
Abstract
Steroid-resistant nephrotic syndrome (SRNS) represents glomerular disease resulting from a number of different etiologies leading to focal segmental glomerulosclerosis (FSGS). Recently, many genes causing SRNS/FSGS have been identified. These genes encode the proteins associated with the formation and/or maintenance of glomerular filtration barrier. Next-generation sequencing is used to analyze large numbers of genes at lower costs. To identify the genetic background of Japanese patients, we studied 26 disease-causing genes using whole-exome sequencing analysis in 24 patients with SRNS and/or FSGS from 22 different Japanese families. We finally found eight causative gene mutations, four recessive and four dominant gene mutations, including three novel mutations, in six patients from five different families, and one novel predisposing mutation in two patients from two different families. Causative gene mutations have only been identified in ~20% of families and further analysis is necessary to identify the unknown disease-causing gene. Identification of the disease-causing gene would support clinical practices, including the diagnosis, understanding of pathogenesis and treatment.
Similar content being viewed by others


Introduction
Idiopathic nephrotic syndrome (NS) is a kidney disorder characterized by high excretion of protein in the urine, hypoalbuminemia and edema. In Europe and the United States, 2 in 100 000 children will develop idiopathic NS in a single year.1, 2 Approximately 80% of patients are steroid responsive and have a favorable prognosis, and the other 20% of children with NS are resistant to steroid therapy and have an unfavorable prognosis.2, 3 Steroid-resistant NS (SRNS) represents glomerular disease resulting from a number of different etiologies leading to focal segmental glomerulosclerosis (FSGS).4
Recently, many genes causing SRNS/FSGS have been identified. These genes encode the proteins associated with formation and/or maintenance of the glomerular filtration barrier.5, 6, 7, 8, 9, 10 Genetic backgrounds of SRNS/FSGS may be different in each ethnic group. There are currently no detailed reports of genotype–phenotype correlations, including the responsibility to the ordinal treatment, complication, prognosis and post-transplant recurrence of SRNS/FSGS. These data will provide useful information for clinical practice and genetic counseling.11 Next-generation sequencing is used to analyze a large number of genes associated with SRNS/FSGS and can be carried out at lower costs compared with the individual sequence analysis of many genes.12 To identify the genetic background of Japanese patients, we studied 26 disease-causing genes using whole-exome sequencing analysis in 24 patients with SRNS and/or FSGS from 22 different families. We finally identified eight causative gene mutations in six patients from five different families and one predisposing gene mutation in two patients from two different families.
Materials and methods
Subjects
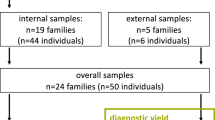
We studied 24 patients with SRNS and/or FSGS (male-to-female ratio, 12:12; ages from 2 to 42 years; median age, 12 years) from 22 different Japanese families (Supplementary Table 1). The institutional review board of the Yamagata University School of Medicine study approved this study, and written informed consent was obtained from the patients or parents of all children. The definitions and criteria for NS and steroid resistance were those used by the International Study of Kidney Disease in Children.13, 14 NS was defined as heavy proteinuria (urinary protein excretion ⩾40 mg m−2 h−1) with hypoalbuminemia ⩽25 g l−1. Steroid resistance was defined as a failure to achieve a response despite 4 weeks of prednisolone therapy (2 mg kg−1 per day prednisolone).
Molecular analysis
We extracted genomic DNA from peripheral blood using a standard method.
We analyzed all the exons of 26 genes: NPHS1, NPHS2, NPHS3 (PLCE1), CD2AP, TRPC6, ACTN4, MYH9, ARHGAP24, MYO1E, IFN2, WT1, LMX1B, SMARCAL1, LAMB2, MT-TL1, COQ6, COQ2, PDSS2, SCARB2, ZMPSTE24, PMM2, ALG1, PTPRO, GPC5, APOL1 and ITGB4,9, 10, 11, 15 using a next-generation sequencer. Exon capture was performed with the SureSelect Human All Exon Kit v.5 (Agilent Technologies, Santa Clara, CA, USA). Exon libraries were sequenced with the HiSeq 2000 platform (Illumina, San Diego, CA, USA) according to the manufacturer’s instructions. Paired 100-base pair reads were aligned to the reference human genome (UCSC hg19) using the Burrows–Wheeler Aligner (version 0.7.3a).16 Probably, PCR duplicates were removed with the Picard 1.88 (http://picard.sourceforge.net/). We picked up the sequences of 26 genes previously identified as the responsible genes for SRSN. Single-nucleotide variants and indels were identified using the Unified Genotyper module of the Genome Analysis Tool Kit (GATK) 2.5.2 (Broad Institute of MIT and Harvard, Cambridge, MA, USA),17 with the Best Practice Variant Detection with the GATK v.3 (https://www.broadinstitute.org/gatk/index.php). Single-nucleotide variants and indels were annotated against the set of RefSeq database, NHLBI-ESP 6500 exomes and dbSNP135 with the ANNOVAR (8 May 2013).18 We predicted the functional effects of mutations using two PolyPhen scores (HumDiv and HumVar models) provided by PolyPhen-2 version 2.2.2,19 Grantham,20 PhastCons21 and GERP22 scores provided by SeattleSeq SNP annotation 137 (http://snp.gs.washington.edu/SeattleSeqAnnotation137/). We performed direct sequencing to confirm the mutations identified by exome sequencing in the patients and some of family members and to find additional gene mutations detected in cases 10, 11, 12 and 15. PCR was performed with the primers designed based on the genome database. The sequence reactions were analyzed on an ABI PRISM 3100 Genetic Analyzer (PE Applied Biosystems, Foster City, CA, USA) with the BigDye Terminator Cycle Sequencing Ready Reaction Kit (PE Applied Biosystems).
Results
We studied 26 disease-causing genes using whole-exome sequencing analysis in 24 patients with SRNS and/or FSGS from 22 different families and found 10 nucleotide changes in eight genes (Supplementary Table 1 and Table 1). Based on the database and functional prediction analyses, we finally identified eight causative gene mutations in six patients from five different families and one predisposing gene mutation in two patients from two different families (Table 2).
For the genes of recessive types, we detected a heterozygous NPHS2 p.R168H mutation23 in Case 15 and a heterozygous LAMB2 c.1405+1g>a mutation (splicing mutation)24 in Case 12 (Table 2). We also detected three polymorphic nucleotide changes as follows: PTPRO p.S338F (rs.200478856), NPHS1 p.A219V (rs.757417823) and NPHS2 p. Q287R (rs.200042397). Direct sequencing revealed that Cases 15 and 12 were a compound heterozygote of NPHS2 p.[R138X];[R168H] mutation25, 26 and a compound heterozygote of LAMB2 p.[abnormal splicing];[G699R] mutation, respectively. Case 15 presented as congenital NS, and steadily progressed to end-stage renal failure (ESRF) at age 11 years. Case 12 manifested the symptoms in infantile period and progressed rapidly to ESRF at age 2 years, but he did not have any ocular symptoms. Cases 15 and 12 had no relapse of FSGS after renal transplantation.
In the analysis of the genes of dominant types, we found five gene mutations in six cases from five families. Cases 10 and 11 carrying a novel CD2AP p.R74M mutation were unrelated and presented different clinical features. Case 10 partially responded to steroid and cyclosporin A therapy; however, he progressed to ESRF. He received cadaveric renal transplantation, but had a recurrence of FSGS. Case 11 frequently relapsed, but he was responsive to therapy and had a complete remission. We performed direct sequencing of all coding regions of CD2AP in Cases 10 and 11, but we did not find any additional mutations.
Case 16 carrying a novel heterozygous TRPC6 p.E875V mutation presented FSGS at age 12 years, did not respond to steroid and cyclosporin A and progressed to ESRF. Her grandfather died of renal disease at age 30 years and her father presented proteinuria.
Case 19 was a daughter of Case 20, and Cases 19 and 20 presented the symptoms at school age and progressed to ESRF. They were complicated with bicornuate uterus and had a novel heterozygous WT1 p.[K141Q; P249T] mutation.
Case 24 carrying a heterozygous IFN2 p.E184K mutation presented FSGS at age 10 years, did not respond to steroid, cyclosporin A and plasma exchange and progressed to ESRF at age 16 years.27 He did not have any signs of Charcot-Marie-Tooth disease (CMT).
The patients carrying those dominant gene mutations manifested the symptoms in childhood period; they were older than the patients carrying the genes of recessive types. The patients carrying the mutations except CD2AP mutation did not respond to therapy and did not relapse FSGS after renal transplantation (Supplementary Table 1 and Table 2).
Discussion
We studied 26 disease-causing genes using whole-exome sequencing analysis in 24 patients with SRNS and/or FSGS from 22 different families and finally found eight causative gene mutations, four recessive and four dominant gene mutations, in six patients from five different families, and one predisposing mutation in two patients from two different families (Table 2). Recently, Sadowski et al. studied 1783 families with SRNS that manifested before 25 years of age and detected a single-gene cause in 526 families (29.5%).10 Our study was very small, but detected gene mutations in 22.7% of families.
Cases 15 and 12 carrying the recessive gene mutations manifested the symptoms during infantile period. Case 15 was a compound heterozygote of NPHS2 p.[R138X];[R168H] mutation25, 26 (Table 2). Podocin encoded by NPHS2 is expressed mainly in the podocytes and localized to the intercellular junction of the podocytes foot processes. Podocin is a lipid raft component of the slit diaphragm that is essential for the localization of nephrin and other components. Most missense mutations including the p.R168H mutation alter the trafficking to the plasma membrane25 and the p.R138X mutation was demonstrated to be present at the plasma membrane but fail to recruit nephrin in lipid rafts.26 Case 15 was resistant to therapy, progressed to ESRF and had no recurrence of FSGS after renal transplantation as reported previously.28 Case 12 was a compound heterozygote of LAMB2 c.[1405+1g>a];[2095g>c]; that is, p.[abnormal splicing];[G699R] mutation24 (Table 2). Laminins are the major noncollagenous constituent of basement membranes and laminin β-chain encoded by LAMB2, a component of laminins, is highly expressed in the basement membrane of muscles at the neuromuscular junctions, kidney glomerulus and vascular smooth muscle. Mutations with complete loss of function are associated with the features of Pierson syndrome and missense mutations cause mild and variable phenotypes.29 The homozygous c.[1405+1g>a] mutation was reported in the patient showing Pierson syndrome.24 The p.G699R is registered as a rare variation (frequency is 0.00274), but is predicted to have a deleterious functional effect using several prediction programs, PolyPhen-2 (HumDiv), MutationTaster score (data not shown) and MutationAssessor score (data not shown). These suggest that a compound heterozygous LAMB2 p.[abnormal splicing];[G699R] mutation is a causative mutation of FSGS.
For the dominant type, Cases 10 and 11 were heterozygous for CD2AP p.R74M mutation. CD2AP is an adapter protein with an SH3 domain, is expressed in lymphoid cells, and interacts with the T-cell adhesion protein CD2.30 CD2AP is also expressed in the slit diaphragm of the podocytes and interacts with podocin and nephrin to form a signaling complex.31, 32 Mice with null mutation die of massive proteinuria shortly after birth and mice with heterozygous null mutation developed glomerular change similar to FSGS at 9 months of age.30 For the human, only one case carrying homozygous p.R612X mutation was clearly demonstrated in association with renal disease.33 Several patients with heterozygous CD2AP mutations have been reported in association with adult onset FSGS.30, 34 However, segregation data was not available in all mutations and the heterozygous mutations were also detected even in unaffected individuals. Case 10 presented FSGS and relapsed FSGS after renal transplantation. In contrast, Case 11 frequently relapsed, but he was responsive to therapy and had a complete remission. In addition to their clinical difference, Cases 10 and 11 had no affected family members. These suggested that a heterozygous CD2AP p.R74M mutation is not a disease-causing mutation, but is a predisposing factor towards glomerular disease.
Case 16 had a novel heterozygous TRPC6 p.E875V mutation. TRPC6 is a cation channel and is widely distributed in the body including the podocytes cell bodies and the slit diaphragms. The majority of mutations associated with FSGS are gain-of-function mutations, which can increase intracellular calcium influx and lead to the apoptosis of the podocyte.35 Case 16 had similarly affected family members, but we could not confirm the linkage because their specimens were not available for analysis.
Cases 19 and 20 had heterozygous WT1 p.[K141Q; P249T] mutation. These mutations were located to exons 1 and 2. Nearly all mutations of WT1 associated with FSGS are located in exons 8 and 9. However, SNPs located in WT1 (promoter to exon 5) and WITI (gene immediately 5′ to WT1) were significantly associated with FSGS.36 and Gebeshuber et al.37 reported that transgenic expression of the microRNA miR-193a in mice rapidly induced FSGS via downregulation of WT1. Those reports suggest that a WT1 mutation located in other than exons 8 and 9 will cause FSGS. Cases 19 and 20 had bicornuate uterus as genitourinary malformations, which may be associated with the sexuality and/or the type of the WT1 mutation.38
Case 24 had a heterozygous IFN2 p.E184K mutation and was not complicated with CMT as reported previously.27 IFN2 is a member of the formin family and has a domain structure similar to the diaphanous formins: a diaphanous-inhibitory domain (DID), formin homology 1 and 2 domains and a diaphanous autoregulatory domain. IFN2 is strongly expressed in podocytes and Schwann cells. IFN2 mutations were detected in the patients with isolated FSGS and the patients with FSGS associated with CMT. All mutations are distributed in different parts of the DID; however, the mutations detected in patients with FSGS and CMT are located in the inner face of the central core of the DID.39, 40 The p.E184K mutation is located in the DID, but not in the inner face of the central core of the DID. The clinical phenotype of the IFN2 mutation may depend on the site and/or type of the mutation.
Detection of an SRSN-causing mutation will support clinical practices, including the diagnosis, understanding of pathogenesis and treatment. It will also avoid an unnecessary steroid therapy, introduce supplement therapy for the patients with a defect in CoQ biosynthesis41, 42 and provide a prenatal diagnosis in severe cases. However, causative gene mutations have been identified only in ~20% of families and further analysis is necessary to identify the unknown disease-causing gene.
References
Schlesinger, E. R., Sultz, H. A., Mosher, W. E. & Feldman, J. G. The nephrotic syndrome. Its incidence and implications for the community. Am. J. Dis. Child. 116, 623–632 (1968).
Niaudet, P., Boyer, O. in Pediatric Nephrology 6th edn (eds Avner, E., Harmon, W., Niaudet, P. & Yoshikawa, N.) 667–702 (Springer: New York, NY, USA, 2009).
International Study on Kidney Disease in Children The primary nephrotic syndrome in children. Identification of patients with minimal change nephrotic syndrome from initial response to prednisone. A report of the International Study of Kidney Disease in Children. J. Pediatr. 98, 561–564 (1981).
Banaszak, B. & Banaszak, P. The increasing incidence of initial steroid resistance in childhood nephrotic syndrome. Pediatr. Nephrol. 27, 927–932 (2012).
Kriz, W. The pathogenesis of ‘classic’ focal segmental glomerulosclerosis-lessons from rat models. Nephrol. Dial. Transplant 18, vi39–vi44 (2003).
Wharram, B. L., Goyal, M., Wiggins, J. E., Sanden, S. K., Hussain, S., Filipiak, W. E. et al. Podocyte depletion causes glomerulosclerosis: diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J. Am. Soc. Nephrol. 16, 2941–2952 (2005).
Sato, Y., Wharram, B. L., Lee, S. K., Wickman, L., Goyal, M., Venkatareddy, M. et al. Urine podocyte mRNAs mark progression of renal disease. J. Am. Soc. Nephrol. 20, 1041–1052 (2009).
Patrakka, J. & Tryggvason, K. New insights into the role of podocytes in proteinuria. Nat. Rev. Nephrol. 5, 463–468 (2009).
Gbadegesin, R., Lavin, P., Foreman, J. & Winn, M. Pathogenesis and therapy of focal segmental glomerulosclerosis: an update. Pediatr. Nephrol. 26, 1001–1015 (2011).
Sadowski, C. E., Lovric, S., Ashraf, S., Pabst, W. L., Gee, H. Y., Kohl, S. et al. A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J. Am. Soc. Nephrol. 26, 1279–1289 (2015); pii: ASN.2014050489.
Rood, I. M., Deegens, J. K. & Wetzels, J. F. Genetic causes of focal segmental glomerulosclerosis: implications for clinical practice. Nephrol. Dial. Transplant. 27, 882–890 (2012).
Brown, E. J., Pollak, M. R. & Barua, M. Genetic testing for nephrotic syndrome and FSGS in the era of next-generation sequencing. Kidney Int. 85, 1030–1038 (2014).
van Husen, M. & Kemper, M. J. New therapies in steroid-sensitive and steroid-resistant idiopathic nephrotic syndrome. Pediatr. Nephrol. 26, 881–892 (2011).
Tarshish, P., Tobin, J. N., Bernstein, J. & Edelmann, C. M. Jr. Prognostic significance of the early course of minimal change nephrotic syndrome: report of the International Study of Kidney Disease in Children. J. Am. Soc. Nephrol. 8, 769–776 (1997).
Saleem, M. A. New developments in steroid-resistant nephrotic syndrome. Pediatr. Nephrol. 28, 699–709 (2013).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 754–760 (2009).
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic. Acids Res. 38, e164 (2010).
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P. et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010).
Grantham, R. Amino acid difference formula to help explain protein evolution. Science 185, 862–864 (1974).
Siepel, A., Bejerano, G., Pedersen, J. S., Hinrichs, A. S., Hou, M., Rosenbloom, K. et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 15, 1034–1050 (2005).
Cooper, G. M., Stone, E. A., Asimenos, G. ; NISC Comparative Sequencing Program, Green, E. D., Batzoglou, S. & Sidow, A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 15, 901–913 (2005).
Weber, S., Gribouval, O., Esquivel, E. L., Morinière, V., Tête, M. J., Legendre, C. et al. NPHS2 mutation analysis shows genetic heterogeneity of steroid-resistant nephrotic syndrome and low post-transplant recurrence. Kidney Int. 66, 571–579 (2004).
Bredrup, C., Matejas, V., Barrow, M., Bláhová, K., Bockenhauer, D., Fowler, D. J. et al. Ophthalmological aspects of Pierson syndrome. Am. J. Ophthalmol. 146, 602–611 (2008).
Roselli, S., Moutkine, I., Gribouvalm, O., Benmerah, A. & Antignac, C. Plasma membrane targeting of podocin through the classical exocytic pathway: effect of NPHS2 mutations. Traffic 5, 37–44 (2004).
Huber, T. B., Simons, M., Hartleben, B., Sernetz, L., Schmidts, M., Gundlach, E. et al. Molecular basis of the functional podocin–nephrin complex: mutations in the NPHS2 gene disrupt nephrin targeting to lipid raft microdomains. Hum. Mol. Genet. 12, 3397–3405 (2003).
Brown, E. J., Schlöndorff, J. S, Becker, D. J, Tsukaguchi, H., Tonna, S. J., Uscinski, A. L. et al. Mutations in the formin gene INF2 cause focal segmental glomerulosclerosis. Nat. Genet. 42, 72–76 (2010).
Ruf, R. G., Lichtenberger, A., Karle, S. M., Haas, J. P., Anacleto, F. E., Schultheiss, M. et al. Patients with mutations in NPHS2 (podocin) do not respond to standard steroid treatment of nephrotic syndrome. J. Am. Soc. Nephrol. 15, 722–732 (2004).
Matejas, V., Hinkes, B., Alkandari, F., Al-Gazali, L., Annexstad, E., Aytac, M. B. et al. Mutations in the human laminin beta 2 (LAMB2) gene and the associated phenotypic spectrum. Hum. Mutat. 31, 992–1002 (2010).
Kim, J. M., Wu, H., Green, G., Winkler, C. A., Kopp, J. B., Miner, J. H. et al. CD2-associated protein haploinsufficiency is linked to glomerular disease susceptibility. Science 300, 1298–1300 (2003).
Schwarz, K., Simons, M., Reiser, J., Saleem, M. A., Faul, C., Kriz, W. et al. Podocin, a raft-associated component of the glomerular slit diaphragm, interacts with CD2AP and nephrin. J. Clin. Invest. 108, 1621–1629 (2001).
Wolf, G. & Stahl, R. A. CD2-associated protein and glomerular disease. Lancet 362, 1746–1748 (2003).
Löwik, M. M., Groenen, P. J., Pronk, I., Lilien, M. R., Goldschmeding, R., Dijkman, H. B. et al. Focal segmental glomerulosclerosis in a patient homozygous for a CD2AP mutation. Kidney Int. 72, 1198–1203 (2007).
Gigante, M., Pontrelli, P., Montemurno, E., Roca, L., Aucella, F., Penza, R. et al. CD2AP mutations are associated with sporadic nephrotic syndrome and focal segmental glomerulosclerosis (FSGS). Nephrol. Dial. Transplant. 24, 1858–1864 (2009).
Winn, M. P. 2007 Young Investigator Award: TRP’ing into a new era for glomerular disease. J. Am. Soc. Nephrol. 19, 1071–1075 (2008).
Orloff, M. S., Iyengar, S. K., Winkler, C. A., Goddard, K. A., Dart, R. A., Ahuja, T. S. et al. Variants in the Wilms' tumor gene are associated with focal segmental glomerulosclerosis in the African American population. Physiol. Genom. 21, 212–221 (2005).
Gebeshuber, C. A., Kornauth, C., Dong, L., Sierig, R., Seibler, J., Reiss, M. et al. Focal segmental glomerulosclerosis is induced by microRNA-193a and its downregulation of WT1. Nat. Med. 19, 481–487 (2013).
Niaudet, P. & Gubler, M. C. WT1 and glomerular diseases. Pediatr. Nephrol. 21, 1653–1660 (2006).
Boyer, O., Nevo, F., Plaisier, E., Funalot, B., Gribouval, O., Benoit, G. et al. INF2 mutations in Charcot–Marie–Tooth disease with glomerulopathy. N. Engl. J. Med. 365, 2377–2388 (2011).
Toyota, K., Ogino, D., Hayashi, M., Taki, M., Saito, K., Abe, A. et al. INF2 mutations in Charcot–Marie–Tooth disease complicated with focal segmental glomerulosclerosis. J. Peripher. Nerv. Syst. 18, 97–98 (2013).
Heeringa, S. F., Chernin, G., Chaki, M., Zhou, W., Sloan, A. J., Ji, Z. et al. COQ6 mutations in human patients produce nephritic syndrome with sensorineural deafness. J. Clin. Invest. 121, 2013–2024 (2011).
Montini, G., Malaventura, C. & Salviati, L. Early coenzyme Q10 supplementation in primary coenzyme Q10 deficiency. N. Engl. J. Med. 358, 2849–2850 (2008).
Acknowledgements
This research was supported in part by a Grant-in-Aid for Scientific Research (C) from the Ministry of Education, Science, Culture and Sports of Japan. We thank all patients and families who participated in this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Ogino, D., Hashimoto, T., Hattori, M. et al. Analysis of the genes responsible for steroid-resistant nephrotic syndrome and/or focal segmental glomerulosclerosis in Japanese patients by whole-exome sequencing analysis. J Hum Genet 61, 137–141 (2016). https://doi.org/10.1038/jhg.2015.122
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2015.122
This article is cited by
-
Precise clinicopathologic findings for application of genetic testing in pediatric kidney transplant recipients with focal segmental glomerulosclerosis/steroid-resistant nephrotic syndrome
Pediatric Nephrology (2023)
-
An inactivating human TRPC6 channel mutation without focal segmental glomerulosclerosis
Cellular and Molecular Life Sciences (2023)
-
Mutational landscape of TRPC6, WT1, LMX1B, APOL1, PTPRO, PMM2, LAMB2 and WT1 genes associated with Steroid resistant nephrotic syndrome
Molecular Biology Reports (2021)
-
The formin INF2 in disease: progress from 10 years of research
Cellular and Molecular Life Sciences (2020)
-
Pair analysis and custom array CGH can detect a small copy number variation in COQ6 gene
Clinical and Experimental Nephrology (2019)
