell tumors or multiple cysts in the pancreas.1 CNS HABs and RCCs are considered the major causes of morbidity and mortality. From the aspect of genetic pathogenesis, VHL disease is caused by mutations in the VHL tumor suppressor gene that is located on chromosome 3p25-26. It is inherited in an autosomal dominant manner with an extremely high penetrance rate, as 97% of carriers who have the VHL disease-causing mutation develop clinical symptoms by the age of 60.2, 3
Identification of pathological genetic mutations is helpful in determining an accurate presymptomatic diagnosis. Furthermore, studies on genotype–phenotype correlations for genetic disease are essential to provide management plans for patients and carriers. To date, the majority of guidelines for VHL disease have been provided from large clinical data sets of Western populations, which have shown some genotype–phenotype correlations and patterns of time course in the development of clinical symptoms.4, 5, 6 However, there are distinct differences in genotype–phenotype characteristics of genetic diseases according to ethnicity. For example, Yoshida et al.7 found that the incidence of nonsense mutations and large deletions among Japanese VHL patients was significantly lower than that in Western VHL type 1 families. Wu et al.8 reported that a family history of VHL disease was uncommon in Chinese VHL patients. However, there have been a few studies from Korea showing VHL gene mutations, most of which are case reports or series with a limited number of patients.
To determine the nature of VHL germline mutations in Koreans, we confirmed 26 VHL germline mutations in the present study and, together with 29 VHL germline mutations previously reported in Korea,9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21 analyzed the molecular patterns of 55 VHL gene mutations, including detection rates and so-called ‘mutation hotspots’. We also compared the data with those from previously reported studies from other East Asian and Western countries. Because of the ethnic characteristics of Korea, which largely consists of a single ethnic group and is geographically isolated, this study could identify distinct molecular characteristics of VHL disease. This study forms the largest and most comprehensive analysis of Korean VHL disease ever performed.
Materials and methods
Patients
We retrospectively reviewed the medical records of 26 patients with germline mutations of the VHL gene who had been diagnosed with VHL disease in Severance Hospital (Seoul, Republic of Korea) and Samsung Medical Center (Seoul, Republic of Korea) between 2003 and 2012. The criteria for diagnosis of VHL disease were: more than one HAB in the CNS or eye, a single HAB in the CNS or retina plus a visceral manifestation (multiple renal, pancreatic or hepatic cysts, pheochromocytoma and RCC) and positive family history plus any one of the above manifestations.1
We also extensively studied VHL germline mutations in Korean populations. We evaluated the extended data from 55 unrelated patients, 26 from the present study and 29 from previous reports from Korea,9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21 and compared the data with three studies from the literature that provided a spectrum of VHL gene mutations in Japan7, 22 and China.8 The data for the phenotype of 17 patients previously reported were also reviewed for this study.11, 16 We also compared the data from East Asian countries with those from Western countries, including North America and Europe.7, 23, 24 With regard to its clinical manifestations, VHL is classified into two types: type 1 is without pheochromocytoma and type 2 is with pheochromocytoma.25 In addition, type 2 VHL disease is subdivided into three categories depending on the absence of RCC (type 2A), presence of RCC (type 2B) and sole manifestation of pheochromocytoma without HAB or RCC (type 2C).26
DNA sequencing
Genomic DNA was purified from peripheral blood leukocytes using a QIAamp DNA Blood Mini Kit (Qiagen GmbH, Hilden, Germany) according to the manufacturer’s instructions. The three exons of the VHL gene as well as their flanking introns were amplified using the following primers: 1F 5′-CGCGAAGACTACGGAGGT-3′ and 1R 5′-CTTCAGACCGTGCTATCGTC-3′; 2F 5′-CTCCCAAAGTGCTGGGATTA-3′ and 2R 5′-TGGGCTTAATTTTTCAAGTGG-3′; 3F 5′-AGTTGTTGGCAAAGCCTCTT-3′ and 3R 5′-AAGGAAGGAACCAGTCCTGT-3′. We confirmed mutations in both strands by direct sequencing with the primers that were also used in the PCR. PCR was carried out using a thermal cycler (Model 9700; Applied Biosystems, Foster City, CA, USA) as follows: 35 cycles of denaturation at 94 °C for 30 s, annealing at 60 °C for 30 s, and extension at 72 °C for 1 min. The amplicon was purified using Agencourt AMPure XP (Beckman Coulter Genomics, Danvers, MA, USA) and direct sequencing was performed using the BigDye Terminator Cycle Sequencing Ready Reaction kit on an ABI Prism 3130 genetic analyzer (Applied Biosystems). All variants were confirmed based on the National Center for Biotechnology Information single-nucleotide polymorphism database and the Human Gene Mutation Database. The method for detection of VHL germline mutations, including that for multiple ligation-dependent probe amplification, used in Samsung Hospital was previously reported.11
Statistics
For each phenotype of interest (CNS HAB, RCC, pheochromocytoma and pancreatic cyst or tumor), the proportions affected within different mutation classes were compared by means of a standard χ2-test or Fisher’s exact test. We separately analyzed the mutation rate of VHL for large deletions (4–380 kb) and point mutations because the frequency of the tests is different. A value of P<0.05 was considered significant. Analyses were performed using SPSS v18.0 software (SPSS, Inc., Chicago, IL, USA).
Results
In the present study, we confirmed 26 VHL germline mutations (bold cases in Table 1) by direct sequencing. We combined these data with an extensive analysis of Korean VHL gene mutations noted in previous reports. There were 41 different mutations in 55 unrelated patients with VHL disease in Korea (Table 1), and the mutations were widely distributed over the three exons of VHL gene (Figure 1). The majority of germline VHL mutations (50/55) were intragenic small alterations identified by direct sequencing, including 36 missense mutations, 4 nonsense mutations, 4 microdeletions, 3 microinsertions and 3 splice-site mutations, and 5 were large deletions detected by multiple ligation-dependent probe amplification. Of 55 unrelated patients, 44 (80%) patients were categorized as VHL type 1 (without pheochromocytoma) and 11 (20%) as type 2 (with pheochromocytoma) (Table 1). Notably, the frequency of missense mutations was significantly higher in VHL type 2 patients (P=0.01, Figure 2).

Summary of the germline mutations including point mutations and small insertions/deletions in the VHL gene from 50 unrelated Korean patients with VHL disease. Five cases with large deletions are not included in this figure. Location and codon are shown. The bars on the upper part of the exon consist of VHL type 1 and those on the lower part consist of VHL type 2. *Mutations reported as novel in Korea. aOne patient has concurrent missense and splicing mutations (208G>A and IVS1+5G>C). bMutations showed different phenotypes as type 2A in patients and type 2C in affected family members. •Known hotspot at codon 167.28

Summary of genotype–phenotype correlations in 55 unrelated Korean patients with VHL disease. Of 55 unrelated VHL patients, 44 (77%) patients were categorized as VHL type 1 (without pheochromocytoma) and 11 (20%) as VHL type 2 (with pheochromocytoma). Forty-four germline mutations in VHL type 1 consisted of 25 missense, 4 nonsense, 4microdeletion, 3 insertion, 3 splice-site and 5 large deletion mutations, but all mutations in VHL type 2 were missense mutations. When we classified the mutations into two groups, missense substitutions and non-missense mutations (nonsense, insertion/deletion and splice-site), we found that all VHL type 2 patients had missense mutations (P=0.010).
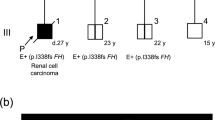
All intragenic mutations were located between codons 58 and 198. Similar to previous reports,23, 27 no mutations were identified in the first 57 or the last 13 codons of the VHL gene (Figure 1). Two regions of frequent germline mutations in the VHL gene were between codons 70–88 and between codons 157–167. Among these mutations, the most common germline mutation in Korean patients was Glu70Lys (found in nine unrelated patients, 16.4%). Of interest, all of these nine patients showed a limited range of VHL type 1 phenotypes: four patients had a single CNS HAB or retinal HAB alone (cases 3 and 4 in Table 1) or combined with renal cysts (cases 5 and 6), four patients had VHL disease with multiple CNS or retinal HABs (cases 7–10) and one patient who showed RCC and retinal HAB had two mutations: both Glu70Lys and a splicing mutation at intron 1 (case 24 in Table 1).
In Korea, the prevalence of codon 70 mutations seems to be higher than that of mutations in codon 167, which was previously known as the ‘mutation hotspot’ of the VHL gene.28 However, the frequency of codon 167 mutations was constant, appearing in 5.5% of Korean VHL patients in this study and nearly 7% of VHL families in Japan, China, France and the United Kingdom.8, 14 In addition, we identified a novel mutation in the Korean population: c.417_418insT, pLeu140fs. This novel frameshift mutation in codon 140, which changes the amino acid from leucine to serine and introduces a premature stop at codon 143, leading to a truncated VHL protein, resulted in a type 1 phenotype (case 33 in Table 1). Patients with the novel mutation had no family history of VHL disease, indicating that this may be a de novo mutation.
Discussion
In this study, we identified a unique hotspot for VHL germline mutations at codon 70 (Glu70Lys) that had a frequency of 16.4% in Korean VHL patients. This rate is similar to that of already known hotspots of the VHL gene including Leu178, Cys162, Arg167, Asn78, Pro86 and Tyr98, which showed a frequency of 3–17%.28 Thus, Glu70Lys may be a mutation hotspot in Korean VHL patients. This missense mutation seems to be associated with limited manifestations of HABs in CNS and retina. The VHL protein (pVHL) has a critical role in tumorigenesis, and mutations in specific domains of the VHL gene correlate with specific phenotypes.29 The mutations that result in the disruption of hypoxia-inducible factor (HIF)-1α binding, directly or through destabilization of the binding domain, usually cause HAB in the CNS, retinal HAB and RCC.29
According to the VHL databases and other studies, mutations in codon 70 have been described in four cases: Glu70Lys and Glu70Stop were each described in two cases, and all four cases showed a type 1 VHL phenotype.5, 23, 24 Glu70Lys was reported in two patients with a retinal HAB.5, 24 As Glu70Lys lies on the pVHL L1 loop linker region in the β-domain distal to the HIF-1α-binding pocket,28 the functional impact of this mutation on tumorigenesis is doubtful. However, Miller et al.30 suggested a significant role for the L1 loop residues in coordinating dynamic motions that are required for the specific interaction between pVHL and HIF-1α. Glu70Lys in the L1 loop results in a modest reduction of binding affinity between pVHL and HIF-1α.30 This mutation lies in the first part of the translated area of the VHL gene, and thus may possibly have a mild effect on the protein.30 According to a structural mutation analysis using a bioinformatics prediction method, Glu70Lys was predicted to act as a destabilizing mutation in the HIF-1α-binding domain, but the effect of this mutation was too mild to disrupt HIF binding, which might lead to RCC.29 Therefore, the mild effect of Glu70Lys could lead to the development of only a single HAB. However, in the present study, the clinical features associated with this mutation varied from single to multiple HABs, and 44% of patients (4/9) with this mutation subsequently developed a phenotype with multiple HABs in the CNS or retina with/without minor visceral manifestations. This missense mutation in codon 70 seems to be associated in Korean patients with sporadic VHL disease with various manifestations of HABs from single to multiple HABs in the CNS or retina, but not with pheochromocytoma, RCC or pancreatic tumors.
One patient (case 24 in Table 1), who had concurrent Glu70Lys and an intronic mutation with c.340+5G>C, showed clinical manifestations with retinal HAB as well as unilateral RCC. In most patients, Glu70Lys seems to be associated with HAB and a limited phenotype of VHL disease, thus the other manifestations including RCC in this case are possibly associated with an additional effect of c.340+5G>C on pVHL. This intronic mutation was previously suggested as a benign polymorphism that caused no major changes in the splicing pattern in in silico analysis.31, 32 However, in our study, c.340+5G>C was found in familial VHL disease (case 25 in Table 1) and two of this patient’s family members had multiple RCCs. Therefore, this intronic mutation could be related to the phenotype of RCCs in VHL disease, a relationship that was also previously described in a sporadic conventional RCC.33
Generally, it is known that most VHL type 1 patients have deletions or premature truncation mutations, whereas most VHL type 2 families are associated with missense mutations. Similarly, we also found that missense mutations are significantly associated with VHL type 2 patients in this study. However, we identified that the proportion of missense mutations was higher in Asian VHL type 1 (104/174, 60% in Table 2) compared with that in Western countries (119/248, 48%) (P=0.017). One possible explanation for this result might be that the prevalence of large deletions was low in Asian VHL type 1 patients. However, when we considered the number of tests for large deletions, the rate of large deletions (4–380 kb) in East Asian type 1 VHL patients was similar to that in Western type 1 VHL patients (Table 2). The prevalence of large deletions is 11–40% in VHL patients in general.11, 34, 35, 36, 37, 38 In Korean patients, large deletions were detected in 5 VHL type 1 patients of 15 tested, indicating an overall detection rate of 33.3% (5/15).11 Therefore, the presence of large deletions should be considered in mutation-negative patients.
Correlation of point mutations with phenotypes in different countries indicated that the major pattern of VHL mutations was consistent between East Asian countries. However, there were different correlations within countries (Table 2). The rate of nonsense mutations in Chinese VHL type 1 patients was significantly higher than that in Korean or Japanese patients (12/49, P=0.013 vs 4/44 Korean, P<0.001 vs 0/41 Japanese, respectively). The microdeletion rate was also higher in type 1 VHL Japanese patients compared with the other two countries (12/41, P=0.011 vs 4/44 Korean, P=0.009 vs 3/49 Chinese, respectively). Notably, the rates of nonsense and microdeletions in Korean VHL type 1 patients were in between those of Chinese and Japanese VHL 1 patients, indicating that this genetic pattern may be because these populations are closely related and live in the same geographical area, which could influence gene flow from neighboring countries.
Genetic analysis is critical for determining the presence of mutations in asymptomatic family members and allows the development of surveillance protocols followed by early intervention, which may improve prognosis. However, to truly benefit from regular tumor surveillance, we should interpret these results based on molecular characteristics determined by distinct ethnic backgrounds. Herein, we demonstrated the spectrum of VHL germline mutations in Korean VHL patients and identified a unique hotspot at codon 70. Even though the genotype–phenotype correlations were similar in different countries, we may need to consider the detection of large deletions of VHL, which have a relatively high mutation rate in East Asian countries. Further studies will help confirm the molecular profiles of patients and assist in genetic counseling and screening programs for better outcomes in countries with different ethnic backgrounds.
References
Maher, E. R. & Kaelin, W. G. von Hippel–Lindau disease. Medicine 76, 381–391 (1997).
Latif, F., Tory, K., Gnarra, J., Yao, M., Duh, F. M., Orcutt, M. L. et al. Identification of the von Hippel–Lindau disease tumor suppressor gene. Science 260, 1317–1320 (1993).
Maher, E. R., Yates, J. R., Harries, R., Benjamin, C., Harris, R., Moore, A. T. et al. Clinical features and natural history of von Hippel–Lindau disease. Q. J. Med. 77, 1151–1163 (1990).
Nielsen, S. M., Rubinstein, W. S., Thull, D. L., Armstrong, M. J., Feingold, E., Stang, M. T. et al. Genotype–phenotype correlations of pheochromocytoma in two large von Hippel–Lindau (VHL) type 2A kindreds with different missense mutations. Am. J. Med. Genet. A 155A, 168–173 (2011).
Mettu, P., Agron, E., Samtani, S., Chew, E. Y. & Wong, W. T. Genotype–phenotype correlation in ocular von Hippel–Lindau (VHL) disease: the effect of missense mutation position on ocular VHL phenotype. Invest. Ophthalmol. Vis. Sci. 51, 4464–4470 (2010).
Huang, J. S., Huang, C. J., Chen, S. K., Chien, C. C., Chen, C. W. & Lin, C. M. Associations between VHL genotype and clinical phenotype in familial von Hippel–Lindau disease. Eur. J. Clin. Invest. 37, 492–500 (2007).
Yoshida, M., Ashida, S., Kondo, K., Kobayashi, K., Kanno, H., Shinohara, N. et al. Germ-line mutation analysis in patients with von Hippel–Lindau disease in Japan: an extended study of 77 families. Jpn J. Cancer Res. 91, 204–212 (2000).
Wu, P., Zhang, N., Wang, X., Ning, X., Li, T., Bu, D. et al. Family history of von Hippel–Lindau disease was uncommon in Chinese patients: suggesting the higher frequency of de novo mutations in VHL gene in these patients. J. Hum. Genet. 57, 238–243 (2012).
Kim, J. H., Cheon, S. H., Oh, C. S., Kim, Y. M. & Park, R. J. Cystic renal cell carcinoma associated with von Hippel–Lindau disease treated by nephron sparing Surgery. Korean J. Urol. 46, 93–95 (2005).
Sohn, B. S., Cho, C. E. & Park, I. Y. Pancreatic cyst in von Hippel–Lindau disease. J. Korean Surg. Soc. 77, 287–290 (2009).
Cho, H. J., Ki, C. S. & Kim, J. W. Improved detection of germline mutations in Korean VHL patients by multiple ligation-dependent probe amplification analysis. J. Korean Med. Sci. 24, 77–83 (2009).
Kim, H. S., Han, B. H., Kim, S. J., Lee, J. S., Park, S. J., Lee, S. U. et al. A case of pancreatic dual Wirsung ducts in a patient with von Hippel–Lindau disease. Korean J. Gastrointest. Endosc. 37, 384–388 (2008).
Seo, J. H., Yang, J. H., Choi, P. L., Kim, Y. L., Choi, Y. S., Park, Y. H. et al. A case of type 1 von Hippel–Lindau (VHL) disease associated with VHL germline mutation. J. Korean Soc. Endocrinol. 21, 239–244 (2006).
Kang, H. C., Kim, I. J., Park, J. H., Shin, Y., Jang, S. G., Ahn, S. A. et al. Three novel VHL germline mutations in Korean patients with von Hippel–Lindau disease and pheochromocytomas. Oncol. Rep. 14, 879–883 (2005).
Kim, J. W., Choi, S. J., Kim, Y. K., Ahn, S. M., Song, K. E., Jung, S. H. et al. A case report of von Hippel–Lindau disease manifested in a monozygous twin. J. Korean Soc. Endocrinol. 20, 395–400 (2005).
Kim, W. T., Ham, W. S., Ju, H. J., Lee, J. S. & Choi, Y. D. Clinical characteristics of renal cell carcinoma in Korean patients with von Hippel–Lindau disease compared to sporadic bilateral or multifocal renal cell carcinoma. J. Korean Med. Sci. 24, 1145–1149 (2009).
Lee, S. H., Park, B. J., Kim, T. S. & Lim, Y. J. Long-term follow-up clinical courses of cerebellar hemangioblastoma in von Hippel–Lindau disease: two case reports and a literature review. J. Korean Neurosurg. Soc. 48, 263–267 (2010).
Myong, N. H. & Park, B. J. Malignant glioma arising at the site of an excised cerebellar hemangioblastoma after irradiation in a von Hippel–Lindau disease patient. Yonsei Med. J. 50, 576–581 (2009).
Shin, K. H., Park, K. J., Kim, S. W., Lee, S. H., Lee, S.-E., Jung, H.-W. et al. Germline mutations of VHL gene in Korean von Hippel–Lindau disease patients. J. Korean Cancer 28, 544–554 (1996).
Yang, J. H., Choi, Y. S., Park, Y. H., Oh, K. S., Chun, B. K., Lee, S. J. et al. A case of von Hippel–Lindau (VHL) germline mutation associated with familial bilateral pheochromocytoma in the VHL disease. Korean J. Med 69, S873–S878 (2005).
Ohn, J. H., Kim, J., Lee, H. J., Seo, W. W., Hwang-Bo, Y., Hong, E. S. et al. A case of von Hippel–Lindau disease presenting with pancreatic neuroendocrine tumor. Endocrinol. Metab. 26, 89–91 (2011).
Clinical Research Group for VHL in Japan. Germline mutations in the von Hippel–Lindau disease (VHL) gene in Japanese VHL. Hum. Mol. Genet. 4, 2233–2237 (1995).
Zbar, B., Kishida, T., Chen, F., Schmidt, L., Maher, E. R., Richards, F. M. et al. Germline mutations in the Von Hippel–Lindau disease (VHL) gene in families from North America, Europe, and Japan. Hum. Mutat. 8, 348–357 (1996).
Olschwang, S., Richard, S., Boisson, C., Giraud, S., Laurent-Puig, P., Resche, F. et al. Germline mutation profile of the VHL gene in von Hippel–Lindau disease and in sporadic hemangioblastoma. Hum. Mutat. 12, 424–430 (1998).
Chen, F., Kishida, T., Yao, M., Hustad, T., Glavac, D., Dean, M. et al. Germline mutations in the von Hippel–Lindau disease tumor suppressor gene: correlations with phenotype. Hum. Mutat. 5, 66–75 (1995).
Ritter, M. M., Frilling, A., Crossey, P. A., Hoppner, W., Maher, E. R., Mulligan, L. et al. Isolated familial pheochromocytoma as a variant of von Hippel–Lindau disease. J. Clin. Endocrinol. Metab. 81, 1035–1037 (1996).
Crossey, P. A., Richards, F. M., Foster, K., Green, J. S., Prowse, A., Latif, F. et al. Identification of intragenic mutations in the von Hippel–Lindau disease tumour suppressor gene and correlation with disease phenotype. Hum. Mol. Genet. 3, 1303–1308 (1994).
Stebbins, C. E., Kaelin, W. G. Jr & Pavletich, N. P. Structure of the VHL–ElonginC–ElonginB complex: implications for VHL tumor suppressor function. Science 284, 455–461 (1999).
Forman, J. R., Worth, C. L., Bickerton, G. R., Eisen, T. G. & Blundell, T. L. Structural bioinformatics mutation analysis reveals genotype–phenotype correlations in von Hippel–Lindau disease and suggests molecular mechanisms of tumorigenesis. Proteins 77, 84–96 (2009).
Miller, F., Kentsis, A., Osman, R. & Pan, Z. Q. Inactivation of VHL by tumorigenic mutations that disrupt dynamic coupling of the pVHL–hypoxia-inducible transcription factor-1alpha complex. J. Biol. Chem. 280, 7985–7996 (2005).
Erlic, Z., Hoffmann, M. M., Sullivan, M., Franke, G., Peczkowska, M., Harsch, I. et al. Pathogenicity of DNA variants and double mutations in multiple endocrine neoplasia type 2 and von Hippel–Lindau syndrome. J. Clin. Endocrinol. Metab. 95, 308–313 (2010).
Dandanell, M., Friis-Hansen, L., Sunde, L., Nielsen, F. C. & Hansen, T. V. Identification of 3 novel VHL germ-line mutations in Danish VHL patients. BMC Med. Genet. 13, 54 (2012).
Young, A. C., Craven, R. A., Cohen, D., Taylor, C., Booth, C., Harnden, P. et al. Analysis of VHL gene alterations and their relationship to clinical parameters in sporadic conventional renal cell carcinoma. Clin. Cancer Res. 15, 7582–7592 (2009).
Stolle, C., Glenn, G., Zbar, B., Humphrey, J. S., Choyke, P., Walther, M. et al. Improved detection of germline mutations in the von Hippel–Lindau disease tumor suppressor gene. Hum. Mutat. 12, 417–423 (1998).
Rocha, J. C., Silva, R. L., Mendonca, B. B., Marui, S., Simpson, A. J. & Camargo, A. A. High frequency of novel germline mutations in the VHL gene in the heterogeneous population of Brazil. J. Med. Genet. 40, e31 (2003).
Cybulski, C., Krzystolik, K., Murgia, A., Gorski, B., Debniak, T., Jakubowska, A. et al. Germline mutations in the von Hippel–Lindau (VHL) gene in patients from Poland: disease presentation in patients with deletions of the entire VHL gene. J. Med. Genet. 39, E38 (2002).
Zhang, J., Huang, Y., Pan, J., Liu, D., Zhou, L., Xue, W. et al. Germline mutations in the von Hippel–Lindau disease (VHL) gene in mainland Chinese families. J. Cancer Res. Clin. Oncol. 134, 1211–1218 (2008).
Siu, W. K., Ma, R. C., Lam, C. W., Mak, C. M., Yuen, Y. P., Lo, F. M. et al. Molecular basis of von Hippel–Lindau syndrome in Chinese patients. Chin. Med. J. (Engl) 124, 237–241 (2011).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Hwang, S., Ku, C., Lee, J. et al. Germline mutation of Glu70Lys is highly frequent in Korean patients with von Hippel–Lindau (VHL) disease. J Hum Genet 59, 488–493 (2014). https://doi.org/10.1038/jhg.2014.61
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2014.61
This article is cited by
-
Von Hipple-Lindau disease complicated with central retinal vein occlusion: a case report
BMC Ophthalmology (2022)
-
Genotype phenotype correlation in Asian Indian von Hippel–Lindau (VHL) syndrome patients with pheochromocytoma/paraganglioma
Familial Cancer (2018)
-
Genotype-phenotype analysis of von Hippel-Lindau syndrome in Korean families: HIF-α binding site missense mutations elevate age-specific risk for CNS hemangioblastoma
BMC Medical Genetics (2016)
